

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Эта процедура была создана для использования для разработки передовых 3D печеночным культур in vitro, которые могут обеспечить более физиологически релевантную оценку генотоксических опасностей, связанных с наноматериальным воздействием как на острые, так и долгосрочные, повторяющиеся режимы дозы.
Abstract
В связи с быстрым развитием и внедрением широкого спектра инженерных наноматериалов (ENM), воздействие ENM неизбежно и разработка надежных, прогностический in vitro тестовых систем имеет важное значение. Гепатическая токсикология является ключевым при рассмотрении воздействия ЭНМ, так как печень играет жизненно важную роль в метаболическом гомеостазе и детоксикации, а также является основным местом накопления ЭНМ после воздействия. Основываясь на этом и общепринятом понимании того, что 2D-модели гепатоцитов не точно имитируют сложности сложных многоклеточных взаимодействий и метаболической активности, наблюдаемой in vivo, больше внимания уделяется разработке физиологически релевантных 3D моделей печени, адаптированных для целей оценки опасности ENM in vitro. В соответствии с принципами 3Rs для замены, сокращения и уточнения экспериментов на животных, 3D HepG2 клеточной линии на основе модели печени была разработана, которая является удобной, экономически эффективной системой, которая может поддерживать как расширенные, так и повторяющиеся режимы воздействия ENM (≤14 дней). Эти модели сфероидов (≥500 мкм в диаметре) сохраняют свою пролиферативную способность (т.е. деление клеточных моделей), позволяя им быть в сочетании с микронуклейным анализом «золотого стандарта» для эффективной оценки генотоксичности в пробирке. Их способность сообщать о ряде токсикологических конечных точек (например, функции печени, (про-) воспалительные реакции, цитотоксия и генотоксичность) характеризовалась использованием нескольких ЭНМ как в острых (24 ч), так и в долгосрочных (120 ч) режимах воздействия. Эта 3D-печеночная печеночная модель может быть использована для оценки более реалистичного воздействия ENM, обеспечивая тем самым будущий подход in vitro для лучшей поддержки оценки опасности ENM в обычном и легко доступном виде.
Introduction
В связи с быстрым развитием и внедрением широкого спектра инженерных наноматериалов (ENM) в изобилии человеческих приложений (например, продуктов питания, косметики, одежды, спортивного оборудования, электроники, транспорта и медицины), неизбежно, что люди будут подвергаться воздействию ЕМ на регулярной основе. При этом, Есть повышенная озабоченность тем, что роман, размер конкретных физио-химических характеристик, которые считают эти материалы выгодными в многочисленных приложениях может вызвать неблагоприятные последствия для здоровья человека и окружающей среды сопутствующих. В настоящее время проводятся многочисленные международные мероприятия, с тем чтобы активно отражать более физиологически релевантное воздействие этих ЭНМ и оценивать потенциальную токсичность этих материалов в течение острых, долгосрочных и повторяющихся сценариев воздействия низких доз.
Гепатическая токсикология является ключевым при рассмотрении воздействия ENM, так как широко известно, что печень является основным местом накопления ENMпосле воздействия 1,2. Кроме того, печень является основной системой органа для метаболизма и детоксикации веществ, которые входят в системное кровообращение3. Основываясь на общепринятом понимании того, что 2D модели гепатоцитов не точно имитируют сложности сложных многоклеточных взаимодействий или надлежащим образом представляют метаболическую активность наблюдается in vivo, больший акцент на разработку надежных и физиологически актуальных моделей в пробирке 3D печени для in vivo заменитьтехнологии была создана 4,5. Использование передовых технологий 3D-культуры улучшает долговечность печеночными моделями in vitro, что позволяет исследовало долгосрочные, повторяющиеся режимы воздействия. Кроме того, этот передовой формат культуры способствует формированию расширенных физиологических, органотипических функций, таких как желчные каналикулы, активные процессы транспортировки и улучшенные возможности метаболизма препарата CYP450, тем самым улучшая прогностику моделей6. Современные 3D-печеночные модели, состоящие из монокультур (только гепатоцитов) или со-культур (гепатоциты с непаренхимальными клетками) существуют в нескольких форматах, начиная от микротыссов или сфероидов в ультранизких адгезионых пластинах, висячих капли сфероидов, клеток, встроенных в матрицы и / или леса и микрофлюидных клеточных культур платформ, все из которых считаются эффективными передовыми моделями in vitro для оценкипеченочной токсичности 6,7. Тем не менее, большинство из этих модельных систем высокого технического обслуживания, требуют специального оборудования и стоят дорого. Кроме того, эти модели часто являются статичными (т.е. моделями неразделимых клеток), что препятствует их использованию при оценке конечных точек опасности, таких как тестирование генотоксичности с использованием методов количественной оценки фиксированного повреждения ДНК. Генотоксичность является основным условием в регулятивной токсикологии, и это жизненно важный компонент оценки риска любого токсиканта8. Существует не один анализ, который может быть применен для количественной оценки всех форм повреждения ДНК, которые могут возникнуть после воздействия экзогенного агента. Тем не менее, основным компонентом in vitro генотоксичность тестирования батареи микронуклид анализа, который является надежным и многогранным методом, который измеряет валовой хромосомныйущерб 9. Это золотой стандарт техники, описанной ВЭСР Тест Руководство 487, для оценки повреждения ДНК in vitro и генотоксичность и является частью требования испытательной батареи длянормативной оценки опасности 10,11.
