

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Denne prosedyren ble etablert for å brukes til å utvikle avanserte 3D leverkulturer in vitro, som kan gi en mer fysiologisk relevant vurdering av gentoksiske farer forbundet med nanomaterial eksponering over både akutte eller langsiktige, gjentatte doseregimer.
Abstract
På grunn av den raske utviklingen og implementeringen av et mangfoldig utvalg av konstruerte nanomaterialer (ENM), er eksponering for ENM uunngåelig, og utviklingen av robuste, prediktive in vitro-testsystemer er avgjørende. Levertoksikologi er nøkkelen når man vurderer ENM-eksponering, da leveren tjener en viktig rolle i metabolsk homeostase og avgiftning, samt å være et viktig sted for ENM-akkumulering etter eksponering. Basert på dette og den aksepterte forståelsen av at 2D-hepatocyttmodeller ikke nøyaktig etterligner kompleksiteten i intrikate multi-cellulære interaksjoner og metabolsk aktivitet observert in vivo, er det større fokus på utvikling av fysiologisk relevante 3D-levermodeller skreddersydd for ENM-farevurderingsformål in vitro. I tråd med prinsippene til 3R-ene for å erstatte, redusere og foredle dyreforsøk, er det utviklet en 3D HepG2 cellelinjebasert levermodell, som er et brukervennlig, kostnadseffektivt system som kan støtte både utvidede og gjentatte ENM-eksponeringsregimer (≤ 14 dager). Disse sfæroidmodellene (≥500 μm i diameter) beholder sin proliferative kapasitet (dvs. delecellemodeller) slik at de kan kobles sammen med "gullstandarden" mikronukleusanalyse for effektivt å vurdere gentoksisitet i vitro. Deres evne til å rapportere om en rekke toksikologiske endepunkter (f.eks. leverfunksjon, (pro-)inflammatorisk respons, cytotoksisitet og gentoksisitet) har blitt karakterisert ved hjelp av flere ENMs på tvers av både akutte (24 timer) og langsiktige (120 h) eksponeringsregimer. Denne 3D in vitro levermodellen har kapasitet til å bli brukt til å evaluere mer realistiske ENM-eksponeringer, og gir dermed en fremtidig in vitro-tilnærming for bedre å støtte ENM-farevurdering på en rutinemessig og lett tilgjengelig måte.
Introduction
På grunn av den raske utviklingen og implementeringen av et mangfoldig utvalg av konstruerte nanomaterialer (ENM) på tvers av en mengde menneskebaserte applikasjoner (f.eks. mat, kosmetikk, klær, sportsutstyr, elektronikk, transport og medisin), er det uunngåelig at mennesker vil bli utsatt for ENM med jevne mellomrom. Med dette er det økte bekymringer for at de nye, størrelsesspesifikke fysiokjemiske egenskapene som anser disse materialene fordelaktige i mange applikasjoner, kan forårsake negative effekter på menneskers helse og miljøet samtidig. For tiden er mange internasjonale aktiviteter på plass for aktivt å reflektere mer fysiologisk relevante eksponeringer for disse ENM og vurdere den potensielle toksisiteten til disse materialene over akutte, langsiktige og gjentatte lavdoseeksponeringsscenarier.
Levertoksikologi er nøkkelen når man vurderer ENM-eksponering, da det er allment kjent at leveren er et viktig sted for ENM-akkumulering etter eksponering1,2. Videre er leveren det primære organsystemet for metabolisme og avgiftning av stoffer som går inn i systemisksirkulasjon 3. Basert på den aksepterte forståelsen av at 2D-hepatocyttmodeller ikke nøyaktig etterligner kompleksiteten i intrikate multicellulære interaksjoner eller på riktig måte representerer metabolsk aktivitet observert in vivo, er det etablert et større fokus på å utvikle robuste og fysiologisk relevante in vitro 3D levermodeller for in vivo-erstatningsteknologier er etablert4,5. Bruk av avanserte 3D-kulturteknologier forbedrer levetiden til in vitro levermodeller, noe som gjør det mulig å undersøke gjentatte eksponeringsregimer på lang sikt. I tillegg fremmer dette avanserte kulturformatet dannelsen av forbedrede fysiologiske, organotypiske egenskaper som galle canaliculi, aktive transportørprosesser og forbedrede CYP450-stoffmetaboliseringsevner, og dermed forbedre forutsigbarheten til modellene6. Nåværende 3D in vitro levermodeller bestående av monokulturer (kun hepatocytter) eller medkulturer (hepatocytter med ikke-parenchymale celler) finnes i flere formater, alt fra mikrotissues eller sfæroider i ultralow adhesjon plater, hengende dråpe sfæroider, celler innebygd i matriser og / eller stillaser og mikrofluidiske cellekulturplattformer, som alle anses effektive avanserte in vitro modeller for hepatisk toksisitet vurdering6,7. Imidlertid er de fleste av disse modellsystemene høyt vedlikehold, krever spesialisert utstyr og er dyre. Videre er disse modellene ofte statiske (dvs. ikke-viderende cellemodeller) som forhindrer bruk i vurderingen av fareendepunkter, for eksempel gentoksisitetstesting ved hjelp av metoder som kvantifiserer fast DNA-skade. Gentoksisitet er en sentral forutsetning i regulatorisk toksikologi, og det er en viktig komponent i risikovurderingen av ethvert toksiskmiddel 8. Det er ingen enkelt analyse som kan brukes til å kvantifisere alle former for DNA-skade som kan oppstå etter eksponering for et eksogent middel. Imidlertid er en kjernekomponent i in vitro genotoxicity testing batteriet mikronukleusanalysen, som er en pålitelig og mangesidig teknikk som måler grov kromosomskade9. Det er en gullstandardteknikk beskrevet av OECDs testretningslinje 487, for å vurdere in vitro DNA-skade og gentoksisitet og er en del av testbatterikravet for regulatorisk farevurdering10,11.
Den humane hepatocellulære karsinomcellelinjen, HepG2, brukes mye til innledende farevurderingsscreening, da cellene er lett tilgjengelige, relativt billige å kilde, enkle å kultur og egnet til høy gjennomstrømningsscreening12,13. Når de dyrkes i 3D sfæriske strukturer, har de vist seg å rekapitulere levermikromiljøet godt og tilby en levermodell med tilstrekkelig proliferative evner til å støtte mikronukleusanalysen3. Videreutvikling av HepG2 sfæroidmodeller ble etablert for å forbedre modellens levetid og leverlignende funksjonalitet for å støtte gentoksisitetsfarevurdering over langsiktige, gjentatte eksponeringsregimer (≤ 14 dager). I tråd med prinsippene til 3R-ene for å erstatte, redusere og foredle dyreeksperimentering, er den nåværende protokollen etablert for å gi en avansert 3D in vitro levermodell som er i stand til pålitelig evaluering av flere toksikologiske endepunkter (f.eks. leverfunksjonalitet, (pro-)inflammatoriske markører, cytotoksisitet og gentoksisitet) etter akutte, langsiktige og gjentatte kjemiske og ENM-eksponeringer i en rutinemessig og lett tilgjengelig måte.
