

Summary
Este es un método para generar virus de vacuna recombinante "sin cicatrices" utilizando la selección del rango de host y la identificación visual de virus recombinantes.
Abstract
El virus de la vacuna (VACV) fue fundamental en la erradicación del virus de la variola (VARV), el agente causante de la viruela, de la naturaleza. Desde su primer uso como vacuna, vacv se ha desarrollado como vector para vacunas terapéuticas y como virus oncolítico. Estas aplicaciones aprovechan el genoma fácilmente manipulado de VACV y la amplia gama de hosts como una plataforma excepcional para generar virus recombinantes con una variedad de aplicaciones terapéuticas. Se han desarrollado varios métodos para generar VACV recombinante, incluyendo métodos de selección de marcadores y selección dominante transitoria. Aquí, presentamos un refinamiento de un método de selección de rango de host junto con la identificación visual de virus recombinantes. Nuestro método aprovecha la presión selectiva generada por la proteína quinasa antiviral huésped R (PKR) junto con un gen de fusión fluorescente que expresa mCherry con la etiqueta E3L, uno de los dos antagonistas de PKR VACV. El casete, incluyendo el gen de interés y la fusión mCherry-E3L está flanqueado por secuencias derivadas del genoma VACV. Entre el gen de interés y mCherry-E3L hay una región más pequeña que es idéntica a los primeros 150 nucleótidos del brazo de 3', para promover la recombinación homóloga y la pérdida del gen mCherry-E3L después de la selección. Demostramos que este método permite una generación eficiente y sin problemas de rVACV en una variedad de tipos de células sin necesidad de selección de fármacos o cribado extensivo de virus mutantes.
Introduction
El virus de la vacuna (VACV) fue fundamental para la primera erradicación exitosa de un patógeno humano, el virus de la variola (VARV), de la naturaleza. Desde el exterminio del virus de la variola, los poxvirus, incluido el VVAC, han seguido siendo virus terapéuticos útiles tanto para la medicina humana como para la medicina animal. Por ejemplo, una vacuna contra el virus de la rabia basada en VCVa ha sido muy eficaz para prevenir la transmisión de la rabia silvática en Europa1 y los Estados Unidos2. Más recientemente, los poxvirus recombinantes que expresan una variedad de moléculas antitumorales (por ejemplo, anticuerpos de cadena única o eritropoyetina humana) han visto un éxito alentador como agentes oncolíticos3,4,5. VACV es particularmente atractivo como vector porque es fácilmente susceptible a la manipulación genética, posee una amplia gama de huéspedes, y es estable en una variedad de condiciones, lo que permite un fácil transporte y viabilidad de la vacuna en el campo6,7. Si bien se han desarrollado múltiples técnicas para generar VACV recombinante para experimentos de laboratorio y generación de vacunas, las estrategias actuales para generar estos virus tienen limitaciones notables.
Debido a la utilidad de VACV, se han desarrollado múltiples estrategias para generar virus recombinantes. La primera estrategia emplea una recombinación homóloga para introducir un casete que incluya el transgén y un gen marcador seleccionable, como un gen de resistencia a los antibióticos. El cassette está flanqueado por dos nucleótidos de 500 o más grandes que dirigen el gen a un sitio específico en el genoma viral, que luego se integra de forma estable por eventos de doble cruce8,9,10. Esta estrategia es rápida y eficiente; sin embargo, resulta en material genético extra en forma del gen marcador que puede producir efectos inesperados. Además, hay un límite superior práctico para el número de transgenes que se pueden introducir limitados por el número de marcadores seleccionables únicos disponibles. Las estrategias de selección dominante transitoria (TDS) han abordado esta cuestión facilitando la generación de virus recombinantes "sin cicatrices"11. Usando esta estrategia, un plásmido que contiene un gen VAV mutante y un gen marcador seleccionable se integran en el genoma viral, pero sin ADN VAV de flanqueo adicional. Este enfoque da como resultado la integración transitoria de todo el plásmido y la duplicación del gen VACV como resultado de la integración por un solo evento cruzado. Este intermedio es estable siempre y cuando se mantenga bajo presión de selección, permitiendo el enriquecimiento de esta construcción. Cuando se elimina la selección, la duplicación vacv permite un segundo evento de cruce que da como resultado la eliminación del plásmido y la posterior formación del tipo salvaje (wt) o del virus recombinante en una proporción aproximada de 50:50. Mientras que tDS genera virus recombinantes sin requerir la introducción estable de ADN extraño, múltiples clones de virus deben ser examinados para detectar la mutación esperada mediante análisis de secuenciación, un paso potencialmente lento y costoso.