Линия клеток гепатоцеллюлярной карциномы человека, HepG2, широко используется для первоначального скрининга оценки опасности, поскольку клетки легко доступны, относительно недороги для источника, просты в культуре и податливы к высокой пропускнойспособности скрининга 12,13. При культуре в 3D сферических структур, они были показаны, чтобы резюмировать микропролезион печени хорошо и предлагают печеночную модель с достаточными возможностями пролиферации для поддержки микронуклидаанализа 3. Для улучшения долговечности и функции модели, подобной печени, была создана дальнейшая разработка шифроидных моделей HepG2 в целях поддержки оценки опасности генотоксичности в течение долгосрочных режимов повторного воздействия (≤14 дней). Таким образом, в соответствии с принципами 3Rs для замены, сокращения и уточнения экспериментов на животных, настоящий протокол был создан, чтобы обеспечить передовые 3D in vitro печеночной модели, способной надежно оценить несколько токсикологических конечных точек (например, функциональность печени, (про-) воспалительные маркеры, цитотоксиситу и генотоксичность) после острой, долгосрочной и повторной химической и ENM воздействия в обычном порядке и легко доступны.
Здесь мы представляем метод создания физиологически релевантной 3D-линии клеток гепатоцитов на основе модельной системы in vitro для оценки опасности генотоксичности после острого или долгосрочного, повторного воздействия ЭНМ. Протокол может быть разбит на 6 ключевых этапов: культивирование криоконсервированных клеток HepG2; Подготовка сфероидов HepG2; HepG2 сфероид передачи от висячих капли к агарозной подвеске; Урожай сфероидов HepG2; микронуклид анализа и скоринга; и анализ данных.
Protocol
1.Культивирование криоконсервированных клеток HepG2
ПРИМЕЧАНИЕ: HepG2 клетки, полученные из американского типа культуры коллекции (ATCC) были отстояны в 1x Dulbecco в модифицированных Eagle Medium (DMEM) с 4,5 г / L D-глюкозы и L-глутамин дополнен 10% плода бычьего рака сыворотки (FBS) и 1% пенициллина / стрептомицин антибиотик.
- Предварительно теплый DMEM клеточной культуры среды (в том числе добавки) в 37 градусов по Цельсию водяной бане в течение 30 мин.
- Удалите один флакон клеток HepG2 из жидкого азота и оттаивать в водяной бане 37 градусов по Цельсию в течение 2-3 минут, в то время как осторожно закрученного флакона, чтобы обеспечить равномерное оттаивание клеточной подвески. Позаботьтесь о том, чтобы не погружать флакон над O-кольцом, чтобы уменьшить потенциал загрязнения.
- После размораживания, удалить флакон с водяной бани и спрей щедро с 70% этанола для обеззараживания внешней поверхности флакона перед размещением под стерильной, класс II ламинарный ткани культуры капюшон.
- Тщательно пипетка содержимое криовиальной клетки HepG2 в центрифугу трубки, содержащей 9 мл предварительно разогретой DMEM клеточной культуры среды (с добавками).
- Используя 10 мл стриптизерши, перенесите 10 мл клеточной подвески в 25 см 2 одноразовыеклеточные культуры колбы и инкубировать культуру в течение 3 дней (от посева) при 5% CO2 и 37 C до тех пор, пока не будет достигнуто 80% слияние, прежде чем пройти суб-культуры в больших 75см 2 одноразовые клетки культуры колбу.
- Как только 80% слияние достигнуто, суб-культурных клеток в стерильных условиях путем трипсинизации с 0,05% трипсина / ЭДТА раствор предварительно нагревается в 37 градусов по Цельсию водяной бане в течение 30 мин. Ни в одной точке клетки не должны быть разрешены для высыхания.
- Как клетки образуют адепт монослой, удалить средства массовой информации, опрокидываясь в дезинфицирующее средство горшок отходов. Затем немедленно вымойте монослой, чтобы удалить все следы существующих средств массовой информации, промыв колбу дважды с 3 мл стерильного раствора 1x PBS при комнатной температуре. Кроме того, отбросить PBS в дезинфицирующее средство горшок отходов.
- После удаления PBS смойка добавьте 5 мл предварительно разогретого раствора 0,05% трипсина-ЭДТА, обеспечивая покрытие всей поверхности клеток и инкубационые клетки в течение 6-8 мин при 37 градусах Цельсия и 5% CO2.
- Аккуратно нажмите колбу, чтобы выбить клетки из нижней части колбы, а затем добавить 5 мл DMEM клеточной культуры среды (с добавками), чтобы нейтрализовать фермент трипсина.
- Перенесите подвеску клетки в трубчатую трубку с центрифугой 50 мл и тщательно свяжь клетки вверх и вниз, чтобы убедиться, что клетки полностью разобщены.
