

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Questa procedura è stata stabilita per essere utilizzata per lo sviluppo di colture epatiche 3D avanzate in vitro, che possono fornire una valutazione più fisiologicamente rilevante dei rischi genotossici associati alle esposizioni di nanomateriali sia su regimi acuti che a lungo termine, a dosi ripetute.
Abstract
A causa del rapido sviluppo e implementazione di una vasta gamma di nanomateriali ingegnerizzati (ENM), l'esposizione all'ENM è inevitabile e lo sviluppo di sistemi di test in vitro robusti e predittivi è essenziale. La tossicologia epatica è fondamentale quando si considera l'esposizione all'ENM, poiché il fegato svolge un ruolo vitale nell'omeostasi metabolica e nella disintossicazione, oltre ad essere un sito importante di accumulo di ENM dopo l'esposizione. Sulla base di questo e della comprensione accettata che i modelli di epatociti 2D non imitano accuratamente le complessità delle intricate interazioni multicellulari e dell'attività metabolica osservate in vivo, c'è una maggiore attenzione allo sviluppo di modelli epatici 3D fisiologicamente rilevanti su misura per scopi di valutazione del rischio ENM in vitro. In linea con i principi dei 3R per sostituire, ridurre e perfezionare la sperimentazione animale, è stato sviluppato un modello epatico basato sulla linea cellulare 3D HepG2, che è un sistema intuitivo ed economico in grado di supportare regimi di esposizione ENM estesi e ripetuti (≤14 giorni). Questi modelli sferoidi (diametro di ≥500 μm) mantengono la loro capacità proliferativa (cioè modelli di cellule divisorie) consentendo loro di essere accoppiati con il saggio micronucleo "gold standard" per valutare efficacemente la genotossicità in vitro. La loro capacità di riferire su una serie di endpoint tossicologici (ad esempio, funzione epatica, risposta (pro)infiammatoria, citotossicità e genotossicità) è stata caratterizzata utilizzando diverse ENM sia nei regimi di esposizione acuta (24 ore) che a lungo termine (120 h). Questo modello epatico in vitro 3D ha la capacità di essere utilizzato per valutare esposizioni ENM più realistiche, fornendo così un futuro approccio in vitro per supportare meglio la valutazione del rischio ENM in modo di routine e facilmente accessibile.
Introduction
A causa del rapido sviluppo e implementazione di una vasta gamma di nanomateriali ingegnerizzati (ENM) in una pletora di applicazioni basate sull'uomo (ad esempio, cibo, cosmetici, abbigliamento, attrezzature sportive, elettronica, trasporto e medicina), è inevitabile che gli esseri umani saranno esposti regolarmente all'ENM. Con questo, ci sono accresciuto problematiche che le nuove caratteristiche fisico-chimiche specifiche delle dimensioni che ritengono questi materiali vantaggiose in numerose applicazioni potrebbero causare effetti negativi sulla salute umana e sull'ambiente in concomitanza. Attualmente sono in atto molte attività internazionali per riflettere attivamente esposizioni più fisiologicamente rilevanti a queste ENM e valutare la potenziale tossicità di questi materiali in scenari di esposizione acuta, a lungo termine e ripetuta a basse dosi.
La tossicologia epatica è fondamentale quando si considera l'esposizione all'ENM, poiché è ampiamente noto che il fegato è un sito importante di accumulo di ENM dopol'esposizione 1,2. Inoltre, il fegato è il sistema di organi primario per il metabolismo e la disintossicazione delle sostanze che entrano nella circolazionesistemica 3. Sulla base della comprensione accettata che i modelli di epatociti 2D non imitano accuratamente le complessità di intricate interazioni multicellulari o rappresentano adeguatamente l'attività metabolica osservata in vivo, è stata stabilita una maggiore attenzione allo sviluppo di modelli epatici 3D robusti e fisiologicamente rilevanti per le tecnologie sostitutive in vivo4,5. L'utilizzo di tecnologie avanzate di coltura 3D migliora la longevità dei modelli epatici in vitro consentendo di esaminare regimi di esposizione ripetuta a lungo termine. Inoltre, questo formato di coltura avanzato promuove la formazione di caratteristiche fisiologiche e organotipiche migliorate come canaliculi biliari, processi di trasporto attivi e migliori capacità di metabolizzazione del farmaco CYP450, migliorando così la prevedibilità deimodelli 6. Gli attuali modelli epatici in vitro 3D costituiti da mono colture (solo epatociti) o co-colture (epatociti con cellule non interpachimali) esistono in diversi formati, che vanno da microtissuali o sferoidi in piastre di adesione ultrabassi, sferoidi a goccia appesi, cellule incorporate in matrici e/o impalcature e piattaforme di coltura cellulare microfluidica, che sono tutti considerati efficaci modelli avanzati in vitro per la valutazione della tossicitàepatica 6,7. Tuttavia, la maggior parte di questi sistemi di modelli sono ad alta manutenzione, richiedono attrezzature specializzate e sono costosi. Inoltre, questi modelli sono spesso statici (cioè modelli cellulari non divisi) che ne impediscono l'uso nella valutazione degli endpoint di pericolo, come i test di genotossicità utilizzando metodi che quantificano i danni fissi al DNA. La genotossicità è un prerequisito fondamentale nella tossicologia normativa ed è una componente vitale della valutazione del rischio di qualsiasi tossico8. Non esiste un singolo saggio che possa essere applicato per quantificare tutte le forme di danno al DNA che possono insorgere a seguito dell'esposizione a un agente esogeno. Tuttavia, un componente centrale della batteria di prova della genotossicità in vitro è il test del micronucleo, che è una tecnica affidabile e sfaccettata che misura il danno cromosomico lordo9. Si tratta di una tecnica gold standard descritta dalla Linea guida 487 del test dell'OCSE, per la valutazione del danno al DNA in vitro e della genotossicità e fa parte del requisito della batteria di provaper la valutazione del rischio normativo 10,11.
La linea cellulare del carcinoma epatocellulare umano, HepG2, è ampiamente utilizzata per lo screening iniziale della valutazione del pericolo in quanto le cellule sono prontamente disponibili, relativamente economiche per la fonte, semplici da coltura e suscettibili di screening ad altaproduttività 12,13. Quando coltivati in strutture sferiche 3D, è stato dimostrato che ricapitolano bene il microambiente epatico e offrono un modello epatico con capacità proliferative sufficienti a supportare il saggio micronucleo3. È stato stabilito un ulteriore sviluppo dei modelli di sferoidi HepG2 per migliorare la longevità e la funzionalità epatica del modello al fine di supportare la valutazione del rischio di genotossicità su regimi di esposizione ripetuta a lungo termine (≤14 giorni). Pertanto, in linea con i principi delle 3R per sostituire, ridurre e perfezionare la sperimentazione animale, è stato stabilito il presente protocollo per fornire un modello epatico 3D avanzato in vitro in grado di valutare in modo affidabile più endpoint tossicologici (ad esempio, funzionalità epatica, marcatori (pro)infiammatori, citotossicità e genotossicità) a seguito di esposizioni chimiche ed ENM acute, a lungo termine e ripetute in modo routine e facilmente accessibile.