Her presenterer vi en metode for å etablere en fysiologisk relevant 3D-hepatocyttcellelinje basert på in vitro-modellsystem for gentoksisitetsfarevurdering etter akutte eller langsiktige, gjentatte ENM-eksponeringer. Protokollen kan deles inn i 6 viktige stadier: kultiverende kryopreserverte HepG2-celler; HepG2 sfæroid forberedelse; HepG2 sfæroid overføring fra hengende dråpe til agarose suspensjon; HepG2 sfæroid høst; micronucleus analyse og scoring; og dataanalyse.
Protocol
1.Culturing cryopreserved HepG2 celler
MERK: HepG2-celler, hentet fra American Type Culture Collection (ATCC) ble dyrket i 1x Dulbeccos Modified Eagle Medium (DMEM) med 4,5 g/L D-glukose og L-glutamin supplert med 10% føtal bovint serum (FBS) og 1% penicillin/streptomy antibiotikacin.
- Forvarm DMEM cellekulturmedium (inkludert kosttilskudd) i et 37 °C vannbad i 30 minutter.
- Fjern ett hetteglass med HepG2-celler fra flytende nitrogen og tin i et 37 °C vannbad i 2-3 min, mens du forsiktig virvler hetteglasset for å muliggjøre jevn opptining av cellefjæringen. Pass på at du ikke senker hetteglasset over O-ringen for å redusere forurensningspotensialet.
- Når hetteglasset er tint, fjern det fra vannbadet og spray sjenerøst med 70% etanol for å dekontaminere den ytre overflaten av hetteglasset før du plasserer under en steril, klasse II laminær vevskulturhette.
- Rør nøye innholdet i krympingen av HepG2-celler i et sentrifugerør som inneholder 9 ml forvarmet DMEM cellekulturmedium (med kosttilskudd).
- Bruk en 10 ml tomgang, overfør 10 ml av cellesuspensjonen til en 25 cm2 engangscellekulturflaske og inkuber kulturen i 3 dager (fra såing) ved 5% CO2 og 37 °C til ~80% samløp nås før den gjennomgår underkultur til en større 75 cm2 engangscellekulturflaske.
- Når 80% samløp er nådd, subkulturceller under sterile forhold ved trypsinisering med 0,05% trypsin / EDTA løsning forvarmet i en 37 ° C vannbad i 30 min. Ikke på noe tidspunkt skal cellene få tørke ut.
- Når celler danner en tilhenger monolayer, fjern mediet ved å tippe inn i en desinfeksjonsmiddel avfallspotte. Vask deretter monolayeren umiddelbart for å fjerne alle spor av eksisterende medier ved å skylle kolben to ganger med 3 ml steril 1x PBS-løsning holdt ved romtemperatur. Kast også PBS i desinfeksjonsmiddelavfallskanne.
- Når PBS-vasken er fjernet, tilsett 5 ml forvarmet 0,05% trypsin-EDTA-løsning, og sørg for å dekke hele overflaten av cellene og inkubere celler i 6-8 min ved 37 °C og 5 % CO2.
- Trykk forsiktig på kolben for å løsne cellene fra bunnen av kolben og tilsett deretter 5 ml DMEM cellekulturmedium (med kosttilskudd) for å nøytralisere trypsinenzymet.
- Overfør cellefjæringen til et 50 ml sentrifugerør og pipette celleopphenget grundig opp og ned for å sikre at cellene er helt disassosiert.
- Sentrifuger den fortynnede cellefjæringen ved 230 x g i 5 min. Kast supernatanten i desinfeksjonsmiddel og re-suspendere cellepellet i 25 ml DMEM cellekulturmedium (med kosttilskudd).
- Overfør cellefjæring til en 75 cm2 engangscellekulturflaske og inkuber ved 37 °C og 5 % CO2 i ytterligere 3 dager før du gjennomgår sfæroidpreparat. Når HepG2s har hatt tid til å akklimatisere og igjen nå ~ 80% samløp, bestemme cellekonsentrasjonen som forberedelse til sfæroid såing.
2. HepG2 sfæroidpreparat
- Gjenta subkulturtrinnene som er angitt ovenfor, bortsett fra etter sentrifugering, re-suspendere cellepellet i 1 ml DMEM kulturmedium forvarmet i et 37 °C vannbad. Pipettecelle suspensjon opp og ned grundig.
- Skår celle levedyktighet ved hjelp av Trypan Blue Exclusion Assay (se OSHA SOP 3.21 Reproduktive toksiner, Mutagens, Teratogens og Embryotoxins – Prosedyrer for sikker håndtering og lagring (2019) for helse- og sikkerhetsveiledning)14 med et 1:1-forhold mellom celleoppheng og forhåndsfiltrert 0,4 % Trypan blå løsning.
- Før celletelling, ta 1 ml Trypan blå oppløsning ved hjelp av en 1 ml sprøyte og filtrer med en 0,45 μm filterenhet i et sterilt, 1 ml rør.
- Overfør 10 μL filtrert, Trypan blå oppløsning til et 0,2 ml rør og tilsett 10 μL cellefjæring. Gjenværende filtrert Trypan blå oppløsning kan lagres opptil 3 måneder ved romtemperatur for fremtidig bruk.
- Spray hemocytometeret grundig med 70% etanol og tørk tørt med et sterilt papirhåndkle før du sikrer dekslene på toppen ved hjelp av pustedamp. Hvis du skyver dekslene over den fuktige overflaten, induserer du sammenhengende krefter ved å generere Newton-ringer.
- Rør forsiktig trypanblå cellefjæring opp og ned ved hjelp av en 1000 μL pipette (for å redusere ren stress) før du legger 10 μL til hemocytometeret. Påse at oppløsningen spres under dekselslippet og dekker hele gitteret uten luftbobler.

Figur 1: Telle celler ved hjelp av et hemocytometer. Diagrammatisk representasjon av et hemocytometer som fremhever hvilken kvadrant det skal telles celler fra. Klikk her for å se en større versjon av denne figuren.
- Under mikroskopet teller du de levende (ikke-arresterte) og døde (fargede blå) cellene som finnes i de fire store hjørneplassene (figur 1). Utelat celler som er funnet å overlappe eller sitte på de indre to kantene av de store hjørne firkantene (dvs. på linjene) i tellingen.
- Ved hjelp av følgende beregning beregner du gjennomsnittlig antall aktive, levedyktige celler (ikke-forankret) som finnes i eksemplet:
Totalt antall celler/ml = Antall aktive celler x x 10 000
x 10 000
hvor fortynning refererer til hvor mange ganger lagerløsningen ble fortynnet i Trypan blå (2x i dette tilfellet) og # av firkanter telt refererer til de fire store hjørne firkantene i hemocytometeret telt - Basert på det levedyktige HepG2-celleantallet og bruk av følgende formel:
C1V1=C2V2
hvor C1 = konsentrasjonen av levedyktige celler for tiden,
V1 = volumet av celle suspensjon for tiden,
C2 = konsentrasjonen av celle suspensjon ønsket,
V2 = volumet av celle suspensjon ønsket - Forbered en 10 ml lagerløsning av HepG2 cellefjæring med DMEM cellekulturmedium i en konsentrasjon på 2,0 x 105 celler / ml for å oppnå 4000 HepG2-celler per 20 μL hengende dråpe. Bland celleopphenget grundig ved å pipettere forsiktig opp og ned ved hjelp av en 1000 μL pipette for å sikre at alle celler er fullstendig suspendert i mediet.