- Центрифуга разбавленной клеточной суспензии при 230 х г в течение 5 мин. Отбросьте супернатант в дезинфицирующее средство и повторно приостанавливайте клеточные гранулы в 25 мл среды культуры клеток DMEM (с добавками).
- Передача клеточной суспензии в 75см 2 одноразовые клетки культуры колбу и инкубировать при 37 градусов по Цельсию и 5% CO2 в течение еще 3 дней, прежде чем пройти фероид подготовки. После того, как HepG2s имели время, чтобы акклиматизироваться и еще раз достичь 80% слияния, определить концентрацию клеток в рамках подготовки к рассаде сфероидов.
2. Подготовка сфероидов HepG2
- Повторите подмухораслевые шаги, о них говорилось выше, за исключением центрифугации, повторно приостанавливайте клеточное гранулирование в 1 мл среды культуры DMEM, предварительно разогретой в водяной бане 37 градусов по Цельсию. Пипетт ячейки подвески вверх и вниз тщательно.
- Оценка жизнеспособности клеток с помощью Trypan Blue Исключение анализ (см. OSHA SOP 3.21 Репродуктивные токсины, Mutagens, Teratogens и embryotoxins - Процедуры для безопасного обращения и хранения (2019)для здоровья и безопасности руководства) 14 с 1:1 отношение клеточной подвески до предварительно отфильтрованного 0,4% Trypan синий раствор.
- Перед подсчетом клеток возьмите 1 мл синего раствора Trypan с помощью шприца 1 мл и профильтруйте с блоком фильтра 0,45 мкм в стерильную трубку 1 мл.
- Перенесите 10 МКЛ отфильтрованного, голубого раствора Трипан в трубку 0,2 мл и добавьте 10 мкл клеточной подвески. Оставшийся отфильтрованный синий раствор Trypan может храниться до 3 месяцев при комнатной температуре для дальнейшего использования.
- Спрей гемоцитометр тщательно с 70% этанола и протрите сухой стерильным бумажным полотенцем, прежде чем обеспечить coverslip на вершине с помощью дыхания пара. Скольжение крышки через дыхание увлажненной поверхности вызывает сплоченные силы, генерируя кольца Ньютона.
- Аккуратно пипетка Trypan синей клеточной подвески вверх и вниз с помощью 1000 МКЛ пипетки (для уменьшения чистого стресса), прежде чем добавить 10 йл гемоцитометра. Убедитесь, что раствор рассеивается под крышкой скольжения и охватывает всю сетку без пузырьков воздуха.

Рисунок 1: Подсчет клеток с помощью гемоцитометра. Схематическое представление гемоцитометра, подчеркивая, из какого квадранта считать клетки. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
- Под микроскопом подсчитайте живые (необлитаемые) и мертвые (окрашенные синие) клетки, найденные в четырех больших угловых квадратах(рисунок 1). Исключите любые ячейки, которые могут перекрываться или сидеть на внутренней стороне двух краев больших угловых квадратов (т.е. на линиях) в графе.
- Используя следующий расчет, вычислите среднее количество живых, жизнеспособных клеток (необлитаемых), присутствующих в образце:
Общее количество ячеек/мл - Live Cell Count x x 10,000
x 10,000
где разбавление относится к тому, сколько раз фондовый раствор был разбавлен в Trypan синий (2x в этом случае) и квадратов считается относится к четырем большим угловым квадратов гемоцитометра считается - На основе жизнеспособного подсчета клеток HepG2 и с использованием следующей формулы:
C1V1Q C2V2
где C1 - концентрация жизнеспособных клеток в настоящее время,
V1 - объем подвески ячейки в настоящее время,
C2 - концентрация клеточной подвески,
V2 - объем клеточной подвески в розыске - Подготовьте 10 мл запас раствора подвески клеток HepG2 с DMEM клеточной культуры среды в концентрации 2,0х 10 5 клеток / мл для того, чтобы достичь 4000 клеток HepG2 на 20 йл висячие капли. Смешайте подвеску клетки тщательно, аккуратно трубы вверх и вниз с помощью 1000 МКЛ пипетки, чтобы обеспечить все клетки полностью приостановлены в средствах массовой информации.
- К колодцам пластины культуры клеток 96-хорошо, добавьте 100 йл стерильной, комнатной температуры PBS, чтобы предотвратить висячие капли от высыхания во время инкубации.
- Возьмите крышку стандартной плоской нижней 96-хорошо пластины клеточной культуры, инвертировать его и тщательно пипетки 20 капель клеточной подвески в центре каждого хорошо паз крышки, как показано на рисунке 2. Используйте многоканаражную пипетку, но добавьте только 2 - 4 капли сразу, так как несколько посевов могут повлиять на точность и размещение капель.
- Центр капель в канавках колодцев, выложенных на крышке; в противном случае они не будут висеть в центре колодцев, когда крышка пластины перевернулась и рискуют упасть в пластину. Аккуратно переверните крышку пластины 96-колодец, так что капли теперь висит и тщательно место на вершине 96-хорошо пластины.
- Поместите всю пластину 96 хорошо с крышкой осторожно в инкубатор при 37 градусов по Цельсию и 5% CO2 в течение 3 дней до передачи сфероидов на агарозу.