Aquí, presentamos un enfoque para generar poxvirus recombinantes combinando los mejores aspectos de cada uno de estos enfoques, similar a un enfoque que se ha descrito para la replicación incompetente modificada vaccinia Ankara12,13,14. Esta estrategia combina la selección de rango visual y de host para generar rápidamente virus recombinantes mediante eventos cruzados dobles, y posteriormente eliminar el gen marcador seleccionable mediante recombinación homóloga. Este enfoque permite la rápida generación de mutantes mediados por la recombinación homóloga, con la naturaleza "sin cicatrices" de los enfoques TDS, sin necesidad de un paso de detección posterior para distinguir el tipo salvaje y los virus mutantes. Nuestro método también utiliza la selección de rango de huésped en lugar de la selección de antibióticos, eliminando el riesgo de cambios fenotípicos inducidos químicamente en la línea celular. Para este enfoque, hemos optado por utilizar la proteína quinasa antiviral huésped R (PKR) como agente selectivo para generar VACV recombinante. PKR se expresa como un monómero inactivo en la mayoría de los tipos de células15. Al enlazar el ARN de doble cadena (dsRNA) en los dominios de enlace dsRNA N-terminal, PKR dimerizes y se autofosforilated16. Esta forma activa de PKR fosforila la subunidad alfa del factor de iniciación eucariota 2 (eIF2), inhibiendo en última instancia la entrega de metaonil-tRNA de iniciador al ribosoma, evitando así la traducción intracelular e inhibiendo ampliamente la replicación de muchas familias de virus17,,18.
En respuesta a la amplia y potente actividad antiviral de PKR, muchos virus han desarrollado al menos una estrategia para prevenir la activación de PKR. La mayoría de los poxvirus expresan dos antagonistas de PKR, codificados por los genes E3L y K3L en VACV, que antagonizan PKR a través de dos mecanismos distintos19. E3 previene la homodimerización pkR mediante la unión de ARN de doble cadena20,21, mientras que K3 actúa como un inhibidor de pseudosustrato mediante la unión directa a PKR activado y, por lo tanto, la inhibición de la interacción con su sustrato eIF222. Es importante destacar que estos dos antagonistas de PKR no inhiben necesariamente pkR de todas las especies. Por ejemplo, el homólogo K3 del virus de la viruela ovina inhibió fuertemente la PKR de las ovejas, mientras que el homólogo e3 de la viruela ovina no mostró una inhibición considerable de PKR23,24. En este estudio, presentamos un método para utilizar la presión selectiva mediada por PKR combinada con la selección de fluorescencia para generar un VACV recombinante eliminado para E3L y K3L (VC-R4), que no puede replicarse en células competentes PKR derivadas de diversas especies. Este virus recombinante proporciona un excelente trasfondo para la rápida generación de virus recombinantes que expresan genes bajo el control del promotor nativo de E3L.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Generación del vector de recombinación
- Diseñe imprimadores para generar el casete de selección. Diseñe cada amplificador individual con secuencias superpuestas con amplicons vecinos y el vector para facilitar el ensamblaje enzimático isotérmico de moléculas de ADN, también llamado ensamblaje Gibson, utilizando cualquiera de varias herramientas de diseño de imprimación en línea.
NOTA: Este protocolo también se puede completar utilizando métodos tradicionales de clonación basados en en endonucleasas de restricción. En ese caso, diseñe imprimaciones con los sitios de restricción apropiados en lugar de con secuencias superpuestas. - Utilizando las imprimaciones diseñadas en el paso 1.1, la PCR amplifica los siguientes elementos en orden de 5' a 3'(Figura 1):500 nucleótidos de la región genómica VACV 5' del brazo E3L (5'), EGFP o el gen de interés, 150 nucleótidos de la región genómica del VAV inmediatamente 3' de E3L (brazo corto de 3'), un sintético promotor de poxvirus temprano/tardío25, el gen de fusión mCherry-E3L, y 500 nucleótidos de la región genómica VACV 3' de E3L incluyendo el brazo corto de 3' (brazo largo de 3').
- En un tubo de PCR, añada los reactivos en el siguiente orden para cada amplificador: 17 l de agua libre de DNase, 1,2 l de cada imprimación (concentración inicial de 10 m, concentración final a 0,5 m), 5 ml de tampón de reacción PCR de 5x, ADN de plantilla (10 ng para amplicons amplificados a partir de plásmidos: EGFP y casete E/L promoter-mCherry-E3L; 100 ng para amplicons amplificado a partir de ADN genómico: 5' y 3' brazos), y 0,5 áml de adn. Ajuste el volumen de agua añadido para un volumen de reacción final de 50 l.
NOTA: La concentración de ADN de la plantilla debe determinarse empíricamente, pero generalmente comenzamos con 10 ng/reacción. - Colocar el tubo o tubos en un termociclador, y derretir el ADN a 98 oC durante 30 s, y luego utilizar 25 rondas de un protocolo pcR de tres pasos: 98 oC para 5 s, 55 oC para 10 s y 72 oC durante 1 min.