- Til brønnene på en 96-brønns cellekulturplate, tilsett 100 μL steril, romtemperatur PBS for å forhindre at hengende dråper tørker ut under inkubasjon.
- Ta lokket på en standard flat bunn 96-brønns cellekulturplate, snu den og forsiktig pipette 20 μL dråper cellefjæring i midten av hvert brønnspor på lokket, som vist i figur 2. Bruk en flerkanals pipette, men tilsett bare 2 - 4 dråper samtidig, da flere sådd kan påvirke nøyaktigheten og plasseringen av dråpene.
- Sentrer dråpene i sporene til brønnene som er lagt ut på lokket; Ellers vil de ikke henge i midten av brønnene når lokket på platen er snudd og står i fare for å falle av i platen. Vend forsiktig lokket på 96-brønnsplaten, slik at dråpene nå henger og plasseres forsiktig på toppen av 96-brønnsplaten.
- Plasser hele 96 brønnplaten med lokket forsiktig inn i en inkubator ved 37 °C og 5 % CO2 i 3 dager før sfæroidoverføring på agarose.
MERK: Det må utvises ekstra forsiktighet ikke bare ved transport av platene til/fra inkubatorene, men ved åpning og lukking av inkubatoren generelt, da overdreven bevegelse kan føre til at platene skifter og sfæroidene enten faller eller dannes feil.
 x 10 000
x 10 000
Figur 2: 3D HepG2 in vitro sfæroid modellpreparat. (A) HepG2-cellene frøet i 20 μL faller på lokket på en 96-brønns plate. (B) HepG2-cellene etter sådd i hengende dråpemodell for å muliggjøre sfæroiddannelse. Klikk her for å se en større versjon av denne figuren.
3. HepG2 sfæroid overføring fra hengende dråpe til agarose suspensjon
MERK: På dag 3 etter sådd i hengende dråper overføres sfæroidene til brønnene på samme 96-brønnsplate som alle tidligere er belagt med et fint lag på 1,5% agarose gel.
- Forbered agarose geler og autoklav (dvs. dag 2 etter sådd) før dagen for platebelegg (dvs. dag 3 etter sådd).
- For å forberede en 1,5% agarose gel, vei 0,30 g agarose i en ren, glassflaske og tilsett deretter 20 ml fenolrødt DMEM-medium. Autoklaver agarose i 1 time ved 230 °C for sterilisering. Agarosebelegget forhindrer HepG2-sfæroidene i å holde seg til bunnen av brønner og danne et cellulært monolag i stedet for å beholde sin 3D-sfæroidstruktur.
- På dag 3 etter sådd fjerner du 96-brønnsplaten som inneholder HepG2 hengende fallsfæroider ut av inkubatoren og vender forsiktig lokket slik at sfæroidene ikke lenger henger.
- Bruk en flerkanals pipette, fjern og kast 100 μL PBS som tidligere er lagt til bunnen av 96-brønnsplaten. La platene lufte i 2-3 min mens du varmer opp agarose som forberedelse til belegg.
FORSIKTIG: Denne prosedyren resulterer i svært varm, flytende agarose som hvis den søles på huden kan brenne og forårsake skade. Videre må det utvises forsiktighet ved håndtering av glassflasken som inneholder væsken, da dette også kan være veldig varmt. - Bruk de 1,5% agarose gelene som tidligere er tilberedt, varm glassflasken som inneholder 20 ml agarose gel i 30 s i en mikrobølgeovn ved maksimal watt (dvs. 900 W). For å belegge to 96-brønns plater, bør en 20 ml flaske ferdiglaget 1,5% agarose gel være tilstrekkelig.
- Når den er smeltet, virvler du forsiktig agarose ved å rotere glassflasken for å fjerne eventuelle bobler og deretter legge til 50 μL agarose i bunnen av hver brønn.
MERK: Når du legger til agarose, må du ikke vinkle platen >45° da agaroseen setter raskt og ikke vil danne et flatt, jevnt lag som kan forstyrre sfæroid vekst. Det er viktig å arbeide effektivt på dette stadiet for å forhindre at agaroseen størkner før platen er helt belagt. - La platen stå i 2 minutter ved romtemperatur før du tilsetter 100 μL forvarmet DMEM cellekulturmedium (med kosttilskudd) på toppen av det faste agaroselaget i hver brønn.
- Vend lokket på 96-brønnsplaten og legg det tilbake på toppen av 96-brønnsplaten slik at sfæroidene nå henger igjen.
- Sentrifuger platen i 3 min ved 200 x g for å overføre sfæroidene fra hengende dråpe inn i de enkelte brønnene på 96-brønnsplaten. Etter overføringen skal HepG2-sfæroidene nå suspenderes i cellekulturmediet. La dem nøye seg med 24 timer i inkubatoren ved 37 °C og 5 % CO2.
- Utsett HepG2-sfæroider av denne størrelsen for enten kjemiske behandlinger eller ENM-behandlinger på dag 4 etter sådd (dvs. 24 timer etter overføring til agarosebelagte plater).
- For å opprettholde celle levedyktighet over lengre kulturperioder, oppdater cellekulturmediet hver tredje dag. For å gjøre dette aspirerer du forsiktig 50 μL av cellekulturmediet fra overflaten av brønnen og erstatter med en frisk 50 μL DMEM cellekulturmedium. Pass på at du ikke fjerner eller forstyrrer sfæroiden når du utfører en middels forandring.
4. Nanomateriale/Kjemisk eksponering
MERK: HepG2 leversfæroidmodellen kan støtte både ENM- og kjemisk baserte eksponeringsregimer, men hovedfokuset i denne protokollen er ENM-eksponeringer. Før eksponering må testen ENM spres på en passende måte; Dette kan utføres som anvist av NanoGenoTox Dispersion Protocol (Grant Agreement No. 20092101, 2018)15.
- Etter dispersjon i henhold til NanoGenoTox Dispersion Protocol, fortynn ENM-suspensjonen fra startkonsentrasjonen på 2,56 mg/ml til den endelige ønskede konsentrasjonen i forvarmet DMEM-cellekulturmedium (inkludert kosttilskudd). Et totalt volum på 5 ml er nødvendig for å dosere en 96 brønnplate.
- For å utsette HepG2-sfæroiden for enten et kjemikalie eller en ENM, ved hjelp av en 200 μL pipette, aspirer 50 μL cellekulturmedium fra overflaten av hver brønn (etterlater 50 μL i brønnen for ikke å forstyrre sfæroidene) og erstatt med 50 μL medium som inneholder testtoksisk middel ved ønsket dose.
- Når testmaterialet er påført, inkuberer du platene for ønsket eksponeringstid ved 37 °C og 5 % CO2.