ПРИМЕЧАНИЕ: Дополнительная осторожность должна быть сделана не только при транспортировке пластин в / из инкубаторов, но при открытии и закрытии инкубатора в целом, как чрезмерное движение может привести к пластины сдвиг и сфероиды либо падать или формировать неправильно.
 x 10,000
x 10,000
Рисунок 2: 3D HepG2 в пробирке фероидной модели подготовки. (A ) HepG2 клетки, посеянные в 20 йл падает на крышку пластины 96-хорошо. (B) Клетки HepG2 пост-посева в висячая модель падения, чтобы обеспечить образование сфероидов. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
3. HepG2 сфероид передачи от висячих капли агарозы подвески
ПРИМЕЧАНИЕ: На 3-й день после посева в висячие капли, сфероиды передаются в колодцы же 96-хорошо пластины все из которых были ранее покрыты тонким слоем 1,5% агарозы гель.
- Подготовка агарозы гели и автоклавы (т.е. день 2 после посева) до дня покрытия пластины (т.е. день 3 после посева).
- Чтобы подготовить 1,5% агарозный гель, весят 0,30 г агарозы в чистую стеклянную бутылку, а затем добавить 20 мл фенол-красной свободной среды DMEM. Автоклав агарозы в течение 1 ч при 230 градусах цельсия для стерилизации. Агарозное покрытие предотвращает присоединение сфероидов HepG2 к основанию скважин и формирование клеточного монослойного вместо сохранения их 3D сфероидной структуры.
- На 3-й день после посева, удалить 96-хорошо пластины, содержащие HepG2 висит падение сфероидов из инкубатора и тщательно перевернуть крышку, чтобы сфероиды больше не висит.
- Используя многокананеловую пипетку, удалите и отбросьте 100 МКЛ PBS, ранее добавленных к основанию пластины из 96 колодец. Разрешить пластины для воздуха в течение 2-3 мин при нагревании агарозы в рамках подготовки к покрытию.
ВНИМАНИЕ: Эта процедура приводит к очень горячей, жидкой агарозы, которые, если пролиты на кожу может сгореть и привести к травмам. Кроме того, следует позаботиться при обработке стеклянной бутылки, содержащей жидкую агарозу, так как это тоже может быть очень жарко. - Используя ранее подготовленные геля агарозы 1,5%, нагревайте стеклянную бутылку, содержащую 20 мл агарозного геля в микроволновой печи при максимальной ватте (т.е. 900 Вт). Чтобы покрыть две пластины 96-колодец, одна бутылка 20 мл заранее подготовленного геля 1,5% агарозы должно быть достаточно.
- После того, как расплавленный, осторожно вихрем агарозы, вращая стеклянную бутылку, чтобы удалить любые пузырьки, а затем добавить 50 йл агарозы в основание каждого хорошо.
ПРИМЕЧАНИЕ: При добавлении агарозы, убедитесь, что не угол пластины »gt;45», как агароза устанавливает быстро и не будет образовывать плоский, уровень слоя, который может нарушить рост сфероидов. Важно эффективно работать на данном этапе, чтобы предотвратить агарозу от затвердеть, прежде чем пластина полностью покрыта. - Разрешить пластины стоять в течение 2 мин при комнатной температуре, прежде чем добавить 100 йл предварительно разогретой DMEM клеточной культуры среды (с добавками) на вершине твердого слоя агарозы в каждой хорошо.
- Переверните крышку пластины 96-хорошо и поместите обратно на вершине 96-хорошо пластины так сфероиды теперь висит еще раз.
- Центрифуга пластины в течение 3 мин при 200 х г для того, чтобы передать сфероиды из подвесной капли в отдельных скважин 96-хорошо пластины. После переноса сфероиды HepG2 должны быть приостановлены в среде клеточной культуры. Позвольте им поселиться в инкубаторе на 24 ч при 37 градусах Цельсия и 5% CO2.
- Разоблачить HepG2 сфероиды такого размера либо химических или ENM лечения на день 4 пост посева (т.е. 24 ч после передачи агарозы покрытием пластин).
- Для того, чтобы поддерживать жизнеспособность клеток в течение длительных периодов культуры, обновлять клеточной культуры среды каждые 3 дня. Для этого аккуратно аспирировать 50 МКЛ среды клеточной культуры с поверхности хорошо и заменить свежим 50 йл DMEM клеточной культуры среды. Позаботьтесь, чтобы не удалить или нарушить сфероид при выполнении среднего изменения.
4. Наноматериальное/химическое воздействие
ПРИМЕЧАНИЕ: Модель сфероида печени HepG2 может поддерживать как режимы воздействия НАУ, так и химические режимы воздействия, но основное внимание в этом протоколе уделяется воздействию ENM. Перед экспозицией испытательный ЕМ должен быть надлежащим образом рассеян; это может быть выполнено в рамках Протокола о дисперсии NanoGenoTox (Грант-соглашение No 20092101, 2018)15.