NOTA: Determine la temperatura de fusión en función de la Tm sugerida por el fabricante para cada conjunto de imprimación. Determine el tiempo de extensión adecuado en función de la longitud de cada amplificador (1 minuto/kb).
- En un tubo de PCR, añada los reactivos en el siguiente orden para cada amplificador: 17 l de agua libre de DNase, 1,2 l de cada imprimación (concentración inicial de 10 m, concentración final a 0,5 m), 5 ml de tampón de reacción PCR de 5x, ADN de plantilla (10 ng para amplicons amplificados a partir de plásmidos: EGFP y casete E/L promoter-mCherry-E3L; 100 ng para amplicons amplificado a partir de ADN genómico: 5' y 3' brazos), y 0,5 áml de adn. Ajuste el volumen de agua añadido para un volumen de reacción final de 50 l.
- Visualice los productos de amplificación en un gel de agarosa del 1%. Añadir 10 l de cada producto de ADN y 2 l de tampón de carga a cada poca, y correr a 8 V/cm durante 1 h.
- Gel purifica cada amplicon usando un kit de extracción de gel de ADN y el protocolo del fabricante. Eluye los amplicons de la columna añadiendo 50 sl de agua libre de DNase e inmediatamente centrifugando.
- Linealizar el vector de clonación pUC19 utilizando la digestión endonucleasa EcoRI. A un tubo, añadir 1 g de pUC19, agua a un volumen de 17 l, 2 L de tampón de reacción y 1 l (20 unidades) de EcoRI. Incubar a 37oC durante 1 h.
- Visualizar los productos de amplificación en un gel de agarosa del 1% a 8 V/cm durante 1 h. Exituir la banda del gel, y purificar el producto utilizando el kit de extracción de gel de ADN como se describe en el paso 1.4.
- Ligar todos los amplicons individuales, gel purificado y el vector linealizado utilizando un kit de mezcla maestro.
- A un tubo PCR, añadir 0.2 pmol de pUC19 linealizado y cada amplicon (brazo de 5', EGFP, brazo corto de 3', e/L promotor-mCherry-E3L cassette, 3'brazo). Agregue agua libre de DNase a un volumen final de 10 l y, a continuación, agregue 10 ml de mezcla maestra de ensamblaje de ADN. Incubar muestras a 50oC durante 1 h.
- Transformar E. coli químicamente competente con 2 ml del producto ensamblado a partir del paso 1.6 como se describió anteriormente26,27. Placa 100 l de las células transformadas en placas de agarosa LB que contienen 100 g/ml de ampicilina. Incubar las placas durante la noche a 37oC.
- Recoger colonias bien aisladas y transferir colonias individuales a tubos que contengan caldo de Luria con ampicilina de 100 g/ml. Incubar los tubos durante la noche a 37 oC mientras agita a 225 rpm.
- Aísle los plásmidos del cultivo nocturno utilizando un kit de minipreparación de plásmido. Compruebe la concentración y pureza del ADN utilizando un espectrofotómetro. Una relación A260/A280 entre 1,8 y 2,0 es aceptable.
- Envíe los plásmidos para la secuenciación de Sanger para determinar si el producto de clonación deseado es correcto. Almacenar el ADN a -20 oC.
2. Generación del virus recombinante
- Infectar una monocapa confluente de células adecuadas con el virus para ser recombinado en una multiplicidad de infección de 1.0 (MOI 1.0) en una placa de 6 pocillos. Incubar las células infectadas a 37oC y 5% deCO2 durante 1 h. A continuación, aspirar el medio y reemplazarlo con DMEM fresco. Incubar las células infectadas a 37oC y 5% deCO2.
NOTA: Para la replicación de virus competentes como un virus de la vacuna que carece de K3L22, una línea celular como la línea de células renales de conejo europeo RK13 (ATCC #CCL-37) o BSC-40 es apropiado. Sin embargo, para los virus deficientes de replicación, como el virus descrito en este artículo que carece de los antagonistas de PKR E3L y K3L, se requiere una línea celular complementaria que exprese estos dos genes en células de derribo o desconexión trans o PKR. - Transfectar las células infectadas con 500 ng del vector generado y validado en el paso 1.10 utilizando un reactivo de transfección disponible comercialmente siguiendo el protocolo del fabricante. Incubar las células a 37oC y 5%co2 durante 48 h.
NOTA: Si se utiliza un virus de la vacuna que carece de E3L y K3L, la presión selectiva mediada por PKR impulsará la selección de virus recombinados y mantendrá la expresión de la proteína de fusión mCherry-E3L en estas células. Si se desea, también debería ser posible amplificar PCR sólo el inserto para usar para la transfección en lugar de todo el plásmido. - 48 horas después de la infección, cosechar la monocapa infectada. En algunos casos, las células se pueden cosechar pipeteando, pero si todavía están bien adheridas, cosecharlas con un rascador de células. Congele las células tres veces y luego sonice los lysates durante 15 s a una amplitud del 50%. Conservar este lisado a -80oC hasta que esté listo para usar.