Qui presentiamo un metodo per stabilire una linea cellulare di epatociti 3D fisiologicamente rilevante basata sul sistema di modelli in vitro per la valutazione del rischio di genotossicità a seguito di esposizioni ENM acute o ripetute a lungo termine. Il protocollo può essere suddiviso in 6 fasi chiave: coltivare cellule HepG2 crioconservate; Preparazione sferoide HepG2; Trasferimento sferoide HepG2 dalla caduta sospesa alla sospensione dell'agarosio; Raccolta sferoide HepG2; test e punteggio del micronucleo; analisi dei dati.
Protocol
1.Coltivare cellule HepG2 crioconservate
NOTA: Le cellule HepG2, ottenute dalla American Type Culture Collection (ATCC) sono state coltivate in 1x Modified Eagle Medium (DMEM) di Dulbecco con 4,5g/L D-glucosio e L-glutammina integrati con il 10% di siero bovino fetale (FBS) e l'1% di penicillina/streptomicina antibiotica.
- Mezzo di coltura cellulare DMEM pre-caldo (compresi gli integratori) in un bagno d'acqua a 37 °C per 30 minuti.
- Rimuovere una fiala di cellule HepG2 dall'azoto liquido e scongelare in un bagno d'acqua a 37 °C per 2-3 minuti, ruotando delicatamente il flaconcino per consentire lo scongelamento uniforme della sospensione cellulare. Fare attenzione a non immergere il flaconcino sopra l'O-ring al fine di ridurre il potenziale di contaminazione.
- Una volta scongelato, rimuovere il flaconcino dal bagno d'acqua e spruzzare generosamente con 70% di etanolo per decontaminare la superficie esterna del flaconcino prima di posizionare sotto una cappa sterile di coltura del tessuto laminare di classe II.
- Pipettare con cura il contenuto del crioviale delle cellule HepG2 in un tubo di centrifuga contenente 9 ml di mezzo di coltura cellulare DMEM prerifapidto (con integratori).
- Utilizzando una strippette da 10 ml, trasferire 10 ml della sospensione cellulare in un pallone da coltura cellulare usa e gettada 25 cm 2 e incubare la coltura per 3 giorni (dalla semina) a 5% CO2 e 37 °C fino a raggiungere ~80% di confluenza prima di sottoporsi a sottocoltura in un pallone da coltura cellulare usa e getta di 75 cm2 più grande.
- Una volta raggiunta la confluenza dell'80%, le cellule di sottocoltura in condizioni sterili mediante tripsicinazione con soluzione di tripsidenza/EDTA dello 0,05% preri warmed in un bagno d'acqua a 37 °C per 30 minuti. In nessun momento le cellule dovrebbero essere autorizzate ad asciugarsi.
- Quando le cellule formano un monostrato aderente, rimuovere il supporto ribaltandolo in un vaso di scarto disinfettante. Quindi lavare immediatamente il monostrato per rimuovere tutte le tracce di mezzi esistenti risciacquando il pallone due volte con 3 ml di soluzione sterile 1x PBS mantenuta a temperatura ambiente. Inoltre, scartare pbs in pentola di scarto disinfettante.
- Una volta rimosso il lavaggio PBS, aggiungere 5 mL di soluzione di tripsiderina-EDTA prerimpita allo 0,05%, assicurandosi di coprire l'intera superficie delle cellule e incubare le cellule per 6-8 minuti a 37 °C e 5% CO2.
- Toccare delicatamente il pallone per rimuovere le cellule dal fondo del pallone e quindi aggiungere 5 ml di terreno di coltura cellulare DMEM (con integratori) per neutralizzare l'enzima tripside.
- Trasferire la sospensione cellulare in un tubo di centrifuga da 50 ml e pipettare accuratamente la sospensione cellulare su e giù per garantire che le cellule siano completamente dissociate.
- Centrifugare la sospensione cellulare diluita a 230 x g per 5 min. Scartare il supernatante in disinfettante e sospendere di nuovo il pellet cellulare in 25 mL di mezzo di coltura cellulare DMEM (con integratori).
- Trasferire la sospensione cellulare in un pallone da coltura cellulare monouso da 75 cm2 e incubare a 37 °C e 5% co2 per altri 3 giorni prima di sottoporsi alla preparazione dello sferoide. Una volta che gli HepG2 hanno avuto il tempo di acclimatarsi e raggiungere ancora una volta una confluenza di ~ 80%, determinare la concentrazione cellulare in preparazione per la semina sferoide.
2. Preparazione sferoide HepG2
- Ripetere i passaggi di sottocoltura sopra indicati, tranne dopo la centrifugazione, sospendere di nuovo il pellet di cellule in 1 mL di mezzo di coltura DMEM prerifuoco in un bagno d'acqua a 37 °C. Sospensione della cella pipetta su e giù accuratamente.
- Ottieni il punteggio di vitalità delle cellule utilizzando il test di esclusione blu di Trypan (vedi OSHA SOP 3.21 Tossine riproduttive, Mutageni, Teratogeni ed Embriotossine - Procedure per la manipolazione e lo stoccaggio sicuri (2019) per la guida alla salute e allasicurezza) 14 con un rapporto 1:1 di sospensione cellulare alla soluzione blu trypan prefiltrata allo 0,4%.
- Prima del conteggio delle cellule, prendere 1 ml di soluzione blu Trypan utilizzando una siringa da 1 ml e filtrare con un'unità filtrante da 0,45 μm in un tubo sterile da 1 ml.
- Trasferire 10 μL di soluzione blu trypan filtrata in un tubo da 0,2 ml e aggiungere 10 μL di sospensione cellulare. La soluzione blu Trypan filtrata rimanente può essere conservata fino a 3 mesi a temperatura ambiente per un uso futuro.
- Spruzzare accuratamente l'emocitometro con etanolo al 70% e asciugare con un tovagliolo di carta sterile prima di fissare il copricapo sulla parte superiore utilizzando vapore del respiro. Far scorrere il coverslip attraverso la superficie inumidita del respiro induce forze coese generando anelli di Newton.
- Pipettare delicatamente la sospensione a celle blu Trypan su e giù utilizzando una pipetta da 1000 μL (per ridurre lo stress a pura lucentezza) prima di aggiungere 10 μL all'emocitometro. Assicurarsi che la soluzione sia dispersa sotto lo slittamento del coperchio e copra l'intera griglia senza bolle d'aria.

Figura 1: Conteggio delle cellule mediante emocitometro. Rappresentazione diagrammatica di un emocitometro che evidenzia da quale quadrante contare le cellule. Clicca qui per visualizzare una versione più grande di questa figura.
- Al microscopio, contare le cellule vive (non macchiate) e morte (blu macchiato) che si trovano nei quattro grandi quadrati d'angolo(Figura 1). Escludere tutte le celle che si sovrappongono o si trovano all'interno di due bordi dei grandi quadrati d'angolo (cioè sulle linee) nel conteggio.
- Utilizzando il seguente calcolo, calcolare il numero medio di cellule vive e vitali (non contaminate) presenti nel campione:
Numero totale di celle/mL = Conteggio celle vive x x 10.000
x x 10.000
dove la diluizione si riferisce a quante volte la soluzione stock è stata diluita in blu di Trypan (2x in questo caso) e il numero di quadrati contati si riferisce ai quattro grandi quadrati d'angolo dell'emocitometro contati - In base al numero di celle HepG2 praticabile e usando la formula seguente:
C1V1=C2V2
dove C1 = la concentrazione di cellule vitali attualmente,
V1 = il volume della sospensione cellulare attualmente,
C2 = la concentrazione di sospensione cellulare voluta,
V2 = il volume della sospensione cellulare voluto - Preparare una soluzione stock da 10 mL di sospensione cellulare HepG2 con terreno di coltura cellulare DMEM a una concentrazione di 2,0 x 105 celle/mL al fine di ottenere 4000 cellule HepG2 per 20 μL di caduta sospesa. Mescolare accuratamente la sospensione cellulare tubazionando delicatamente su e giù utilizzando una pipetta da 1000 μL per assicurarsi che tutte le celle siano completamente sospese all'interno del supporto.