- После дисперсии в соответствии с Протоколом дисперсии NanoGenoTox разбавляют подвеску ENM от стартовой концентрации 2,56 мг/мл до конечной желаемой концентрации в предварительно разогретой среде культуры клеток DMEM (включая добавки). Общий объем 5 мл требуется для дозы 1 96 пластины хорошо.
- Чтобы подвергать HepG2 сфероид либо химических или ENM, используя 200 МКЛ пипетки, аспирировать 50 МКЛ клеточной культуры среды с поверхности каждой хорошо (оставляя 50 йл в хорошо, чтобы не беспокоить сфероидов) и заменить 50 йл среды, содержащей тест токсиканта в требуемой дозе.
- После того, как испытательный материал был применен, инкубировать пластины для желаемого времени экспозиции при 37 градусов по Цельсию и 5% CO2.
- Если проводится долгосрочный (≥24 ч) режим воздействия, то сразу же после того, как истекли желаемые сроки воздействия, урожай сфероидов для анализа конечных точек микронукля, описанный ниже в шагах 6.1 - 6.4.
- Однако при режимах острого воздействия (например, ≤24 ч), после окончания периода воздействия, урожай, бассейн и хранение 50 МКЛ супернатанта из каждой пластины 96 хорошо при -80 градусов по Цельсию для дальнейшего биохимического анализа позже. Замените среду клеточной культуры на 50 МКЛ свежей среды, содержащей 6 мкг/мл циточалазин B, и оставьте инкубировать в течение 1 - 1,5 клеточных циклов (т.е. 24 - 26 ч для HepG2) в рамках подготовки к сбору микронуклидов блока цитокинезии.
ПРИМЕЧАНИЕ: Для острых (≤24 ч) режимов воздействия, цитокинезис блок микронуклея анализа с cytochalasin B может быть применен, но для долгосрочных (≥24 ч) режимы воздействия, моноядерной версии (без цитохалазин B) анализа должны быть использованы, как описано ниже на рисунке 4.
5. Сбор сфероидов HepG2
ПРИМЕЧАНИЕ: После химического или ENM воздействия лечения, как клеточной культуры среды или сфероидной ткани могут быть собраны для анализа нескольких конечных точек. В зависимости от анализа конечных точек сфероиды могут быть собраны индивидуально (например, для анализа изображений) или объединиться (например, для анализа микронуклея блока цитокинезиса).
- Удалите пластину из 96-х колодец из инкубатора.
- Используя трубу из 200 мкл, аспирировать 100 МКЛ среды клеточной культуры, включая сфероидную ткань из каждой хорошо и собирать в стерильной, 15 мл центрифуги трубки. Позаботьтесь, чтобы избежать контакта с агарозой.
- После сбора центрифуга суспензии сфероида на 230 х г в течение 5 мин. Удалите супернатант и храните при -80 градусов по Цельсию для дальнейшего анализа конечных точек (например, тесты функции печени) позже.
- Повторно приостанавливайте гранулы сфероидов в 1 мл стерильной, комнатной температуры PBS (1x).
- После мытья, центрифуга сфероидной суспензии снова на 230 х г в течение 3 мин. Откажитесь от супернатанта, повторно приостанавливайте в 500 МКЛ 0,05% трипсина-ЭДТА раствора и инкубировать в течение 6-8 мин при 37 градусов по Цельсию и 5% CO2.
- После инкубации, осторожно пипетки трипсинизированных клеток вверх и вниз, чтобы полностью отмежеваться и повторно приостановить HepG2 клеток до нейтрализации с 1 мл DMEM клеточной культуры среды.
- Центрифуга разбавленной клеточной суспензии при 230 х г в течение 5 мин. Откажитесь от супернатанта в дезинфицирующее средство и повторно приостанавливайте гранулы клеток в 2 мл комнатной температуры PBS (1x).
- Центрифуга клеточной подвески при 230 x g в течение 5 мин. Отбросьте супернатант в дезинфицирующее средство, а затем повторно приостановить гранулы клетки еще раз в 2 мл холодного PBS (1x). Убедитесь, что клетки хорошо рассеяны, чтобы предотвратить скопления клеток, заслоняющих поле зрения при установке на слайды микроскопа.
6. Микронуклид анализ и скоринг
Для ручного метода анализа микронуклей требуется цитоцентрифуг для производства цитодота (определенной, концентрированной области клеток) в центре слайда микроскопа. Этот процесс поддерживает более эффективный скоринг слайда, поскольку он позволяет бомбардиру легко найти ячейки, представляющие интерес, в отличие от оценки целого слайда, где клетки могут быть широко распространены.
- Опустите матовые слайды микроскопа (три за дозу) в 70% этанола с последующим ddH2O и оставьте в воздухе сухой в течение 5 минут.
- Поместите подготовленный микроскоп слайды в воронку куветта, как показано на рисунке 3A, где стеклянная горка (iii) помещается в металлическую опору (iv) с фильтровальной картой (ii) и воронкой кюветта (i) закрепленной сверху.
- Упорядочить куветт воронки в цитоцентрифуге с воронкой лицом вверх, так что 100 йл клеточной подвески могут быть непосредственно добавлены в каждый из них.
- Цитоспина в течение 5 мин при 500 x г, чтобы обеспечить равномерное распределение ячеек на поверхности слайда.