- Serialmente 10 veces diluir el lisato cosechado en el paso 2.3 de 10-1 a 10-6 añadiendo 120 l del lisado a 1080 l de DMEM (10-1), y luego agregando 120 ol de esta dilución a 1080 s de DMEM (10-2), y repitiendo este proceso cuatro veces más. Agregue 1 ml de cada dilución a un individuo, pozo confluente de una línea celular competente PKR, en este caso células RK13.
- Incubar las células infectadas a 37oC y 5% deCO2 durante 1 h. A continuación, aspirar el medio y reemplazarlo con DMEM fresco Incubar las células infectadas a 37 oC y 5% CO2.
- 24 a 48 horas después de la infección, identificar virus recombinantes mediante microscopía de fluorescencia. Las placas de los virus recombinantes expresan la fluorescencia roja debido a la integración del gen de fusión mCherry-E3L(Figura 2). Si inicialmente se utilizó un virus carente de inhibidores de PKR, todas las placas contendrán virus recombinantes.
- La placa purifica los virus recombinantes tres veces en las células RK13. Después de la ronda final de purificación de la placa, todas las placas deben expresar fluorescencia roja.
- Infectar una placa confluente de 6 pocillos de células RK13 que expresan los inhibidores de LA PKR VACV E3L y K3L (células RK13+E3L+K3L28)con el virus de fluorescencia roja purificado por placa del paso 2.6. Apunta a aproximadamente 50-100 placas por pozo.
NOTA: Estas células proporcionan a los antagonistas de LA PKR VACV en trans y alivian la presión selectiva mediada por PKR para mantener el gen de fusión mCherry-E3L, promoviendo así la generación "sin cicatrices" del virus recombinante. - Identifique los virus colapsados mediante microscopía de fluorescencia utilizando un microscopio EVOS2 y un cubo de filtro GFP (Excitación: 470/22, Emisión: 525/50) y un cubo de filtro RFP (Excitación: 531/40, Emisión: 593/40).
NOTA: La frecuencia con la que se pierde el gen de fusión mCherry-E3L es de aproximadamente 2,5%(Tabla 2). Si EGFP no se incluye como un gen marcador, las placas de virus mutantes que han perdido el gen de fusión mCherry-E3L serán incoloras. - La placa purifica las placas de solo verde (VC-R4) o incoloras (E3L) tres veces en las células RK13+E3L+K3L. Asegúrese de que no haya placas de color rojo fluorescencia.
- Confirmar la pérdida de mCherry-E3L y la presencia de la mutación esperada por PCR y secuenciación de Sanger.
NOTA: Si el gen o la mutación de interés no tiene actividad inhibitoria de PKR, se deben cultivar virus recombinantes en células RK13+E3L+K3L o en una línea celular deficiente o equivalente inhibida por PKR(Figura 3).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Usamos el procedimiento diagramado en la Figura 1 para generar un VACV que carece de ambos antagonistas PKR E3L y K3L, reemplazando E3L por EGFP en un virus ya eliminado para K3L (vP872). La Figura 2 muestra placas fluorescentes rojas en células RK13 competentes de PKR indicativas de la expresión viral de mCherry-E3L, así como EGFP expresada en células RK13+E3L+K3L que confirman la pérdida de E3L y el colapso del marcador de selección mCherry-E3L. El cuadro 3 confirma que este virus recombinante, VC-R4, que carece de ambos antagonistas PKR no puede replicar en las células RK13 competentes PKR, mientras que el virus primario, vP872 que expresa E3L, es la replicación competente. Para confirmar que esta incapacidad de replicar en las células RK13 fue solamente debido a la pérdida de E3L, reemplazamos el EGFP en el VC-R4 con el E3L, para generar un virus revertante usando el mismo protocolo de selección. La Figura 3 también valida que este virus revertante se replique tan eficientemente como vP872 en las células RK13. Curiosamente, las placas incoloras consistentes con el colapso del marcador de selección mCherry-E3L se identificaron antes de la selección en las células RK13+E3+K3 que generalmente se requieren para seleccionar recombinantes "sin cicatrices", probablemente debido a la identidad de secuencia extendida entre la recombinación mCherry-E3L y el gen E3L que se inserta en VC-R4. Por lo tanto, para determinar la eficiencia de la recombinación y la tasa de colapso elegimos producir virus que expresan el poxvirus PKR antagonista K3L para evitar el problema del colapso temprano23. La Figura 4 indica la aparición de placas incoloras (puntas de flecha) después de la infección de las células RK13+E3L+K3L. El Cuadro 1 muestra los resultados de tres experimentos independientes, en los que, en promedio, el 12,6% de los viriones progenie se habían recombinado con el plásmido transinfectado, similar a las frecuencias notificadas anteriormente29,,30,,31. La Tabla 2 detalla la frecuencia de las placas incoloras en relación con las placas totales en las células RK13+E3L+K3L, lo que demuestra la tasa de colapso y pérdida del marcador de selección mCherry-E3L ocurrió a una frecuencia de aproximadamente 1.8%.