- Ai pozzi di una piastra di coltura cellulare a 96 po ', aggiungere 100 μL di PBS sterile a temperatura ambiente per evitare che le gocce appese si asciughino durante l'incubazione.
- Prendere il coperchio di una piastra di coltura cellulare piatta standard da 96 porcile, invertirla e pipettare con cura 20 μL gocce della sospensione cellulare al centro di ogni scanalatura del poggiagli del coperchio, come mostrato nella figura 2. Utilizzare una pipetta multicanale ma aggiungere solo 2 - 4 gocce contemporaneamente poiché la semina multipla può influire sulla precisione e sul posizionamento delle gocce.
- Centrare le gocce all'interno delle scanalature dei pozzi disposti sul coperchio; altrimenti non si appenderanno al centro dei pozzi quando il coperchio della piastra viene capovolto e rischiano di cadere nella piastra. Capovolgere delicatamente il coperchio della piastra da 96 punti, quindi le gocce sono ora appese e posizionare con cura sopra la piastra da 96 pozzi.
- Posizionare l'intero piatto di 96 pozzi con coperchio delicatamente in un'incubatrice a 37 °C e 5% di CO2 per 3 giorni prima del trasferimento dello sferoide sull'agarosio.
NOTA: Occorre prestare particolare attenzione non solo quando si trasportano le piastre da/verso gli incubatori, ma quando si apre e si chiude l'incubatore in generale, poiché un movimento eccessivo può causare lo spostamento delle piastre e la caduta o la forma errata degli sferoidi.
 x x 10.000
x x 10.000
Figura 2: Preparazione del modello di sferoide 3D HepG2 in vitro. (A) Le cellule HepG2 sementi in 20 μL cade sul coperchio di una piastra da 96 po' . (B) Le cellule HepG2 dopo la semina nel modello a goccia appesa per consentire la formazione di sferoidi. Clicca qui per visualizzare una versione più grande di questa figura.
3. Trasferimento sferoide HepG2 dalla caduta sospesa alla sospensione dell'agarosio
NOTA: Il giorno 3 dopo la semina in gocce appese, gli sferoidi vengono trasferiti nei pozzi della stessa piastra da 96 po ', tutti precedentemente rivestiti con uno strato fine di gel di agarosio all'1,5%.
- Preparare gel di agarosio e autoclave (cioè il giorno 2 dopo la semina) prima del giorno del rivestimento della piastra (cioè il giorno 3 dopo la semina).
- Per preparare un gel di agarosio all'1,5%, pesare 0,30 g di agarosio in una bottiglia di vetro pulita e quindi aggiungere 20 ml di mezzo DMEM privo di fenolo-rosso. Autoclave l'agarosio per 1 h a 230 °C per la sterilizzazione. Il rivestimento in agarosio impedisce agli sferoidi HepG2 di aderire alla base dei pozzi e formare un monostrato cellulare invece di mantenere la loro struttura sferoide 3D.
- Il giorno 3 post semina, rimuovere la piastra da 96 po 'contenente gli sferoidi a goccia appesa HepG2 dall'incubatrice e capovolgere accuratamente il coperchio in modo che gli sferoidi non siano più appesi.
- Utilizzando una pipetta multicanale, rimuovere e scartare i 100 μL di PBS precedentemente aggiunti alla base della piastra da 96 pozza. Lasciare ariere le piastre per 2-3 minuti riscaldando l'agarosio in preparazione del rivestimento.
ATTENZIONE: Questa procedura si traduce in agarosio liquido molto caldo che se versato sulla pelle può bruciare e causare lesioni. Inoltre, è necessario prestare attenzione quando si maneggia la bottiglia di vetro contenente l'agarosio liquido in quanto anche questo può essere molto caldo. - Utilizzando i gel di agarosio all'1,5% precedentemente preparati, riscaldare la bottiglia di vetro contenente il gel di agarosio da 20 ml per 30 s in un forno a microonde al watt massimo (cioè 900 W). Per rivestire due piatti da 96 po ', dovrebbe essere sufficiente una bottiglia da 20 ml di gel di agarosio pre-preparato all'1,5%.
- Una volta sciolto, ruotare delicatamente l'agarosio ruotando la bottiglia di vetro per rimuovere eventuali bolle e quindi aggiungere 50 μL di agarosio nella base di ogni pozzo.
NOTA: Quando si aggiunge l'agarosio, assicurarsi di non inclinare la piastra >45° poiché l'agarosio si imposta rapidamente e non formerà uno strato piatto e livellato che può interrompere la crescita dello sferoide. È importante lavorare in modo efficiente in questa fase per evitare che l'agarosio si solidifica prima che la piastra sia completamente rivestita. - Lasciare riposare la piastra per 2 minuti a temperatura ambiente prima di aggiungere 100 μL di terreno di coltura cellulare DMEM prerifapidto (con integratori) sopra lo strato solido di agarosio in ogni pozzo.
- Capovolgere il coperchio della piastra da 96 po 'e riposizionare sopra la piastra da 96 po ', quindi gli sferoidi sono ora appesi ancora una volta.
- Centrifugare la piastra per 3 minuti a 200 x g per trasferire gli sferoidi dalla goccia appesa nei singoli pozzi della piastra da 96 pozzetti. Dopo il trasferimento, gli sferoidi HepG2 dovrebbero ora essere sospesi nel mezzo di coltura cellulare. Consentire loro di accontentarsi di 24 ore nell'incubatore a 37 °C e 5% CO2.
- Esporre sferoidi HepG2 di queste dimensioni a trattamenti chimici o ENM il giorno 4 dopo la semina (cioè 24 ore dopo il trasferimento in piastre rivestite di agarosio).
- Per mantenere la vitalità delle celle per lunghi periodi di coltura, aggiornare il mezzo di coltura cellulare ogni 3 giorni. Per fare questo, aspirare delicatamente 50 μL del mezzo di coltura cellulare dalla superficie del pozzo e sostituire con un nuovo mezzo di coltura cellulare DMEM 50 μL. Fare attenzione a non rimuovere o disturbare lo sferoide quando si esegue un cambio medio.
4. Esposizione nanomateriale/chimica
NOTA: Il modello di sferoide epatico HepG2 può supportare sia i regimi di esposizione ENM che chimici, ma l'obiettivo principale di questo protocollo sono le esposizioni ENM. Prima dell'esposizione, l'ENM di prova deve essere adeguatamente disperso; questo può essere eseguito come indicato dal Protocollo di dispersione NanoGenoTox (Accordo di sovvenzione n. 20092101, 2018)15.
- A seguito della dispersione secondo il protocollo di dispersione NanoGenoTox, diluire la sospensione ENM dalla concentrazione iniziale di 2,56 mg/mL alla concentrazione finale desiderata nel mezzo di coltura cellulare DMEM preriscaldato (compresi gli integratori). È necessario un volume totale di 5 mL per dosare una piastra da 96 pozione.