Рисунок 3: Цитоскопина установки для подготовки обработанных клеток на слайдах микроскопа. (A) Отображает отдельные компоненты, (i) кюветт воронка, (ii) фильтр карты, (iii) стеклянный микроскоп слайд и (iv) металлическая поддержка, необходимая для цитозпина HepG2 клетки на микроскоп слайды. (B)Окончательная воронка cuvette создана. (C)Правильное размещение воронки кюветта в цитоцентрифуге. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
- Оставьте горки в воздух сухими перед фиксацией в ледяном, 90% метанол в течение 10 мин.
- После того, как фиксированной, оставить слайды в воздух сухой на ночь при комнатной температуре перед хранением при -20 градусов по Цельсию на срок до 6 месяцев.
- При необходимости удалите заранее подготовленные слайды микроскопа из морозильной камеры -20 градусов по Цельсию и дайте прогреться до комнатной температуры, прежде чем проводить окрашивание Giemsa.
ВНИМАНИЕ: В соответствии с Правилами (EC) No 1272/2008 (CLP), раствор окрашивания Giemsa является легковоспламеняющейся жидкостью, которая может быть токсичной при проглатывании и причинить вред при контакте с глазами, кожей или при вдыхании. Обратитесь к связанному листу SDS для детального хранения, обработки и здоровья и безопасности советы по этому химическому веществу до использования. - В то время как слайды размораживается, подготовьте 20% раствора окрашивания Giemsa (25 мл, необходимых для окрашивания слайдов No30), разбавленных в буфере фосфатазы (рН 6,8). Тщательно смешайте, аккуратно закрученных решение перед фильтрацией с помощью сложенной фильтровальной бумаги, помещенной в воронку.
- Используя пипетку Pasteur, добавьте 3 - 5 капель отфильтрованного раствора Giemsa в цитот на каждом слайде и оставьте на 8 - 10 минут.
- Вымойте слайды в двух последовательных фосфатазы буфер моет перед кратко промывки под холодной водой, чтобы удалить излишки пятна остатки. Оставьте горки в воздух сухим.
- После высыхания, в дым капот, окунуть окрашенные слайды в ксилен в течение 10 с, прежде чем добавить каплю монтажа среды в центре цитоты и место стеклянной крышкой сверху.
- Оставьте микроскоп слайды в капоте дыма на ночь, чтобы высохнуть перед ручным скоринга; они могут храниться бесконечно при комнатной температуре.
7. Анализ данных
- Как описано в Руководящих принципах ОЭСР по тестированию 487 (2014)11, для оценки и количественной оценки повреждений ДНК, вызванных воздействием ЭНМ или химического агента, используйте световой микроскоп (100x цель с погружением нефти) 2000 мононуклеированных или 1000 бинуклеированных клеток на биологическую репликацию, чтобы забить наличие микронуклидов, как показано на рисунке 4.

Рисунок 4: Микронуклид анализа скоринга решение дерева. Схематическое дерево решения, чтобы подчеркнуть необходимость различных процедур оценки скоринга и цитотоксии при использовании микронуклидного анализа с 3D-моделями после острых или долгосрочных режимов воздействия. Острые (≤24 ч) воздействия позволяют использовать цитокинезис заблокирован микронуклей анализа, в то время как долгосрочные (≥24 ч) воздействия требуют моноядерной версии анализа; оба из них описаны в Руководстве по тестированию ОЭСР 487. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
- На основе доли микронуклидов, присутствующих на количество мононуклеированных или бинуклеированных клеток, вычисляйте процент от значения генотоксичности.
- Для того, чтобы оценить повреждения ДНК наблюдается не в результате клеточного мусора, вызванного высокой долей апоптотических клеток, принять меру цитотоксичности рядом. В этом случае, в зависимости от наличия цитохалазин B, используйте либо CPBI или RVCC расчета (как описано на рисунке 4). Генотоксичность должна оцениваться только в образцах, где цитотоксичность составляет менее 55% ± 5%, как определено в Руководстве по тестированию ОЭСР 48711.
Representative Results
Пригодность этой клеточной линии на основе 3D-модели сфероида печени для долгосрочной культуры и оценки генотоксической опасности была оценена путем проведения базовой характеристики для определения жизнеспособности и печени, как функциональность модели в течение 14 дней в культуре, а также его применимость для микронуклида анализа.
Базовая характеристика 3D-модели сфероида печени HepG2
До любой токсикологической оценки in vitro важно проверить, что 3D сфероиды HepG2 сформировались должным образом перед выполнением переноса агарозы или химической/ЭНМ-обработки. Сфероиды HepG2, произведенные с использованием метода подвешивания капли, обычно принимают 2 - 3 дня после посева (4000 клеток/сфероидов) для формирования компактных сфероидов сферической формы со средним диаметром 495,52 мкм Вт х 482,69 мкм H, как показано на рисунке 5A-5C. Сфероиды HepG2, которые сформировались правильно и приемлемы для использования для токсикологической оценки in vitro, должны иметь компактную сферическую структуру с гладкой поверхностью и без визуальных проекций. Рисунок 5 приводит примеры хорошегокачества (рисунок 5D-F) и низкогокачества (рисунок 5G-I) сфероидов. Последний из которых должен быть отброшен. Как правило, 90-95% сфероидов, образоваваемых на тарелку, образуются правильно и жизнеспособны для дальнейших экспериментов.