- Hvis det gjennomføres et langsiktig (≥24 h) eksponeringsregime, må du umiddelbart etter at ønsket eksponeringstidsramme er utløpt, høste sfæroidene for mikronukleusendepunktanalyse som beskrevet nedenfor i trinn 6.1 – 6.4.
- Men med akutte eksponeringsregimer (f.eks. ≤24 h), når eksponeringsperioden er avsluttet, høster, basseng og lagrer 50 μL supernatant fra hver brønn i 96 brønnplaten ved -80 °C for videre biokjemisk analyse senere. Erstatt cellekulturmediet med 50 μL ferskt medium som inneholder 6 μg/ml Cytochalasin B og la det inkuberes i 1 – 1,5 cellesykluser (dvs. 24 – 26 timer for HepG2) som forberedelse til cytokinesis blokkmikronukleusanalysen.
MERK: For akutte (≤24 h) eksponeringsregimer kan cytokinesis-blokkmikronukleusanalysen med Cytochalasin B brukes, men for langsiktige (≥24 h) eksponeringsregimer må mononukleærversjonen (uten Cytochalasin B) av analysen brukes som beskrevet nedenfor i figur 4.
5. HepG2 sfæroid høsting
MERK: Etter enten kjemiske eller ENM-eksponeringsbehandlinger kan både cellekulturmedium eller sfæroidvev høstes for analyse av flere endepunkter. Avhengig av endepunktanalysen kan sfæroider enten høstes individuelt (f.eks. for bildeanalyse) eller samles sammen (f.eks. for cytokinesis blokkmikronukleusanalyse).
- Fjern 96-brønnsplaten fra inkubatoren.
- Ved hjelp av en 200 μL pipette aspirerer du 100 μL cellekulturmedium, inkludert sfæroidvevet fra hver brønn og samler i et sterilt, 15 ml sentrifugerør. Pass på å unngå kontakt med agarose.
- Når den er samlet inn, sentrifugerer du sfæroidfjæringen ved 230 x g i 5 minutter. Fjern supernatanten og oppbevar den ved -80 °C for videre endepunktanalyse (f.eks. leverfunksjonstester) senere.
- Re-suspendere pellet av sfæroider i 1 ml steril, romtemperatur PBS (1x).
- Når den er vasket, sentrifugerer du sfæroidfjæringen igjen på 230 x g i 3 minutter. Kast den supernatante, re-suspendere i 500 μL av 0,05% trypsin-EDTA-løsning og inkuber i 6-8 min ved 37 °C og 5 % CO2.
- Etter inkubasjon, pipette forsiktig de trypsiniserte cellene opp og ned for å fullstendig fjerne tilknytningen og suspendere HepG2-cellene på nytt før nøytralisering med 1 ml DMEM-cellekulturmedium.
- Sentrifuger den fortynnede cellefjæringen ved 230 x g i 5 min. Kast supernatanten i desinfeksjonsmiddel og re-suspendere cellepellet i 2 ml romtemperatur PBS (1x).
- Sentrifuger cellesuspensjonen ved 230 x g i 5 min. Kast supernatanten i desinfeksjonsmiddelet og sett deretter cellepelleten på nytt i 2 ml kald PBS (1x). Forsikre deg om at cellene er godt spredt for å forhindre klumper av celler som skjuler synsfeltet når de er montert på mikroskopsklier.
6. Micronucleus analyse og scoring
For den manuelle metoden for mikronukleusanalysen er det nødvendig med en cytocentrifuge for å produsere en cytodot (en definert, konsentrert celleregion) i midten av mikroskopskredet. Denne prosessen støtter mer effektiv skåring av lysbildet, da det gjør det mulig for scoreren å enkelt finne cellene av interesse, i motsetning til å evaluere et helt lysbilde der cellene kan spres bredt.
- Dypp frostede mikroskopsklier (tre per dose) i 70% etanol etterfulgt av ddH2O og la det lufttørke i 5 min.
- Plasser klargjorte mikroskop glir inn i cuvettetrakten som vist i figur 3A, der glassskuffen (iii) er plassert i metallstøtten (iv) med et filterkort (ii) og cuvettetrakt (i) festet på toppen.
- Ordne cuvette trakter i cytocentrifuge med trakten vendt opp, slik at 100 μL celle suspensjon kan direkte legges inn i hver enkelt.
- Cytospin i 5 min ved 500 x g for å sikre at cellene er jevnt fordelt på overflaten av lysbildet.

Figur 3: Cytospin-oppsett for å klargjøre behandlede celler på mikroskopskred. (A) Viser de enkelte komponentene, (i) cuvettetrakten, (ii) filterkortet, (iii) glassmikroskopet og (iv) metallstøtte som kreves for å cytospin HepG2-celler på mikroskopet. (B) Den endelige cuvettetrakten som er satt opp. (C) Riktig plassering av cuvettetrakten i cytocentrifugen. Klikk her for å se en større versjon av denne figuren.
- La skredene lufttørke før fiksering i iskald, 90 % metanol i 10 minutter.
- Når de er festet, la lysbildene lufttørke over natten ved romtemperatur før lagring ved -20 °C i opptil 6 måneder.
- Fjern eventuelt det ferdiglagde mikroskopet fra -20 °C fryseren og la det varmes opp til romtemperatur før du foretar Giemsa-farging.
FORSIKTIG: I henhold til forordning (EF) nr. 1272/2008 [CLP] er Giemsa fargingsløsning en svært brannfarlig væske som kan være giftig ved svelging og forårsake skade ved kontakt med øyne, hud eller ved innånding. Se det tilhørende SDS-arket for detaljert lagring, håndtering og helse- og sikkerhetsråd om dette kjemikaliet før bruk. - Mens lysbildene avriming, forberede en 20% Giemsa farging løsning (25 ml kreves for å flekk ~ 30 lysbilder) fortynnet i fosfatase buffer (pH 6.8). Bland grundig ved å virvle oppløsningen forsiktig før filtrering ved hjelp av brettet filterpapir plassert i en trakt.
- Bruk en Pasteur pipette, tilsett 3 - 5 dråper filtrert Giemsa-løsning til cytodoten på hvert lysbilde og la det stå i 8 - 10 min.
- Vask lysbilder i to påfølgende fosfatasebuffervasker før du kort skyller under kaldt vann for å fjerne overflødig flekkrester. La skredene være lufttørke.
- Når den er tørr, dypp fargede sklier i xylen i 10 s i en avtrekkshette, før du legger til en dråpe monteringsmedium i midten av cytodoten og et sted et glassdeksler på toppen.
- La mikroskopet glir i avtrekkshetten over natten for å tørke før manuell scoring; de kan lagres på ubestemt tid ved romtemperatur.
7. Dataanalyse
- Som beskrevet i OECDs testretningslinjer 487 (2014)11, for å vurdere og kvantifisere DNA-skade forårsaket som følge av eksponering for et ENM- eller kjemisk middel, bruk et lysmikroskop (100x mål med nedsenkningsolje) 2000 mononukleerte eller 1000 binucleated celler per biologisk replikering for å score for tilstedeværelse av mikronuklei, som vist i figur 4.