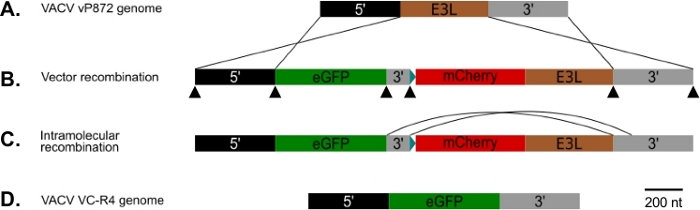
Figura 1: Diagrama de p837-GOI-mCherry-E3L, así como la estrategia de recombinación visual y de rango de host. (A) brazo de 5' (negro) y 3' brazo (gris) flanquean el locus E3L (marrón) en VACV. (B) En p837-GOI-mCherry-E3L, estos brazos flanquean un casete que contiene el gen de interés (GOI), en este caso EGFP, (verde) separado de un gen de fusión mCherry-E3L (rojo) bajo control del promotor sintético del poxvirus temprano/tardío25 azul) por un brazo corto de 3' (gris). Estos brazos externos impulsan la recombinación homóloga entre vacV y el p837-GOI-mCherry-E3L. Las puntas de flecha negras indican los sitios de las imprimaciones superpuestas utilizadas para generar este plásmido por la clonación de Gibson. (C) Cuando se elimina la presión selectiva PKR, se pueden seleccionar virus que han sufrido recombinación intramolecular entre los brazos cortos y largos de 3'. (D) Resultando en un virus (VC-R4) que contiene únicamente el gen de interés en el locus E3L. Haga clic aquí para ver una versión más grande de esta figura.

Figura 2: Micrografías fluorescentes de (arriba) una placa de virus recombinante 24 horas después de la recombinación con p837-GOI-mCherry-E3L expresando tanto mCherry (izquierda) como EGFP (derecha) en células RK13. (Abajo) Micrografía de una placa de virus recombinante 48 horas después de que se haya eliminado la presión selectiva mediada por PKR en las células RK13++, expresando EGFP (derecha) pero no mCherry (izquierda). La barra de escala indica 650 m para todos los paneles. Haga clic aquí para ver una versión más grande de esta figura.
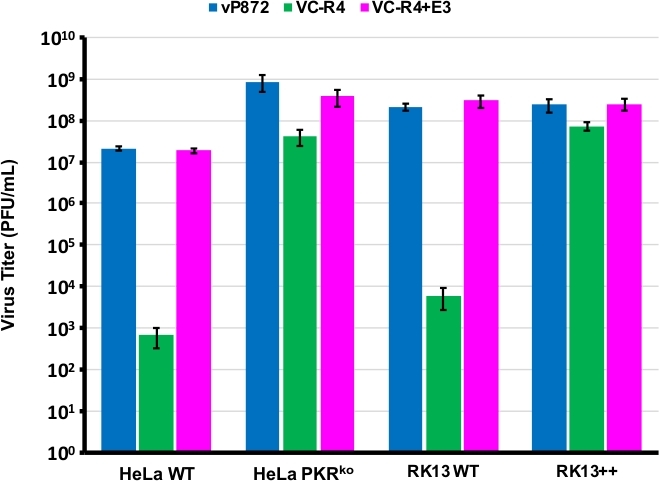
Figura 3: VC-R4 no puede replicar en las células competentes PKR. Las líneas celulares indicadas se infectaron con vP872 (azul), VC-R4 (verde) o VC-R4+E3L (magenta) en MOI a 0,1. 48 horas después de la infección las células infectadas fueron cosechadas y mitadas por dilución en serie en células RK13+E3L+K3L. Los titers se notifican en PFU/mL, las barras de errores representan la desviación estándar de tres experimentos de réplica. Haga clic aquí para ver una versión más grande de esta figura.