- Esporre lo sferoide HepG2 a una sostanza chimica o all'ENM, utilizzando una pipetta da 200 μL, aspirare 50 μL di mezzo di coltura cellulare dalla superficie di ciascun pozzo (lasciando 50 μL nel pozzo in modo da non disturbare gli sferoidi) e sostituire con un mezzo da 50 μL contenente il tossico in esame alla dose richiesta.
- Una volta applicato il materiale di prova, incubare le piastre per il tempo di esposizione desiderato a 37 °C e 5% CO2.
- Se viene condotto un regime di esposizione a lungo termine (≥24 ore), immediatamente dopo che è trascorso il periodo di esposizione desiderato, raccogliere gli sferoidi per l'analisi dell'endpoint micronucleo come descritto di seguito nei passaggi da 6.1 a 6.4.
- Tuttavia, con regimi di esposizione acuta (ad esempio, ≤24 ore), una volta terminato il periodo di esposizione, raccogliere, mettere in comune e immagazzinare 50 μL di supernatante da ogni pozzo nella piastra del pozzo 96 a -80 °C per ulteriori analisi biochimiche in seguito. Sostituire il mezzo di coltura cellulare con 50 μL di mezzo fresco contenente 6 μg/mL di citocalcasina B e lasciare incubare per cicli cellulari da 1 a 1,5 (cioè 24 -26 ore per HepG2) in preparazione per il raccolto del test del micronucleo del blocco di citocinesi.
NOTA: Per i regimi di esposizione acuta (≤24 h), è possibile applicare il saggio di micronucleo del blocco di citocinesi con citocalcasina B, ma per i regimi di esposizione a lungo termine (≥24 ore), la versione mononucleare (senza citocalcasina B) del saggio deve essere utilizzata come descritto di seguito nella figura 4.
5. Raccolta sferoide HepG2
NOTA: A seguito di trattamenti chimici o di esposizione enm, sia il mezzo di coltura cellulare che il tessuto sferoide possono essere raccolti per l'analisi di più endpoint. A seconda dell'analisi degli endpoint, gli sferoidi possono essere raccolti singolarmente (ad esempio, per l'analisi delle immagini) o raggruppati insieme (ad esempio, per il test del micronucleo del blocco di citocinesi).
- Rimuovere la piastra da 96 pozzi dall'incubatore.
- Utilizzando una pipetta da 200 μL, aspirare i 100 μL del mezzo di coltura cellulare, incluso il tessuto sferoide di ciascun pozzo, e raccogliere in un tubo di centrifuga sterile da 15 ml. Fare attenzione a evitare il contatto con l'agarosio.
- Una volta raccolto, centrifugare la sospensione dello sferoide a 230 x g per 5 minuti. Rimuovere il supernatante e conservare a -80 °C per ulteriori analisi degli endpoint (ad esempio, test di funzionalità epatica) in un secondo momento.
- Sospendere di nuovo il pellet di sferoidi in 1 mL di PBS sterile a temperatura ambiente (1x).
- Una volta lavato, centrifugare nuovamente la sospensione dello sferoide a 230 x g per 3 minuti. Scartare il supernatante, sospendere di nuovo in 500 μL della soluzione di tripsiderina-EDTA allo 0,05% e incubare per 6-8 min a 37 °C e 5% CO2.
- Dopo l'incubazione, pipettare delicatamente le cellule tripsinizzate su e giù per dissociare completamente e sospendere di nuovo le cellule HepG2 prima di neutralizzare con 1 mL di terreno di coltura cellulare DMEM.
- Centrifugare la sospensione cellulare diluita a 230 x g per 5 min. Scartare il supernatante in disinfettante e sospendere di nuovo il pellet cellulare in 2 mL di PBS a temperatura ambiente (1x).
- Centrifugare la sospensione cellulare a 230 x g per 5 min. Scartare il supernatante in disinfettante e quindi sospendere nuovamente il pellet cellulare in 2 mL di PBS freddo (1x). Assicurarsi che le celle siano ben disperse per evitare che ciuffi di cellule oscurano il campo visivo quando montate su vetrini al microscopio.
6. Test e punteggio del micronucleo
Per il metodo manuale del saggio del micronucleo, è necessario un citocentrifugo per produrre un citodot (una regione di cellule definita e concentrata) al centro dello scivolo del microscopio. Questo processo supporta un punteggio più efficiente della diapositiva in quanto consente al marcatore di individuare facilmente le celle di interesse, anziché valutare un'intera diapositiva in cui le celle possono essere ampiamente diffuse.
- Vetrini al microscopio smerigliato a immersione (tre per dose) in etanolo al 70% seguito da ddH2O e lasciare asciugare all'aria per 5 minuti.
- Posizionare i vetrini preparati in imbuto di cuvette, come illustrato nella figura 3A, dove lo scivolo di vetro (iii) è posto nel supporto metallico (iv) con una scheda filtrante (ii) e un imbuto di cuvette (i) fissato in alto.
- Disporre gli imbuti di cuvette nel citocentrifugo con l'imbuto rivolto verso l'alto, in modo che 100 μL di sospensione cellulare possano essere aggiunti direttamente in ciascuno di essi.
- Citospino per 5 minuti a 500 x g per garantire che le cellule siano distribuite uniformemente sulla superficie dello scivolo.

Figura 3: Configurazione dei citospini per preparare le cellule trattate su vetrini al microscopio. (A) Visualizza i singoli componenti, (i) imbuto di cuvette, (ii) scheda filtrante, (iii) vetrino al microscopio in vetro e (iv) supporto metallico necessario per citospino cellule HepG2 su vetrini al microscopio. (B)L'imbuto finale della cuvette allestito. (C) Il corretto posizionamento dell'imbuto della cuvetta all'interno del citocentrifugo. Clicca qui per visualizzare una versione più grande di questa figura.
- Lasciare asciugare i vetrini all'aria prima della fissazione in ghiaccio-freddo, 90% metanolo per 10 min.
- Una volta fissati, lasciare asciugare i vetrini all'aria durante la notte a temperatura ambiente prima di conservare a -20 °C per un massimo di 6 mesi.
- Se necessario, rimuovere i vetrini del microscopio pre-preparati dal congelatore a -20 °C e lasciare riscaldare a temperatura ambiente prima di effettuare la colorazione Giemsa.
ATTENZIONE: Ai sensi del regolamento (CE) n. 1272/2008 [CLP], la soluzione di colorazione Giemsa è un liquido altamente infiammabile che può essere tossico se ingerito e causare danni a contatto con gli occhi, la pelle o se inalato. Fare riferimento alla scheda SDS associata per informazioni dettagliate sullo stoccaggio, la manipolazione e la salute e la sicurezza di questa sostanza chimica prima dell'uso. - Mentre le diapositive si stanno scongelando, preparare una soluzione di colorazione Giemsa al 20% (25 mL necessari per macchiare ~30 diapositive) diluita in tampone di fosfatasi (pH 6.8). Mescolare accuratamente ruotando delicatamente la soluzione prima di filtrare utilizzando carta filtrante piegata posta in un imbuto.
- Utilizzando una pipetta Pasteur, aggiungere 3 - 5 gocce di soluzione Giemsa filtrata al citodot su ogni diapositiva e lasciare per 8 - 10 minuti.