Figur 4: Micronucleus assay scoring beslutningstre. Skjematisk beslutningstre for å markere nødvendigheten av ulike vurderingsprosedyrer for skåring og cytotoksisitet ved bruk av mikronukleusanalysen med 3D-modeller etter akutte eller langsiktige eksponeringsregimer. Akutte (≤24 h) eksponeringer tillater bruk av cytokinesis blokkert mikronukleusanalyse, mens langsiktige (≥24 h) eksponeringer krever mononukleær versjon av analysen; begge er beskrevet i OECDs testretningslinje 487. Klikk her for å se en større versjon av denne figuren.
- Basert på andelen mikronuklei tilstede per antall mononukleerte eller binuklede celler scoret, beregne en prosentandel av gentoksisitetsverdien.
- For å vurdere DNA-skaden som observeres er ikke som følge av celleavfall forårsaket av en høy andel apoptotiske celler, ta et mål på cytotoksisitet ved siden av. I dette tilfellet, avhengig av tilstedeværelsen av Cytochalasin B, bruk enten CPBI- eller RVCC-beregning (som beskrevet i figur 4). Gentoksisitet må bare evalueres i prøver der cytotoksisitet er mindre enn 55% ± 5% som definert i OECDs testretningslinje 48711.
Representative Results
Egnetheten til denne cellelinjebaserte 3D leversfæroidmodellen for langsiktig kultur og gentoksisk farevurdering ble evaluert ved å gjennomføre baseline karakterisering for å bestemme levedyktigheten og leverlignende funksjonaliteten til modellen i løpet av 14 dager i kulturen, samt dens anvendbarhet for mikronukleusanalysen.
Baseline karakterisering av 3D HepG2 Lever sfæroid modell
Før en in vitro toksikologiske vurdering er det viktig å kontrollere at 3D HepG2-sfæroidene har dannet seg riktig før du utfører agaroseoverføringen eller kjemisk / ENM-behandling. HepG2 sfæroider produsert ved hjelp av hengende dråpemetode tar vanligvis 2 - 3 dager etter sådd (4000 celler / sfæroid) for å danne kompakte, sfæriske formede sfæroider med en gjennomsnittlig diameter på 495,52 μm B x 482,69 μm H som vist i figur 5A-5C. HepG2 sfæroider som har dannet seg riktig og er akseptable å brukes til in vitro toksikologiske vurdering, må ha en kompakt, sfærisk formet struktur med en jevn overflate og ingen visuelle projeksjoner. Figur 5 gir eksempler på god kvalitet (Figur 5D-F) og dårlig kvalitet ( Figur5G-I) sfæroider. Sistnevnte skal kastes. Vanligvis vil 90-95% av sfæroider dannet per plate dannes riktig og være levedyktige for videre eksperimentering.

Figur 5: Lyse mikroskopibilder som viser den naturlige morfologien til HepG2-sfæroidene som dannes via hengende dråpemetode. (A-C) viser dag 2 og (D-I) Dag 4 HepG2 lever sfæroider etter sådd. (D-F) er eksempler på hepG2-sfæroider av god kvalitet, mens (G-I) viser dårlig dannede sfæroider. Alle bilder ble tatt på et X20-mål ved hjelp av et mikroskop. Skalalinjen representerer 20 μm. Klikk her for å se en større versjon av denne figuren.
For ytterligere å bekrefte HepG2 sfæroid levedyktighet, kan en grunnleggende kolorimetrisk Bromocresol Green Albumin (BCG) Assay eller Urea Assay utføres for å vurdere deres leverlignende funksjonalitet. Leverlignende funksjonalitet ble vurdert i tråd med levedyktigheten ved hjelp av Trypan Blue Exclusion Assay over en 14-dagers kulturperiode for å bestemme levetiden til leversfæroidmodellen og fastslå om den kunne støtte langsiktig eller gjentatt ENM / kjemisk basert farevurdering (Figur 6). Albuminkonsentrasjonen forble konsistent i løpet av kulturperiodens varighet. Urea-produksjonen viser en økning i konsentrasjonen av urea produsert per sfæroid over en uke i kulturen før den når et platå ved dag 7. Det er viktig å merke seg at nivåene av albumin og urea produsert i 3D HepG2 sfæroider er vesentlig høyere enn det som ble observert i samme cellelinje dyrket i et 2D-format. Faktisk var 2D-kulturer av HepG2-celler, toppalbumin- og ureanivåer henholdsvis 0,001 mg / ml og 0,010 ng / μL. Videre, i tidligere arbeid publisert av Shah et al. ved hjelp av et nesten identisk HepG2 sfæroidsystem, fremhever forfatterne en bemerkelsesverdig forbedring i metabolsk aktivitet (CYP1A1 og CYP1A2) i 3D HepG2 in vitro-modellsystemene sammenlignet med de 2D-dyrkede HepG2-cellene5.

Figur 6: 14-dagers baseline karakteriseringsdata for HepG2 leversfæroider. Etter overføring fra hengende slipp fremhever (A) levedyktigheten til HepG2 sfæroidmodellen over en 14-dagers periode mens (B) og (C) fremhever henholdsvis leverlignende albumin- og ureafunksjonalitet. Gjennomsnittlige data ± SEM presentert, n = 4. Klikk her for å se en større versjon av denne figuren.
Med den uunngåelige utviklingen av en nekrotisk kjerne, en kjent begrensning av 3D leversfæroidkulturer, måtte levedyktigheten til denne HepG2-baserte modellen etableres for å demonstrere at den var i stand til å opprettholde langsiktige (5-10 dagers) eksponeringsregimer samtidig som de opprettholder den proliferative evnen som kreves for å støtte mikronukleusanalysen5. Faktisk har denne 3D lever sfæroid modellen vist seg å beholde > 70% levedyktighet over 10 dager i kulturen. Basert på dette og i forbindelse med den vedvarende leverlignende funksjonaliteten som observeres i løpet av den ≥14-dagers kulturperioden, kan denne 3D-leversfæroidmodellen dermed støtte langsiktige, gjentatte ENM-eksponeringsregimer opptil 10 dager lange (dvs. før levedyktigheten til sfæroidene faller under 70%). For referanse anbefales det at albuminnivåer for HepG2 sfæroider sådd ved 4000 celler / sfæroid skal være ≥20,0 ng / μL mens ureaproduksjon skal være ≥0,25 ng / μL før du foretar en in vitro toksikologisk vurdering med denne modellen.