Рисунок 5: Свет микроскопии изображения, отображающие естественную морфологию сфероидов HepG2 формируется с помощью метода подвешивания падение. (A-C) показать День 2 и (D-I) День 4 HepG2 печени сфероиды после посева. (D-F) являются примерами хорошего качества сфероидов HepG2 в то время как (G-I) показывает плохо сформированные сфероиды. Все снимки были сделаны на цели X20 с помощью микроскопа. Планка масштаба составляет 20 мкм. Пожалуйста, нажмите здесь, чтобы просмотреть более широкую версию этой цифры.
Для дальнейшего подтверждения жизнеспособность сфероидов HepG2, основной колоритный бромокрезол Зеленый Альбумин (BCG) Анализ или асей мочевины могут быть выполнены для оценки их печени, как функциональность. Печень,как функциональность была оценена в соответствии с жизнеспособностью с помощью Trypan Blue Исключение анализа в течение 14 дней культуры период, чтобы определить долговечность модели сфероидов печени и установить, если он может поддерживать долгосрочные или повторяющиеся ENM / химической оценки опасности (Рисунок 6). Концентрация альбумин оставалась последовательной в течение периода культуры. Производство мочевины показывает увеличение концентрации мочевины, производимой на сфероид в течение недели в культуре, прежде чем достичь плато к 7-му дню. Важно отметить, что уровни альбумин и мочевины, производимые в сфероидах 3D HepG2, значительно выше, чем уровни, наблюдаемые в той же клеточной линии, культурной в 2D формате. Действительно, 2D культуры клеток HepG2, пиковые уровни альбумин и мочевины были 0,001 мг/мл и 0,010 нг/Л соответственно. Кроме того, в предыдущей работе, опубликованной Shah et al. с использованием почти идентичной сфероидной системы HepG2, авторы подчеркивают заметное улучшение метаболической активности (CYP1A1 и CYP1A2) в 3D системах модели HepG2 in vitro по сравнению с 2D культурными клетками HepG25.

Рисунок 6: 14-дневные базовые данные характеристики для сфероидов печени HepG2. После передачи от висячих капель, (A) подчеркивает жизнеспособность hepG2 сфероидной модели в течение 14-дневного периода в то время как (B) и (C) выделить печени, как альбумин и мочевины функциональность соответственно. Средние данные ± представлены SEM, n No 4. Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
С неизбежным развитием некротического ядра, известное ограничение 3D культур сфероидной печени, жизнеспособность этой модели на основе HepG2 должна была быть создана, чтобы продемонстрировать, что она была в состоянии поддерживать долгосрочные (5-10 дней) режимы воздействия при сохранении пролиферативной способности, необходимой дляподдержки микронуклида анализа 5. Действительно, эта 3D модель сфероидной печени было показано, чтобы сохранить йgt;70% жизнеспособности в течение 10 дней в культуре. Основываясь на этом и в сочетании с устойчивой печени, как функциональность наблюдается в течение ≥ 14 день культуры период, это 3D печени сфероидная модель может таким образом поддерживать долгосрочные, повторяющиеся режимы воздействия ENM до 10 дней (т.е., до жизнеспособности сфероидов падение ниже 70%). Для справки, рекомендуется, что уровни альбумин для сфероидов HepG2 посеяны на 4000 клеток / сфероидов должно быть ≥20,0 нг / йл в то время как производство мочевины должно быть ≥0,25 нг / йл до проведения в пробирке токсикологической оценки с этой моделью.
Оценка генотоксичности инженерных наноматериалов
Для оценки генотоксичности анализ микронуклидов использовался для определения наличия микронуклеев после острых (24 ч) и долгосрочных (120 ч) воздействий ЭНМ. Афлатоксин B1 является известнымканцерогеном печени 16,17 и рекомендуется положительный контроль для микронуклида анализа. Эксперименты по оптимизации показали, что 0,1 МК Альфатоксина В1 вызывает значительный положительный (увеличение в ≥2,0 раза) генотоксический ответ в 3D сфероидах печени HepG2 и, таким образом, используется в каждом микронуклеусном анализе, проведенном с помощью этой модели. Для обеспечения достоверности результатов анализа микронуклидов с помощью модели сфероидов HepG2 фоновая частота микронуклидов для клеток HepG2, используемых в этой 3D-модели in vitro, должна лежать в пределах 0,6% - 1,2%. В результате, Альфатоксин B1 должен вызвать генотоксический ответ, по крайней мере в два раза выше, чем видели с отрицательным контролем; таким образом, 0,1 МК Альфатоксин B1 должен вызывать частоту микронуклея между 1,5% - 3,0%. Используя эти параметры управления, ENM связанных генотоксичность in vitro может быть надежно оценена. Основываясь на Руководстве по тестированию ОЭСР 487, важно отметить, что при тестировании ENM или химического вещества выбранные концентрации не должны вызывать более 55% ± 5% цитотоксии (указывается снижением значений CPBI или RVCC по отношению к отрицательномуконтролю) 11. Рисунок 7 иллюстрирует данные, полученные при оценке афлатоксина B1 и двух ENM (диоксид титана (TiO2)и щепки (Ag)) после острого и долгосрочного воздействия в сфероидах HepG2, а последующий генотоксический потенциал был проанализирован с помощью микронуклидов. Оба оцениваемых ЭНМ были протестированы на нонцитоксической, низкой дозе 5,00 мкг/мл при остром (24 ч) воздействии и долгосрочном (120 ч) режиме воздействия. Аналогичная тенденция к генотоксичности как в TiO2, так и в Ag ENMs может наблюдаться, в результате чего повышенная генотоксичность ответ, который привел после 24 ч воздействия не было очевидно после длительного воздействия 5 дней. Это было несмотря на устойчивую генотоксичность, вызванную положительным контролем афлатоксина В1 в обеих точках времени.