- Lavare gli scivoli in due lavaggi tampone fosfatasi successivi prima di risciacquare brevemente sotto acqua fredda per rimuovere eventuali avanzi di macchia in eccesso. Lasciare gli scivoli all'aria asciutti.
- Una volta asciutto, in un cappuccio fumi, immergere gli scivoli macchiati in xilene per 10 s prima di aggiungere una goccia di mezzo di montaggio al centro del citodot e un posto un coverslip di vetro sulla parte superiore.
- Lasciare asciugare durante la notte i vetrini del microscopio nella cappa dei fumi prima di segnare manualemente; possono essere conservati a tempo indeterminato a temperatura ambiente.
7. Analisi dei dati
- Come descritto nelle linee guida per i test 487 (2014)11dell'OCSE, per valutare e quantificare i danni al DNA indotti dall'esposizione a un ENM o ad un agente chimico, utilizzare un microscopio a luce (obiettivo 100x con olio ad immersione) 2000 cellule mononucleate o 1000 binucleate per replicazione biologica per ottenere la presenza di micronuclei, come mostrato nella figura 4.

Figura 4: Albero decisionale per la soluzione del test del micronucleo. Albero decisionale schematico per evidenziare la necessità di diverse procedure di valutazione del punteggio e della citotossicità quando si utilizza il saggio di micronucleo con modelli 3D a seguito di regimi di esposizione acuta o a lungo termine. Le esposizioni acute (≤24 ore) consentono l'uso del saggio di micronucleo bloccato dalla citocinesi, mentre le esposizioni a lungo termine (≥24 ore) richiedono la versione mononucleare del saggio; entrambi sono descritti nell'indirizzo 487 del test dell'OCSE. Clicca qui per visualizzare una versione più grande di questa figura.
- Sulla base della proporzione di micronuclei presenti per numero di cellule mononucleate o binucleate valutate, calcolare una percentuale del valore di genotossicità.
- Al fine di valutare il danno al DNA osservato non è il risultato di detriti cellulari causati da un'alta percentuale di cellule apoptotiche, prendere una misura di citotossicità insieme. In questo caso, a seconda della presenza di Citocalcasina B, utilizzare il calcolo CPBI o RVCC (come descritto nella figura 4). La genotossicità deve essere valutata solo in campioni in cui la citotossicità è inferiore al 55% ± 5% come definito nella linea guida 48711 dell'OCSE.
Representative Results
L'idoneità di questo modello di sferoide epatico 3D a base di linea cellulare per la coltura a lungo termine e la valutazione del rischio genotossico è stata valutata conducendo la caratterizzazione di base per determinare la vitalità e la funzionalità epatica del modello per la durata di 14 giorni in coltura e la sua applicabilità per il test del micronucleo.
Caratterizzazione di base del modello sferoide epatico 3D HepG2
Prima di qualsiasi valutazione tossicologica in vitro, è importante verificare che gli sferoidi 3D HepG2 si siano formati correttamente prima di eseguire il trasferimento di agarosio o il trattamento chimico /ENM. Gli sferoidi HepG2 prodotti con il metodo della goccia appesa di solito durano da 2 a 3 giorni dopo la semina (4000 cellule/sferoide) per formare sferoidi compatti e sferici con un diametro medio di 495,52 μm W x 482,69 μm H, come mostrato nella figura 5A-5C. Gli sferoidi HepG2 che si sono formati correttamente e sono accettabili per essere utilizzati per la valutazione tossicologica in vitro devono avere una struttura compatta e sferica con una superficie liscia e senza proiezioni visive. La figura 5 fornisce esempi di sferoidi di buona qualità (figura5D-F)e di scarsa qualità (figura5G-I). Quest'ultimo dovrebbe essere scartato. Tipicamente, il 90-95% degli sferoidi formati per piastra si formerà correttamente ed sarà vitale per ulteriori sperimentazioni.

Figura 5: Immagini di microscopia leggera che mostrano la morfologia naturale degli sferoidi HepG2 formati tramite il metodo della goccia appesa . (A-C) mostrano il giorno 2 e (D-I) Giorno 4 Sferoidi epatici HepG2 dopo la semina. (D-F) sono esempi di sferoidi HepG2 di buona qualità, mentre(G-I)mostra sferoidi mal formati. Tutte le immagini sono state scattate su un obiettivo X20 utilizzando un microscopio. La barra di scala rappresenta 20 μm. Fare clic qui per visualizzare una versione più grande di questa figura.
Per confermare ulteriormente la fattibilità dello sferoide HepG2, è possibile eseguire un saggio colorimetrico di base Bromocresol Green Albumin (BCG) o un saggio di Urea per valutare la loro funzionalità epatica. La funzionalità epatica è stata valutata in linea con la fattibilità utilizzando il test di esclusione blu di Trypan per un periodo di coltura di 14 giorni per determinare la longevità del modello di sferoide epatico e stabilire se poteva supportare la valutazione del rischio a lungo termine o ripetuta basata su ENM / chimica(figura 6). La concentrazione di albumina rimase costante per tutta la durata del periodo della coltura. La produzione di urea mostra un aumento della concentrazione di urea prodotta per sferoide per una settimana di coltura prima di raggiungere un altopiano entro il giorno 7. È importante notare che i livelli di albumina e urea prodotti negli sferoidi 3D HepG2 sono sostanzialmente superiori a quelli osservati nella stessa linea cellulare coltivata in formato 2D. In effetti, le colture 2D delle cellule HepG2, i livelli di picco di albumina e urea erano rispettivamente 0,001 mg/mL e 0,010 ng/μL. Inoltre, in precedenti lavori pubblicati da Shah et al. utilizzando un sistema sferoide HepG2 quasi identico, gli autori evidenziano un notevole miglioramento dell'attività metabolica (CYP1A1 e CYP1A2) nei sistemi modello 3D HepG2 in vitro rispetto alle cellule HepG2 coltivate in 2D5.

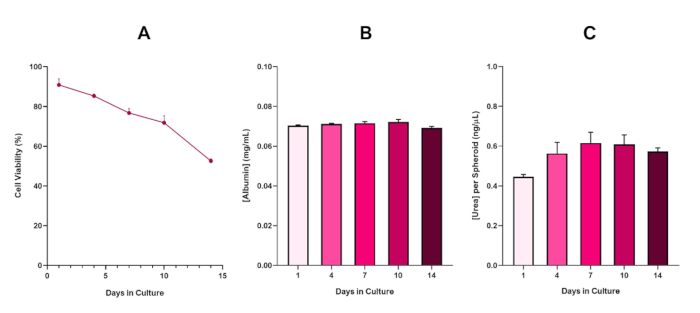
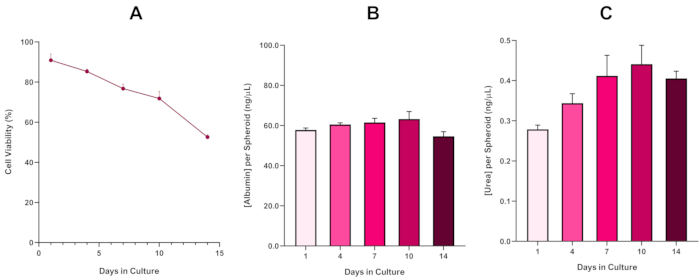
Figura 6: Dati di caratterizzazione di base a 14 giorni per gli sferoidi epatici HepG2. A seguito del trasferimento dalla caduta sospesa, (A) evidenzia la fattibilità del modello sferoide HepG2 per un periodo di 14 giorni, mentre (B) e (C) evidenziano rispettivamente la funzionalità dell'albumina e dell'urea simile al fegato. Dati meschino ± SEM presentati, n = 4. Clicca qui per visualizzare una versione più grande di questa figura.