Gentoksisitetsvurdering av konstruerte nanomaterialer
For gentoksisitetsvurdering ble mikronukleusanalysen brukt til å bestemme tilstedeværelsen av mikronuklei etter både akutte (24 timer) og langsiktige (120 t) ENM-eksponeringer. Aflatoxin B1 er et kjent leverkarsinogen16,17 og er en anbefalt positiv kontroll for mikronukleusanalysen. Optimeringsforsøk har vist at 0,1 μM alfatoksin B1 induserer en betydelig positiv (≥2,0 ganger økning) gentoksisk respons i 3D HepG2 leversfæroider og brukes dermed i alle mikronukleusanalyser som utføres med denne modellen. For å sikre gyldigheten av mikronukleusanalyseresultatene ved hjelp av HepG2 sfæroidmodellen, bør bakgrunnsmikronukleusfrekvensen for HepG2-celler som brukes i denne 3D-in vitro-modellen ligge innenfor et område på 0,6% - 1,2%. Som et resultat bør Alfatoxin B1 indusere en gentoksisk respons på minst to ganger høyere enn det som er sett med negativ kontroll; Dermed bør 0,1 μM alfatoksin B1 indusere en mikronukleifrekvens mellom 1,5% – 3,0%. Ved hjelp av disse kontrollparametrene kan ENM-assosiert gentoksisitet in vitro deretter vurderes pålitelig. Basert på OECDs testretningslinje 487 er det viktig å merke seg at når du tester en ENM eller et kjemikalie, bør de valgte konsentrasjonene ikke indusere mer enn 55% ± 5% cytotoksisitet (indikert ved reduksjon i CPBI- eller RVCC-verdier i forhold til den negative kontrollen)11. Figur 7 illustrerer dataene som genereres da Aflatoxin B1 og to ENMer (titandioksid (TiO2) og sliver (Ag)) ble evaluert etter både akutte og langsiktige eksponeringer i HepG2-sfæroidene, og påfølgende gentoksisk potensial ble analysert ved hjelp av mikronukleusanalysen. Begge ENMs vurderte ble testet ved en ikke-cytotoksisk, lav dose på 5,00 μg/ml over en akutt (24 t) eksponering og langsiktig (120 t) eksponeringsregime. En lignende trend for gentoksisitet på tvers av både TiO2 og Ag ENMs kan observeres, hvorved den forhøyede gentoksisitetsresponsen som resulterte etter 24 timers eksponering ikke var tydelig etter en langsiktig 5-dagers eksponering. Dette til tross for vedvarende gentoksisitet indusert av Aflatoxin B1 positiv kontroll på begge tidspunktene.

Figur 7: Gentoksisitetsvurdering etter TiO2- og Ag ENM-eksponering på HepG2 leversfæroider. Gentoksisitetsvurdering (mikronukleusfrekvens) ved bruk av mikronukleusanalyseinnlegget (A) akutt (24 timer) og (B) langtidseksponering (120 timer) for 5,00 μg/ml TiO2 og Ag ENM. Negativ kontroll er bare et medium, mens den positive kontrollen er 0,1 μM av Aflatoxin B1. Gjennomsnittsdata (n=2) presentert ± SD. Signifikans angitt i forhold til den negative kontrollen: * = p≤ 0,05. Klikk her for å se en større versjon av denne figuren.
Discussion
Bruksområder for 3D-levermodeller varierer betydelig avhengig av det spesifikke biokjemiske endepunktet eller ugunstige utfallsveier som målrettes. Hver modell har sine fordeler og begrensninger, fra interdonorvariasjon i primære menneskelige hepatocyttmodeller (PHH) til redusert cytokrom p450-aktivitet i cellelinjebaserte modeller, men alle er verdifulle i seg selv6,12,18,19. Ved vurdering av gentoksisitet er det begrensninger i modellenes kompatibilitet med regulatoriske godkjente endepunkter som in vitro mikronukleusanalysen, da aktiv spredning er nødvendig. Dette er nødvendig, da gentoksisitetsvurdering krever kvantifisering av fast DNA-skade som skal vurderes etter celledeling når det er mulighet for DNA-reparasjon for å korrigere forbigående lesjoner. Dessverre er svært differensiert hepatocytt (dvs. HepaRG) baserte sfæroider eller PHH-mikrotissues, som anses å vise de mest fysiologisk relevante leverlignende egenskapene, statiske (ikke-proliferative) modeller12,19,20. Som et resultat gir 3D HepG2 sfæroidmodellen som presenteres her en passende, alternativ modell som er i stand til å støtte gentoksisitetstesting. HepG2 cellelinjebaserte sfæroider har tilstrekkelig aktivt dele celler på den ytre overflaten av sfæroidene samtidig som grunnleggende leverlignende egenskaper, for eksempel albumin og urea produksjon og noen CYP450 aktivitet5,12,19. Hovedsakelig er denne in vitro levermodellen utviklet for å utfylle mikronukleusanalysen, da dette er en av de to in vitro-analysene som anbefales i batteriet for gentoksisitetstesting8,10,11,21. Modellen kan imidlertid lett anvendes på DNA-sekvenseringsanalyse og genuttrykksteknologier (RNA), mens den har potensial til å bli ytterligere tilpasset og utnyttet for andre DNA-skadeendepunkter, for eksempel kometanalysen. Likevel er det viktig å vurdere hvilken rolle ENM-interferens spiller i enkelte endepunktanalyser. For eksempel kan strømningscytometribaserte analyser ikke være egnet for ENM gentoksisitetsvurdering spesielt på grunn av partikkelinterferens22.
En begrensende faktor for sfæroidmodeller som aktivt gjennomgår celledeling er deres størrelse. Optimalisering av såingstetthet er kritisk, da det må være nok celler som gjør at modellen kan fortsette å spre seg; men ikke for høyt et cellenummer, noe som resulterer i at sfæroiden blir altfor kompakt, noe som fører til en økt nekrotisk kjerne. Årsaken til denne nekrose antas å være begrenset oksygen og næringsdiffusjon, da grensen for denne diffusjonen antas å være ca. 100 - 150 μm vev23,24. Dette avhenger imidlertid av celletype, cellenummer, stillasinteraksjoner og kulturforhold25. Siden det har vist seg at ca 700 μm diameter er grensen for å unngå for tidlig utbrudd av nekrose i midten av C3A sfæroider, såing 4000 HepG2 celler per sfæroid sikrer diameteren på modellen på tidspunktet for eksponering er ≤ 500 μm26. Videre etablerte Shah et al. at HepG2-celler frøet over 5000 celler per sfæroid viste en 25% reduksjon i levedyktighet etter 7 dager i kultur, noe som kan gjelde gjennomsnittlig diameter på 680 μm og begrenset tilgjengelighet av næringsstoffer i en 20 μL hengende dråpe5. For å overvinne dette gjennomgår modellen som er utviklet i den nåværende protokollen et kritisk skritt der hengende dråpe overføres til agarosebelagte brønner etter innledende dannelse av sfæroidet. Dette sikrer at et større volum av kulturmedium er til stede for å opprettholde det stadig økende antall celler i sfæroidene. Som et resultat forblir HepG2 sfæroidmodellen over 70% levedyktig etter 10 dager i kultur og kan brukes til langsiktig farevurdering in vitro.