Figura 4: Pérdida de la expresión mCherry-E3L en celdas RK13+E3L+K3L. Superposición de micrografías fluorescentes y de contraste de fase de células RK13+E3L infectadas con VC-R4+K3L. Tres placas ya no expresan mCherry (círculos) debido al colapso del casete de selección que produce VC-R4+K3L. Haga clic aquí para ver una versión más grande de esta figura.
| Experimento 1 | Experimento 2 | Experimento 3 | |
| Placas rojas (RK13) | 30 | 11 | 18 |
| Placas totales (RK13+E3L+K3L) | 225 | 64 | 249 |
| Tasa de recombinación | 13.30% | 17.20% | 7.20% |
Tabla 1: Frecuencia de recombinación de VACV con el plásmido p837-K3L-mCherry-E3L.
| Experimento 1 | Experimento 2 | Experimento 3 | |
| Placas totales (RK13+E3L+K3L) | 115 | 44 | 210 |
| Placas incoloras (RK13+E3L+K3L) | 3 | 1 | 1 |
| Tasa de recombinación | 2.60% | 2.30% | 0.50% |
Tabla 2: Frecuencia de pérdida de mCherry-E3L de VC-R4+K3L-mCherry-E3L en células RK13+E3+K3.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Aquí presentamos una variación de una estrategia de selección de marcadores transitorios 32 para generar virus de vacuna recombinante sin retener ADN extraño en el virus recombinante final. Nuestra estrategia utiliza presión selectiva mediada por la proteína antiviral huésped PKR en lugar de otras formas de presión selectiva como los antibióticos. El uso de genes antivirales de huésped elimina la posibilidad de cambios fenotípicos inducidos químicamente en las células, o un mayor riesgo de mutación debido a los medicamentos de selección. Además, a diferencia de la selección de fármacos, no hay fase de retraso para nuestro enfoque, porque PKR se expresa constitutivamente en todas las células. La selección visual secundaria basada en la expresión mCherry también mejora la especificidad de este método al asegurar que sólo las placas que expresan el transgén se seleccionan durante la primera fase, y es igualmente eficiente como un marcador selectivo negativo mientras se seleccionan virus recombinantes maduros que han perdido el gen mCherry-E3L.
Los pasos más críticos para esta estrategia de recombinación son la generación del vector de recombinación adecuado y la purificación de placa adecuada para garantizar que el virus seleccionado sea clonal. En este artículo sugerimos "Ensamblaje Gibson" para generar el vector de recombinación. Esta estrategia es extremadamente eficiente y permite el montaje de todos los fragmentos que componen el vector de recombinación en un solo día. Sin embargo, debido a que el brazo corto de 3' y el brazo largo de 3' comparten secuencias idénticas, estos fragmentos tienen el potencial de unirse durante la reacción de clonación, y algunos vectores pueden no contener el casete mCherry-E3L. En nuestra experiencia esto es raro, pero confirmar la estructura del vector después de la clonación es necesario. También hemos generado vectores de recombinación para esta estrategia utilizando métodos tradicionales de endonucleasa y ligasa. Esta estrategia evita el problema descrito anteriormente, pero puede ser más laborioso. La purificación de placas es generalmente sencilla y depende principalmente del uso de células permisivas apropiadas para la recombinación inicial, las células competentes para PKR para la purificación inicial de la placa para asegurarse de que sólo los virus recombinantes pueden replicarse, y luego las células permisivas de nuevo para facilitar la recombinación intramolecular y la pérdida del marcador seleccionable. Por lo tanto, la atención cercana a las líneas celulares es fundamental para la aplicación exitosa y eficiente de esta estrategia.
En este estudio, demostramos el uso de este método para generar un REcombinante VACV eliminado para los antagonistas PKR E3L y K3L y expresando EGFP bajo el control del promotor E3L. En el futuro, este virus servirá como un fondo eficiente para futuros virus recombinantes, ya que es incapaz de replicar en las células competentes PKR. Por lo tanto, habrá una fuerte presión selectiva mediada por PKR para conducir el casete de recombinación mCherry-E3L en viriones de progenie y, al mismo tiempo, esencialmente prevenir la replicación del virus no recombinante. Además, la pérdida de EGFP por la captación del casete de recombinación es un marcador de selección secundario útil para garantizar que las placas recogidas no estén coinfectadas con un virus no recombinante. Observamos tasas de recombinación consistentes con las tasas previamente reportadas para VACV, pero los marcadores fluorescentes visuales aumentan la eficiencia de generar virus recombinantes al asegurar que se aumenta la probabilidad de que se seleccionen los virus recombinantes adecuados. Nuestra observación de placas incoloras después de dos rondas de selección en células paquisca, presumiblemente debido a la mayor longitud de secuencia idéntica entre E3L y el gen marcador mCherry-E3L, sugiere que la tasa de pérdida de mCherry-E3L puede ser "sintonizada" aumentando o disminuyendo la longitud del brazo corto de 3'. La limitación principal de esta técnica es el uso de PKR como presión selectiva para recombinantes. El uso más eficiente de esta estrategia de recombinación es la generación de estos virus en un fondo que carece de antagonistas PKR. Sin embargo, el marcador de selección colorimétrica permite utilizar esta estrategia de recombinación incluso sin la selección mediada por PKR, simplemente mediante placas que purifican la placa mCherry. Si bien la falta de presión selectiva mediada por PKR reducirá la eficiencia del primer paso de detección, el porcentaje de placas que expresan mCherry sigue siendo lo suficientemente alto como para que la selección basada en color sea viable. Por lo tanto, este método se puede utilizar para insertar casi cualquier gen en el genoma del poxvirus.