Con l'inevitabile sviluppo di un nucleo necrotico, una limitazione nota delle colture di sferoidi epatiche 3D, è stato necessario stabilire la fattibilità di questo modello basato su HepG2 per dimostrare che era in grado di sostenere regimi di esposizione a lungo termine (5-10 giorni) mantenendo la capacità proliferativa necessaria per supportare il test del micronucleo5. In effetti, questo modello di sferoide epatico 3D ha dimostrato di mantenere la vitalità del >70% in 10 giorni in coltura. Sulla base di questo e in combinazione con la funzionalità epatica sostenuta osservata durante il periodo di coltura di ≥14 giorni, questo modello di sferoide epatico 3D può quindi supportare regimi di esposizione ENM ripetuti a lungo termine fino a 10 giorni (cioè, prima che la vitalità degli sferoidi scende al di sotto del 70%). Per riferimento, si consiglia che i livelli di albumina per gli sferoidi HepG2 sementi a 4000 cellule/sferoide siano di ≥20,0 ng/μL, mentre la produzione di urea dovrebbe essere di ≥0,25 ng/μL prima di condurre una valutazione tossicologica in vitro con questo modello.
Valutazione della genotossicità dei nanomateriali ingegnerizzati
Per la valutazione della genotossicità, il saggio del micronucleo è stato utilizzato per determinare la presenza di micronuclei a seguito di esposizioni acute (24 ore) e a lungo termine (120 h) enm. L'aflatossina B1 è un agente cancerogenoepatico noto 16,17 ed è un controllo positivo raccomandato per il test del micronucleo. Esperimenti di ottimizzazione hanno dimostrato che 0,1 μM di Alfatossina B1 induce una risposta genotossica positiva significativa (≥ aumento di 2,0 volte) negli sferoidi epatici 3D HepG2 e quindi viene utilizzata in ogni saggio di micronucleo condotto con questo modello. Per garantire la validità dei risultati del test del micronucleo utilizzando il modello sferoide HepG2, la frequenza micronuclea di fondo per le cellule HepG2 utilizzate in questo modello 3D in vitro dovrebbe trovarsi entro un intervallo dello 0,6% - 1,2%. Di conseguenza, l'alfatossine B1 dovrebbe indurre una risposta genotossica di almeno due volte superiore a quella osservata con il controllo negativo; pertanto, 0,1 μM di alfatossina B1 dovrebbero indurre una frequenza micronuclei compresa tra l'1,5% e il 3,0%. Utilizzando questi parametri di controllo, la genotossicità associata all'ENM in vitro può quindi essere valutata in modo affidabile. Sulla base della linea guida 487 dell'OCSE per i test, è importante notare che durante la prova di un ENM o di una sostanza chimica, le concentrazioni selezionate non devono indurre più del 55% ± 5% di citotossicità (indicata da una riduzione dei valori CPBI o RVCC in relazione al controllo negativo)11. La figura 7 illustra i dati generati quando l'aflatossina B1 e due ENM (biossido di titanio (TiO2) e sliver (Ag)) sono state valutate a seguito di esposizioni sia acute che a lungo termine negli sferoidi HepG2, e il successivo potenziale genotossico è stato analizzato utilizzando il saggio del micronucleo. Entrambe le ENM valutate sono state testate a una dose non citotossica e bassa di 5,00 μg/mL su un'esposizione acuta (24 ore) e un regime di esposizione a lungo termine (120 h). Una tendenza simile per la genotossicità sia nelle ENM TiO2 che ag può essere osservata, per cui l'elevata risposta di genotossicità risultante dall'esposizione di 24 ore non era evidente dopo un'esposizione a lungo termine di 5 giorni. Ciò nonostante la genotossicità sostenuta indotta dal controllo positivo dell'aflatossina B1 in entrambi i punti di tempo.

Figura 7: Valutazione della genotossicità a seguito dell'esposizione di TiO2 e Ag ENM sugli sferoidi epatici HepG2. Valutazione della genotossicità (frequenza micronuclea) mediantel'esposizione a lungo termine (120ore)a 5,00 μg/mL di TiO2 e Ag ENM. Il controllo negativo è solo un supporto, mentre il controllo positivo è di 0,1 μM di aflatossina B1. Dati meschino (n=2) ± SD. Significatività indicata in relazione al controllo negativo: * = p≤ 0,05. Clicca qui per visualizzare una versione più grande di questa figura.
Discussion
Le applicazioni per i modelli epatici 3D variano considerevolmente a seconda del particolare endpoint biochimico o della via di esito avverso presa di mira. Ogni modello ha i suoi benefici e limiti, dalla variazione interdonore nei modelli di epatociti umani primari (PHH) alla ridotta attività del citocromo p450 nei modelli basati sulla linea cellulare, ma tutti sono preziosi a se stessi6,12,18,19. Quando si valuta la genotossicità ci sono limitazioni nei modelli compatibilità con endpoint approvati dalla regolamentazione come il saggio di micronucleo in vitro, poiché è necessaria la proliferazione attiva. Ciò è necessario, poiché la valutazione della genotossicità richiede che la quantificazione dei danni fissi al DNA sia valutata dopo la divisione cellulare quando vi è la possibilità di riparare il DNA per correggere le lesioni transitorie. Sfortunatamente, gli epatociti altamente differenziati (cioè gli sferoidi a base di HepaRG) o le microtissudi PHH, che si ritiene presentino le caratteristiche epatiche più fisiologicamente rilevanti, formano modelli statici (non proliferativi)12,19,20. Di conseguenza, il modello sferoide 3D HepG2 qui presentato fornisce un modello alternativo adatto in grado di supportare i test di genotossicità. Gli sferoidi a base di linea cellulare HepG2 hanno sufficienti cellule che dividono attivamente sulla superficie esterna degli sferoidi pur mantenendo caratteristiche epatiche di base, come la produzione di albumina e urea e alcune attività CYP4505,12,19. Principalmente questo modello epatico in vitro è stato sviluppato per integrare il saggio del micronucleo, poiché questo è uno dei due saggi in vitro raccomandati nella batteria per i test di genotossicità8,10,11,21. Tuttavia, il modello può essere prontamente applicato alle tecnologie di analisi del sequenziamento del DNA e di espressione genica (RNA), mentre ha il potenziale per essere ulteriormente adattato e utilizzato per altri endpoint di danno al DNA, come il saggio della cometa. Tuttavia, è importante considerare il ruolo svolto dalle interferenze ENM in alcune analisi degli endpoint. Ad esempio, le analisi basate sulla citometria del flusso potrebbero non essere adatte per la valutazione della genotossicità ENM specificamente a causa dell'interferenza delleparticelle 22.