Mens HepG2 sfæroid-modellen kan støtte både akutte og langsiktige eksponeringsregimer, er forfriskende cellekulturmedium i lengre kulturperioder begrenset for denne modellen, da fullstendig utskifting av mediet ikke anbefales på grunn av det potensielle tapet av sfæroidene. Det antas at med ENM-eksponeringer er tendensen til homogene ENM-dispersjoner til agglomerat og sediment høy. Det er imidlertid bemerkelsesverdig at hastigheten som et ENM-sedimenter kan variere avhengig av partikkelparametrene (f.eks. størrelse, form og tetthet) og kan bestemmes teoretisk ved hjelp av in vitro sedimentering, diffusjon og dosimetri (ISDD) modell, eller dens nylige derivater, ofte referert til når det gjelder ENM (suspensjon) eksponering nærmer seg27,28. Med dette er det antatt at hvis bare 50% av cellekulturmediet forsiktig fjernes fra overflaten av cellekulturen, bør forstyrrelsen og etterfølgende fjerning av ENM-dosen i teorien være minimal. Men med Brownian bevegelse i spill, kan dette ikke strengt tatt være tilfelle, og videre arbeid i avsetning og sedimentering av hver enkelt ENM som skal testes bør gjennomføres for å sikre at riktig dosimetri beholdes gjennom de langsiktige eksponeringsregimene27. I hovedsak er dette en potensiell begrensning å vurdere når man utfører gjentatte doseringsregimer, da dette kan være avgjørende for den endelige, akkumulerte konsentrasjonen. Kjemisk basert eksponering derimot, mens de ikke uten egne begrensninger å vurdere, tilbyr en mer forenklet tilnærming ved at kjemiske stoffer har en tendens til å forbli i løsning og dermed en direkte erstatning av den opprinnelige kjemiske konsentrasjonen i tillegg til den nylig tilsatte konsentrasjonen sikrer at kjemikalier som går tapt under medieforfriskninger, erstattes tilsvarende29. Fremtidige anvendelser vil omfatte evaluering av modellens egnethet for gjentatte eksponeringsregimer over langsiktige kulturperioder, da gjentatte doseringsstrategier er avgjørende for å vurdere evnen til et bestemt organsystem til å forbedre eller overvinne bivirkningene, hvis noen, indusert av bioakkumulering av et xenobiotisk stoff.
Avslutningsvis har denne 3D in vitro-levermodellen kapasitet til å bli brukt til å evaluere en rekke realistiske eksponeringsscenarier, og dermed gi en fremtidig in vitro-tilnærming for bedre å støtte både ENM og kjemisk farevurdering på en rutinemessig og lett tilgjengelig måte.
Disclosures
Forfatterne har ingenting å avsløre.
Acknowledgments
Forfatterne vil gjerne erkjenne at denne forskningen har fått støtte fra EUs forsknings- og innovasjonsprogram Horizon 2020 for PATROLS-prosjektet, under tilskuddsavtale nr.
Materials
| Name | Company | Catalog Number | Comments |
| Aflotoxin B1 | Sigma Aldrich, UK | A6636-5MG | |
| Agarose | Sigma Aldrich, UK | A9539-50G | |
| Autoclave Tape | |||
| BCG Albumin Assay | Sigma Aldrich, UK | MAK124 | |
| Bovine Serum Albumin Powder | Sigma Aldrich, UK | A9418 | |
| Cell Freezing Aid | Thermo Fisher Scientific, UK | 5100-0001 - Mr Frosty | |
| Centrifuge | Eppendorf | 5810 R | |
| Cytochalasin B | Merck, UK | 250233 | |
| Cytology Metal Clips | |||
| Cytospin 4 Centrifuge | ThermoFisher Scientific, UK | CM00730202 | |
| DMEM with 4.5g/L D-Glucose, L-Glutamine | GIBCO, Paisley, UK | 41965-039 | |
| DMEM, phenol-red free with 4.5g/L D-Glucose, L-Glutamine with Hepes | GIBCO, Paisley, UK | 21063-029 | |
| DPX Mounting Medium | FisherScientific, UK | D/5330/05 | |
| Ethanol | FisherScientific, UK | 10048291 | |
| FBS | GIBCO, Paisley, UK | 10270-106 | |
| Filter Cards for Shandon Cytospin | FisherScientific, UK | 15995742 | |
| Frosted Glass Slides | ThermoFisher Scientific, UK | ||
| Giemsa's Stain Improved R66 Solution, Gurr | VWR Chemicals, UK | MFCD00081642 | |
| Glass Coverslips (24 x 60) | Deckglaser, VWR | ECN631-1575 | |
| Haemocytometer and Coverslip | |||
| Immersion Oil for Microscope | Zeiss, UK | 518F, ISO8034 | |
| Laminar Class II Tissue Culture Hood | Scanlaf Mars | ||
| Light Microscope | Zeiss, UK | Axiovert 40C | |
| Liquid Nitrogen | |||
| Methanol | FisherScientific, UK | 10284580 | |
| Microwave | |||
| Non-Filtered, Sterile 200µl and 1000µl Pipette tips | Greiner-Bio-One, UK | ||
| NuncMicroWell 96-Well Microplates | ThermoFisher Scientific, Denmark | 167008 | |
| P1000 and P200 micropipettes | |||
| P300 and P50 multi-channel pipettes | |||
| PBS pH 7.4 1X, MgCl2 and CaCl2 Free | GIBCO, Paisley, UK | 14190-094 | |
| Pen/Strep | GIBCO, Paisley, UK | 15140-122, Penicillin/Strepmyocin 100X or 10,000U/ml | |
| Phosphatase Buffer Tablets | GIBCO, Paisley, UK | 10582-013 | |
| Pipette Boy | |||
| Simport Scientific CytoSep Funnels for Shandon Cytospin 4 Centrifuges | FisherScientific, UK | 11690581 | |
| Sonifier SFX 550 240V CE 1/2" - Probe | Branson, USA | 101-063-971R | |
| T-25 and T-75 Tissue Culture Flask | Greiner-Bio-One, UK | T-25 (690175) and T-75 (660175) | |
| Trypan Blue Solution | Sigma Aldrich, UK | T8154-100mL | |
| Urea Assay Kit | Sigma Aldrich, UK | MAK006 | |
| Virkon Disinfectant | DuPont, UK | Rely+On Virkon | |
| Water Bath (37?C) | Grant JBNova 18 | ||
| Weighing Balance | |||
| Xylene | FisherScientific, UK | 10588070 | |
| 0.05% Trypsin-EDTA | GIBCO, Paisley, UK | 5300-054 | |
| 0.2mL and 1.0mL Eppendorf Tubes | Greiner-Bio-One, UK | ||
| 0.45µm Filter Unit | Millex HA, MF-Millipore, UK | SLHA033SS | |
| 1.0mL Syringe | BD Plastipak, FisherScientific, UK | 300185 | |
| 20mL LS Scintillation Glass Vials, 22-400 Foil Lined PP Caps | DWK Life Sciences GmbH, Germany | WHEA986581 | |
| 37?C and 5% CO2 ISO Class 5 Hepa Filter Incubator | NUAIRE DHD Autoflow | ||
| 3mL Pasteur Pipette | Greiner-Bio-One, UK | ||
| 50mL Conical Falcon Tubes | Greiner-Bio-One, UK | ||
| 50mL or 100mL Glass Bottles | |||
| 50mL Skirted Falcon Tubes | Greiner-Bio-One, UK | ||
| 5mL, 10mL and 25mL Pipettes | Greiner-Bio-One, UK | ||
| 9.4cm Square, Petri Dish | Greiner-Bio-One, UK | 688161 |
References
- Geiser, M., Kreyling, W. G. Deposition and biokinetics of inhaled nanoparticles. Particle and Fibre Toxicology. 7, 2 (2010).