Como lo demuestra la inserción de EGFP, con este enfoque, cualquier gen puede insertarse rápidamente en el locus E3L bajo el control del promotor nativo, siempre que las células nulas PKR o las líneas celulares de complemento se utilicen para experimentos posteriores si el transgén no es un antagonista PKR. Esta estrategia, combinada con el virus VC-R4 que informamos aquí, añade un nuevo y potente método para generar de forma rápida y confiable virus de vacuna recombinante utilizando presión selectiva mediada por el huésped y la identificación visual de los recombinantes al principio del proceso.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Los autores no declaran intereses financieros en competencia.
Acknowledgments
Este proyecto fue financiado por los Institutos Nacionales de Salud (AI114851) a SR.
Materials
| Name | Company | Catalog Number | Comments |
| 2X-Q5 Master Mix | NEB | M0492L | High-fidelity polymerase used in PCR |
| Ampicillin | ThermoFisher Scientific | 11593027 | Bacterial selective agent |
| Disposable Cell Scrapers | ThermoFisher Scientific | 08-100-242 | Cell scraper to harvest infected cells |
| EVOS FL Auto 2 Cell imaging system | ThermoFisher Scientific | AMAFD2000 | Fluorescent microscope |
| EVOS Light Cube, GFP | ThermoFisher | AMEP4651 | GFP Cube |
| EVOS Light Cube, RFP | ThermoFisher | AMEP4652 | RFP Cube |
| GenJet | SignaGen Laboratories | SL100489 | Transfection reagent |
| Luria Bertani (LB) Broth | Gibco | 10855021 | Bacterial growth medium |
| Monarch DNA gel extraction kit | NEB | T1020L | Gel purification kit used to purify amplicons and linearized vectors |
| Monarch Plasmid Miniprep kit | NEB | T1010L | Miniprep kit ussed to purify plasmids |
| NanoDrop One | ThermoFisher Scientific | ND-ONE-W | Spectrophotometer used to measure RNA and DNA concentration |
| NEBuilder Master Mix | NEB | E2621L | Isothermal enzymatic assembly kit used to generate the recombination vector |
| Q500 Sonicator | Qsonica | Q500-110 | Sonicator for virus lysates |
| RK13 cells | ATCC | CCL-37 | Rabbit kidney cells |
| VWR Multiwell Cell Culture plates | VWR | 10062-892 | Cell culture plates |
References
- Brochier, B., et al. Large-scale eradication of rabies using recombinant vaccinia-rabies vaccine. Nature. 354 (6354), 520-522 (1991).
- Pastoret, P. P., Brochier, B. The development and use of a vaccinia-rabies recombinant oral vaccine for the control of wildlife rabies; a link between Jenner and Pasteur. Epidemiology and Infection. 116 (3), 235-240 (1996).
- Chan, W. M., McFadden, G. Oncolytic Poxviruses. Annual review of virology. 1 (1), 119-141 (2014).
- Nguyen, D. H., et al. Vaccinia virus-mediated expression of human erythropoietin in tumors enhances virotherapy and alleviates cancer-related anemia in mice. Molecular Therapy. 21 (11), 2054-2062 (2013).
- Frentzen, A., et al. Anti-VEGF single-chain antibody GLAF-1 encoded by oncolytic vaccinia virus significantly enhances antitumor therapy. Proceedings of the National Academy of Sciences of the United States of America. 106 (31), 12915-12920 (2009).
- Pastoret, P. P., Vanderplasschen, A. Poxviruses as vaccine vectors. Comparative Immunology, Microbiology and Infectious Diseases. 26 (5-6), 343-355 (2003).
- COLLIER, L. H. The development of a stable smallpox vaccine. The Journal of Hygiene. 53 (1), 76-101 (1955).
- Weir, J. P., Bajszár, G., Moss, B. Mapping of the vaccinia virus thymidine kinase gene by marker rescue and by cell-free translation of selected mRNA. Proceedings of the National Academy of Sciences of the United States of America. 79 (4), 1210-1214 (1982).
- Mackett, M., Smith, G. L., Moss, B. Vaccinia virus: a selectable eukaryotic cloning and expression vector. Proceedings of the National Academy of Sciences of the United States of America. 79 (23), 7415-7419 (1982).
- Nakano, E., Panicali, D., Paoletti, E. Molecular genetics of vaccinia virus: demonstration of marker rescue. Proceedings of the National Academy of Sciences of the United States of America. 79 (5), 1593-1596 (1982).