Un fattore limitante dei modelli di sferoide che subiscono attivamente la divisione cellulare è la loro dimensione. L'ottimizzazione della densità di semina è fondamentale in quanto devono esserci abbastanza cellule che consentano al modello di continuare a proliferare; ma non un numero di cellule troppo alto, il che fa diventare lo sferoide e troppo compatto, portando ad un aumento del nucleo necrotico. Si ritiene che la causa di questa necrosi sia limitata all'ossigeno e alla diffusione dei nutrienti, poiché si pensa che il limite di questa diffusione sia di circa 100 - 150 μm ditessuto 23,24. Tuttavia, questo dipende dal tipo di cella, dal numero di cella, dalle interazioni dell'impalcatura e dalle condizioni dicoltura 25. Poiché, è stato dimostrato che circa 700 μm di diametro è il limite per evitare l'insorgenza prematura della necrosi al centro degli sferoidi C3A, la semina di 4000 cellule HepG2 per sferoide assicura che il diametro del modello al momento dell'esposizione sia di ≤500 μm26. Inoltre, Shah et al. Per superare questo problema, il modello ideato nel presente protocollo subisce un passaggio critico in cui la goccia appesa viene trasferita in pozzi rivestiti di agarosio dopo la formazione iniziale dello sferoide. Ciò garantisce un maggiore volume di mezzo di coltura presente per sostenere il numero sempre crescente di cellule all'interno degli sferoidi. Di conseguenza, il modello sferoide HepG2 rimane percorribile oltre il 70% dopo 10 giorni in coltura e può essere utilizzato per la valutazione del rischio a lungo termine in vitro.
Mentre il modello di sferoide HepG2 può supportare sia regimi di esposizione acuta che a lungo termine, il mezzo di coltura cellulare rinfrescante durante lunghi periodi di coltura è limitato per questo modello in quanto la sostituzione completa del mezzo non è consigliata a causa della potenziale perdita degli sferoidi. Si presume che con le esposizioni enm, la tendenza di dispersioni omogenee dell'ENM ad agglomerare e sedimentare sia elevata. Tuttavia, è degno di nota il fatto che la velocità con cui un sedimento ENM può variare a seconda dei parametri delle particelle (ad esempio, dimensioni, forma e densità) e può essere determinata teoricamente utilizzando il modello di sedimentazione, diffusione e dosimetria in vitro (ISDD), o i suoi derivati recenti, spesso menzionati quando l'esposizione all'ENM (sospensione)si avvicina a 27,28. Con questa mente, si presume che se solo il 50% del mezzo di coltura cellulare viene accuratamente rimosso dalla superficie della coltura cellulare, l'interruzione e la successiva rimozione della dose ENM dovrebbero in teoria essere minime. Tuttavia, con il moto browniano in gioco, questo potrebbe non essere strettamente il caso e ulteriori lavori sulla deposizione e la sedimentazione di ogni particolare ENM da testare dovrebbero essere intrapresi per garantire che la dosimetria corretta sia mantenuta per tutti i regimi di esposizione a lungo termine27. Principalmente questa è una potenziale limitazione da considerare quando si eseguono ripetuti regimi di dosing in quanto ciò potrebbe essere fondamentale per la concentrazione finale accumulata. Le esposizioni a base chimica, d'altra parte, pur non senza i propri limiti da considerare, offrono un approccio più semplicistico in quanto le sostanze chimiche tendono a rimanere in soluzione e quindi una sostituzione diretta della concentrazione chimica originale oltre alla concentrazione appena aggiunta garantisce che qualsiasi sostanza chimica persa durante il ristoro del supporto sia sostituitadi conseguenza 29. Le applicazioni future includerebbero la valutazione dell'idoneità del modello per i regimi di esposizione ripetuta durante i periodi di coltura a lungo termine, poiché le strategie di dosamento ripetute sono di fondamentale importanza per valutare la capacità di un particolare sistema di organi di migliorare o superare gli eventuali effetti avversi indotti dal bioaccumulo di una sostanza xenobiotica.
In conclusione, questo modello epatico in vitro 3D ha la capacità di essere utilizzato per valutare una serie di scenari di esposizione realistici, fornendo così un futuro approccio in vitro per supportare meglio sia l'ENM che la valutazione del rischio chimico in modo di routine e facilmente accessibile.
Disclosures
Gli autori non hanno nulla da rivelare.
Acknowledgments
Gli autori vorrebbero riconoscere che questa ricerca ha ricevuto finanziamenti dal programma di ricerca e innovazione Horizon 2020 dell'Unione Europea per il progetto PATROLS, nell'ambito della sovvenzione n. 760813
Materials
| Name | Company | Catalog Number | Comments |
| Aflotoxin B1 | Sigma Aldrich, UK | A6636-5MG | |
| Agarose | Sigma Aldrich, UK | A9539-50G | |
| Autoclave Tape | |||
| BCG Albumin Assay | Sigma Aldrich, UK | MAK124 | |
| Bovine Serum Albumin Powder | Sigma Aldrich, UK | A9418 | |
| Cell Freezing Aid | Thermo Fisher Scientific, UK | 5100-0001 - Mr Frosty | |
| Centrifuge | Eppendorf | 5810 R | |
| Cytochalasin B | Merck, UK | 250233 | |
| Cytology Metal Clips | |||
| Cytospin 4 Centrifuge | ThermoFisher Scientific, UK | CM00730202 | |
| DMEM with 4.5g/L D-Glucose, L-Glutamine | GIBCO, Paisley, UK | 41965-039 | |
| DMEM, phenol-red free with 4.5g/L D-Glucose, L-Glutamine with Hepes | GIBCO, Paisley, UK | 21063-029 | |
| DPX Mounting Medium | FisherScientific, UK | D/5330/05 | |
| Ethanol | FisherScientific, UK | 10048291 | |
| FBS | GIBCO, Paisley, UK | 10270-106 | |
| Filter Cards for Shandon Cytospin | FisherScientific, UK | 15995742 | |
| Frosted Glass Slides | ThermoFisher Scientific, UK | ||
| Giemsa's Stain Improved R66 Solution, Gurr | VWR Chemicals, UK | MFCD00081642 | |
| Glass Coverslips (24 x 60) | Deckglaser, VWR | ECN631-1575 | |
| Haemocytometer and Coverslip | |||
| Immersion Oil for Microscope | Zeiss, UK | 518F, ISO8034 | |
| Laminar Class II Tissue Culture Hood | Scanlaf Mars | ||
| Light Microscope | Zeiss, UK | Axiovert 40C | |
| Liquid Nitrogen | |||
| Methanol | FisherScientific, UK | 10284580 | |
| Microwave | |||
| Non-Filtered, Sterile 200µl and 1000µl Pipette tips | Greiner-Bio-One, UK | ||
| NuncMicroWell 96-Well Microplates | ThermoFisher Scientific, Denmark | 167008 | |
| P1000 and P200 micropipettes | |||
| P300 and P50 multi-channel pipettes | |||
| PBS pH 7.4 1X, MgCl2 and CaCl2 Free | GIBCO, Paisley, UK | 14190-094 | |
| Pen/Strep | GIBCO, Paisley, UK | 15140-122, Penicillin/Strepmyocin 100X or 10,000U/ml | |
| Phosphatase Buffer Tablets | GIBCO, Paisley, UK | 10582-013 | |
| Pipette Boy | |||
| Simport Scientific CytoSep Funnels for Shandon Cytospin 4 Centrifuges | FisherScientific, UK | 11690581 | |
| Sonifier SFX 550 240V CE 1/2" - Probe | Branson, USA | 101-063-971R | |
| T-25 and T-75 Tissue Culture Flask | Greiner-Bio-One, UK | T-25 (690175) and T-75 (660175) | |
| Trypan Blue Solution | Sigma Aldrich, UK | T8154-100mL | |
| Urea Assay Kit | Sigma Aldrich, UK | MAK006 | |
| Virkon Disinfectant | DuPont, UK | Rely+On Virkon | |
| Water Bath (37?C) | Grant JBNova 18 | ||
| Weighing Balance | |||
| Xylene | FisherScientific, UK | 10588070 | |
| 0.05% Trypsin-EDTA | GIBCO, Paisley, UK | 5300-054 | |
| 0.2mL and 1.0mL Eppendorf Tubes | Greiner-Bio-One, UK | ||
| 0.45µm Filter Unit | Millex HA, MF-Millipore, UK | SLHA033SS | |
| 1.0mL Syringe | BD Plastipak, FisherScientific, UK | 300185 | |
| 20mL LS Scintillation Glass Vials, 22-400 Foil Lined PP Caps | DWK Life Sciences GmbH, Germany | WHEA986581 | |
| 37?C and 5% CO2 ISO Class 5 Hepa Filter Incubator | NUAIRE DHD Autoflow | ||
| 3mL Pasteur Pipette | Greiner-Bio-One, UK | ||
| 50mL Conical Falcon Tubes | Greiner-Bio-One, UK | ||
| 50mL or 100mL Glass Bottles | |||
| 50mL Skirted Falcon Tubes | Greiner-Bio-One, UK | ||
| 5mL, 10mL and 25mL Pipettes | Greiner-Bio-One, UK | ||
| 9.4cm Square, Petri Dish | Greiner-Bio-One, UK | 688161 |
References
- Geiser, M., Kreyling, W. G. Deposition and biokinetics of inhaled nanoparticles. Particle and Fibre Toxicology. 7, 2 (2010).