- Modrzynska, J. Toxicological effects of nanoparticle deposition in the liver. Kgs. Lyngby, Denmark: Technical University of Denmark. , (2018).
- Elje, E., et al. The comet assay applied to HepG2 liver spheroids. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 845, 403033 (2019).
- Breslin, S., O'Driscoll, L. Three-dimensional cell culture: the missing link in drug discovery. Drug Discovery Today. 18, 240-249 (2013).
- Shah, U. -K., et al. A three-dimensional in vitro HepG2 cells liver spheroid model for genotoxicity studies. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 825, 51-58 (2018).
- Lauschke, V. M., Hendriks, D. F. G., Bell, C. C., Andersson, T. B., Ingelman-Sundberg, M. Novel 3D Culture Systems for Studies of Human Liver Function and Assessments of the Hepatotoxicity of Drugs and Drug Candidates. Chemical Research in Toxicology. 29, 1936-1955 (2016).
- van Grunsven, L. A. 3D in vitro models of liver fibrosis. Advanced Drug Delivery Reviews. 121, 133-146 (2017).
- Corvi, R., Madia, F. In vitro genotoxicity testing - can the performance be enhanced. Food and Chemical Toxicology. 106, 600-608 (2017).
- Doak, S. H., Manshian, B., Jenkins, G. J. S., Singh, N. In vitro genotoxicity testing strategy for nanomaterials and the adaptation of current OECD guidelines. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 745, 104-111 (2012).
- Fenech, M. Cytokinesis-block micronucleus cytome assay. Nature Protocols. 2, 1084-1104 (2007).
- OECD. OECD Guidelines. Test 489: In vivo Mammalian Alkaline Comet Assay. , (2016).
- Gerets, H. H. J., et al. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biology and Toxicology. 28, 69-87 (2012).
- Sison-Young, R. L., et al. A multicenter assessment of single-cell models aligned to standard measures of cell health for prediction of acute hepatotoxicity. Archives of Toxicology. 91, 1385-1400 (2017).
- European Guidelines 2019. European Agency for Safety and Health at Work. , Available from: https://osha.europa.eu/en/safety-and-health-legislation/european-guidelines (2019).
- Jensen, K. A. The NANOGENOTOX Dispersion Protocol for NANoREG. European Union Grant Agreement n° 2009. 21, 01 (2014).
- Marchese, S., et al. Aflatoxin B1 and M1: Biological Properties and Their Involvement in Cancer Development. Toxins. 10, 214 (2018).
- Rushing, B. R., Selim, M. I. Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food and Chemical Toxicology. 124, 81-100 (2019).
- Kermanizadeh, A., Brown, D. M., Moritz, W., Stone, V. The importance of inter-individual Kupffer cell variability in the governance of hepatic toxicity in a 3D primary human liver microtissue model. Scientific Reports. 9, 7295 (2019).
- Berger, B., et al. Comparison of Liver Cell Models Using the Basel Phenotyping Cocktail. Frontiers in Pharmacology. 7, 443 (2016).
- Ramaiahgari, S. C., et al. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Archives of Toxicology. , (2014).
- Li, Y., et al. Factors affecting the in vitro micronucleus assay for evaluation of nanomaterials. Mutagenesis. 32 (1), 151-159 (2016).
- Kirkland, D., Reeve, L., Gatehouse, D., Vanparys, P. A core in vitro genotoxicity battery comprising the Ames test plus the in vitro micronucleus test is sufficient to detect rodent carcinogens and in vivo genotoxins. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 721 (1), 27-73 (2011).
- Curcio, E., et al. Mass transfer and metabolic reactions in hepatocyte spheroids cultured in rotating wall gas-permeable membrane system. Biomaterials. 28, 5487-5497 (2007).
- Glicklis, R., Merchuk, J. C., Cohen, S. Modeling mass transfer in hepatocyte spheroids via cell viability, spheroid size, and hepatocellular functions. Biotechnology and Bioengineering. 86, 672-680 (2004).
- Asthana, A., Kisaalita, W. S. Microtissue size and hypoxia in HTS with 3D cultures. Drug Discovery Today. 17, 810-817 (2012).
- Gaskell, H., et al. Characterization of a functional C3A liver spheroid model. Toxicology Research. 5, 1053-1065 (2016).
- Cho, E. C., Zhang, Q., Xia, Y. The effect of sedimentation and diffusion on cellular uptake of gold nanoparticles. Nature Nanotechnology. 6, 385-391 (2011).
- Hinderliter, P. M., et al. ISDD: A computational model of particle sedimentation, diffusion and target cell dosimetry for in vitro toxicity studies. Particle and Fiber Toxicology. 7, 36 (2010).
- Kramer, N. I., di Consiglio, E., Blaauboer, B. J., Testai, E. Biokinetics in repeated-dosing in vitro drug toxicity studies. Toxicology in vitro. 30, 217-224 (2015).
Tags
Bioingeniør Utgave 160 In Vitro levermodeller Nanomaterialer Farevurdering Langtidseksponering Nano(geno)toksikologi DNA-skadeErratum
Formal Correction: Erratum: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure
Posted by JoVE Editors on 01/26/2021.
Citeable Link.
An erratum was issued for: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure. The Representative Results section was updated.
Figure 6 in the Representative Results section was updated from:
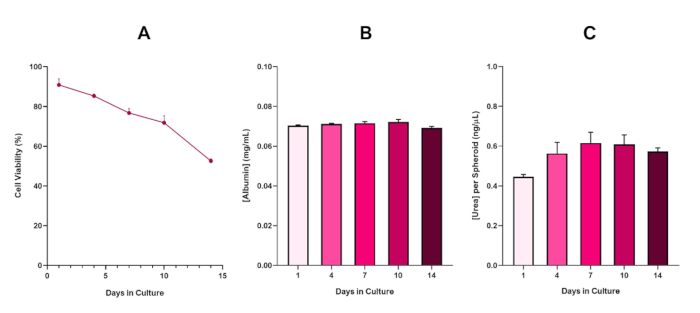
to:
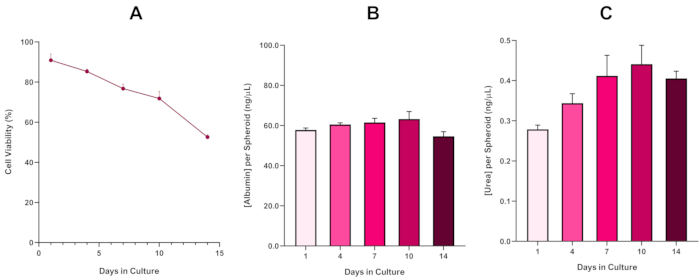
The fourth paragraph in the Representative Results section was updated from:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥0.06 mg/mL whilst urea production should be ≥0.4 ng/µL before conducting an in vitro toxicological assessment with this model.
to:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥50.0 ng/μL whilst urea production should be ≥0.25 ng/µL before conducting an in vitro toxicological assessment with this model.