- Falkner, F. G., Moss, B. Transient dominant selection of recombinant vaccinia viruses. Journal of Virology. 64 (6), 3108-3111 (1990).
- Staib, C., Drexler, I., Ohlmann, M., Wintersperger, S., Erfle, V., Sutter, G. Transient Host Range Selection for Genetic Engineering of Modified Vaccinia Virus Ankara. BioTechniques. 28 (6), 1137-1148 (2000).
- Staib, C., Drexler, I., Sutter, G. Construction and Isolation of Recombinant MVA. Vaccinia Virus and Poxvirology. , 77-99 (2004).
- Di Lullo, G., et al. Marker gene swapping facilitates recombinant Modified Vaccinia Virus Ankara production by host-range selection. Journal of Virological Methods. 156 (1-2), 37-43 (2009).
- Pfaller, C. K., Li, Z., George, C. X., Samuel, C. E. Protein kinase PKR and RNA adenosine deaminase ADAR1: New roles for old players as modulators of the interferon response. Current Opinion in Immunology. 23 (5), 573-582 (2011).
- Bevilacqua, P. C., George, C. X., Samuel, C. E., Cech, T. R. Binding of the protein kinase PKR to RNAs with secondary structure defects: Role of the tandem A - G mismatch and noncontigous helixes. Biochemistry. 37 (18), 6303-6316 (1998).
- Krishnamoorthy, T., Pavitt, G. D., Zhang, F., Dever, T. E., Hinnebusch, A. G. Tight Binding of the Phosphorylated Subunit of Initiation Factor 2 (eIF2) to the Regulatory Subunits of Guanine Nucleotide Exchange Factor eIF2B Is Required for Inhibition of Translation Initiation. Molecular and Cellular Biology. 21 (15), 5018-5030 (2001).
- Rothenburg, S., Georgiadis, M. M., Wek, R. C. Evolution of eIF2α kinases: Adapting translational control to diverse stresses. Evolution of the Protein Synthesis Machinery and Its Regulation. , 235-260 (2016).
- Bratke, K. A., McLysaght, A., Rothenburg, S. A survey of host range genes in poxvirus genomes. Infection, Genetics and Evolution. 14, 406-425 (2013).
- Chang, H. W., Watson, J. C., Jacobs, B. L. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proceedings of the National Academy of Sciences. 89 (11), 4825-4829 (1992).
- Romano, P. R., et al. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: role of complex formation and the E3 N-terminal domain. Molecular and Cellular Biology. 18 (12), 7304-7316 (1998).
- Beattie, E., Tartaglia, J., Paoletti, E. Vaccinia virus-encoded eIF-2 alpha homolog abrogates the antiviral effect of interferon. Virology. 183 (1), 419-422 (1991).
- Park, C., Peng, C., Brennan, G., Rothenburg, S. Species-specific inhibition of antiviral protein kinase R by capripoxviruses and vaccinia virus. Annals of the New York Academy of Sciences. 1438 (1), 18-29 (2019).
- Rothenburg, S., Brennan, G. Species-Specific Host-Virus Interactions: Implications for Viral Host Range and Virulence. Trends in Microbiology. , (2019).
- Chakrabarti, S., Sisler, J. R., Moss, B. Compact, synthetic, vaccinia virus early/late promoter for protein expression. BioTechniques. 23 (6), 1094-1097 (1997).
- Chung, C. T., Niemela, S. L., Miller, R. H. One-step preparation of competent Escherichia coli: Transformation and storage of bacterial cells in the same solution (recombinant DNA). Biochemistry. 86, 2172-2175 (1989).
- Chung, C. T., Miller, R. H. Preparation and storage of competent Escherichia coli cells. Methods in Enzymology. 218, 621-627 (1993).
- Rahman, M. M., Liu, J., Chan, W. M., Rothenburg, S., McFadden, G. Myxoma Virus Protein M029 Is a Dual Function Immunomodulator that Inhibits PKR and Also Conscripts RHA/DHX9 to Promote Expanded Host Tropism and Viral Replication. PLOS Pathogens. 9 (7), 1003465 (2013).
- Evans, D. H., Stuart, D., McFadden, G. High levels of genetic recombination among cotransfected plasmid DNAs in poxvirus-infected mammalian cells. Journal of Virology. 62 (2), 367-375 (1988).
- Ball, L. A. High-frequency homologous recombination in vaccinia virus DNA. Journal of Virology. 61 (6), 1788-1795 (1987).
- Spyropoulos, D. D., Roberts, B. E., Panicali, D. L., Cohen, L. K. Delineation of the viral products of recombination in vaccinia virus-infected cells. Journal of Virology. 62 (3), 1046-1054 (1988).
- Liu, L., et al. Transient dominant host-range selection using Chinese hamster ovary cells to generate marker-free recombinant viral vectors from vaccinia virus. BioTechniques. 62 (4), 183-187 (2017).