- Modrzynska, J. Toxicological effects of nanoparticle deposition in the liver. Kgs. Lyngby, Denmark: Technical University of Denmark. , (2018).
- Elje, E., et al. The comet assay applied to HepG2 liver spheroids. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 845, 403033 (2019).
- Breslin, S., O'Driscoll, L. Three-dimensional cell culture: the missing link in drug discovery. Drug Discovery Today. 18, 240-249 (2013).
- Shah, U. -K., et al. A three-dimensional in vitro HepG2 cells liver spheroid model for genotoxicity studies. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 825, 51-58 (2018).
- Lauschke, V. M., Hendriks, D. F. G., Bell, C. C., Andersson, T. B., Ingelman-Sundberg, M. Novel 3D Culture Systems for Studies of Human Liver Function and Assessments of the Hepatotoxicity of Drugs and Drug Candidates. Chemical Research in Toxicology. 29, 1936-1955 (2016).
- van Grunsven, L. A. 3D in vitro models of liver fibrosis. Advanced Drug Delivery Reviews. 121, 133-146 (2017).
- Corvi, R., Madia, F. In vitro genotoxicity testing - can the performance be enhanced. Food and Chemical Toxicology. 106, 600-608 (2017).
- Doak, S. H., Manshian, B., Jenkins, G. J. S., Singh, N. In vitro genotoxicity testing strategy for nanomaterials and the adaptation of current OECD guidelines. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 745, 104-111 (2012).
- Fenech, M. Cytokinesis-block micronucleus cytome assay. Nature Protocols. 2, 1084-1104 (2007).
- OECD. OECD Guidelines. Test 489: In vivo Mammalian Alkaline Comet Assay. , (2016).
- Gerets, H. H. J., et al. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biology and Toxicology. 28, 69-87 (2012).
- Sison-Young, R. L., et al. A multicenter assessment of single-cell models aligned to standard measures of cell health for prediction of acute hepatotoxicity. Archives of Toxicology. 91, 1385-1400 (2017).
- European Guidelines 2019. European Agency for Safety and Health at Work. , Available from: https://osha.europa.eu/en/safety-and-health-legislation/european-guidelines (2019).
- Jensen, K. A. The NANOGENOTOX Dispersion Protocol for NANoREG. European Union Grant Agreement n° 2009. 21, 01 (2014).
- Marchese, S., et al. Aflatoxin B1 and M1: Biological Properties and Their Involvement in Cancer Development. Toxins. 10, 214 (2018).
- Rushing, B. R., Selim, M. I. Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food and Chemical Toxicology. 124, 81-100 (2019).
- Kermanizadeh, A., Brown, D. M., Moritz, W., Stone, V. The importance of inter-individual Kupffer cell variability in the governance of hepatic toxicity in a 3D primary human liver microtissue model. Scientific Reports. 9, 7295 (2019).
- Berger, B., et al. Comparison of Liver Cell Models Using the Basel Phenotyping Cocktail. Frontiers in Pharmacology. 7, 443 (2016).
- Ramaiahgari, S. C., et al. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Archives of Toxicology. , (2014).
- Li, Y., et al. Factors affecting the in vitro micronucleus assay for evaluation of nanomaterials. Mutagenesis. 32 (1), 151-159 (2016).
- Kirkland, D., Reeve, L., Gatehouse, D., Vanparys, P. A core in vitro genotoxicity battery comprising the Ames test plus the in vitro micronucleus test is sufficient to detect rodent carcinogens and in vivo genotoxins. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 721 (1), 27-73 (2011).
- Curcio, E., et al. Mass transfer and metabolic reactions in hepatocyte spheroids cultured in rotating wall gas-permeable membrane system. Biomaterials. 28, 5487-5497 (2007).
- Glicklis, R., Merchuk, J. C., Cohen, S. Modeling mass transfer in hepatocyte spheroids via cell viability, spheroid size, and hepatocellular functions. Biotechnology and Bioengineering. 86, 672-680 (2004).
- Asthana, A., Kisaalita, W. S. Microtissue size and hypoxia in HTS with 3D cultures. Drug Discovery Today. 17, 810-817 (2012).
- Gaskell, H., et al. Characterization of a functional C3A liver spheroid model. Toxicology Research. 5, 1053-1065 (2016).
- Cho, E. C., Zhang, Q., Xia, Y. The effect of sedimentation and diffusion on cellular uptake of gold nanoparticles. Nature Nanotechnology. 6, 385-391 (2011).
- Hinderliter, P. M., et al. ISDD: A computational model of particle sedimentation, diffusion and target cell dosimetry for in vitro toxicity studies. Particle and Fiber Toxicology. 7, 36 (2010).
- Kramer, N. I., di Consiglio, E., Blaauboer, B. J., Testai, E. Biokinetics in repeated-dosing in vitro drug toxicity studies. Toxicology in vitro. 30, 217-224 (2015).
Tags
Bioingegneria Numero 160 Modelli epatici in vitro Nanomateriali Valutazione del pericolo Esposizione a lungo termine Nano(geno)tossicologia Danni al DNAErratum
Formal Correction: Erratum: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure
Posted by JoVE Editors on 01/26/2021.
Citeable Link.
An erratum was issued for: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure. The Representative Results section was updated.
Figure 6 in the Representative Results section was updated from:
to:
The fourth paragraph in the Representative Results section was updated from:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥0.06 mg/mL whilst urea production should be ≥0.4 ng/µL before conducting an in vitro toxicological assessment with this model.
to:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥50.0 ng/μL whilst urea production should be ≥0.25 ng/µL before conducting an in vitro toxicological assessment with this model.