Рисунок 7: Оценка генотоксичности после TiO2 и Ag ENM воздействия на сфероиды печени HepG2. Оценка генотоксичности (частота микронуклидов) с использованием микронуклидного анализа(A) острого (24 часа) и(B)долгосрочного (120 часов) воздействия 5,00 мкг/мл TiO2 и Ag ENM. Отрицательный контроль является только средством массовой информации, в то время как положительный контроль составляет 0,1 МК афлатоксина B1. Средние данные (n'2), представленные ± SD. Значение, указанное в отношении≤ отрицательного контроля: Пожалуйста, нажмите здесь, чтобы просмотреть большую версию этой цифры.
Discussion
Приложения для 3D печеночными моделями значительно различаются в зависимости от конкретной биохимической конечной точки или неблагоприятного исхода пути мишенью. Каждая модель имеет свои преимущества и ограничения, от междонорных вариаций в первичных человеческих гепатоцитов (PHH) моделей для снижения активности цитохрома p450 в клеточной линии на основе моделей, но всеони являются ценными сами по себе 6,12,18,19. При оценке генотоксичности в моделях существуют ограничения совместимости с утвержденными нормативными конечными точками, такими как анализ микронуклидов in vitro, так как требуется активное распространение. Это необходимо, так как оценка генотоксичности требует количественной оценки фиксированного повреждения ДНК, которая будет оцениваться после клеточного деления, когда есть возможность для восстановления ДНК, чтобы исправить переходные поражения. К сожалению, высокодифференцированные гепатоциты (т.е. HepaRG) на основе сфероидов или микротыссов PHH, которые считаются экспонатом наиболее физиологически релевантных печеночных характеристик, образуют статические(нераспространенные) модели 12,19,20. В результате представленная здесь модель сфероидов 3D HepG2 обеспечивает подходящую альтернативную модель, способную поддерживать тестирование генотоксичности. Сфероиды на основе клеточной линии HepG2 имеют достаточно активное деление клеток на внешней поверхности сфероидов, сохраняя при этом основные функции печени, такие как производство альбумин и мочевины, а также некоторые виды активности CYP4505,12,19. Главным образом эта модель печени in vitro была разработана в дополнение к анализу микронуклидов, так как это один из двух анализов in vitro, рекомендованных в батарее для тестирования генотоксичности8,10,11,21. Тем не менее, модель может быть легко применена к анализу секвенирования ДНК и экспрессии генов (РНК) технологий, в то время как она имеет потенциал для дальнейшей адаптации и использования для других конечных точек повреждения ДНК, таких как анализ кометы. Тем не менее важно учитывать роль, которую играет вмешательство в ЕММ в некоторых анализах конечных точек. Например, анализы на основе цитометрии потока могут не подходить для оценки генотоксичности ENM конкретно из-за вмешательства частиц22.
Одним из ограничивающих факторов сфероидных моделей, которые активно проходят деление клеток, является их размер. Оптимизация плотности посева имеет решающее значение, поскольку должно быть достаточно клеток, которые позволяют модели продолжать размножаться; но не слишком высокое количество клеток, что приводит к слишком компактному сфероиду, что приводит к увеличению некротического ядра. Причиной этого некроза, как полагают, ограничено распространение кислорода и питательных веществ, так как предел этой диффузии, как полагают, составляет примерно 100 - 150мкм ткани 23,24. Тем не менее, это зависит от типа клетки, номер ячейки, эшафот взаимодействий и культурныхусловий 25. С тех пор было показано, что примерно 700 мкм диаметром является пределом для избежания преждевременного начала некроза в центре сфероидов C3A, посев 4000 клеток HepG2 на сфероид обеспечивает диаметр модели на момент экспозиции составляет ≤500мкм 26. Кроме того, Шах и др. установили, что клетки HepG2, посеянные выше 5000 клеток на сфероид, продемонстрировали 25% снижение жизнеспособности после 7 дней в культуре, что может относиться к среднему диаметру 680 мкм и ограниченной доступности питательных веществ в 20 йл, висящейпадение 5. Чтобы преодолеть это, модель, разработанная в настоящем протоколе, проходит критический шаг, когда висячие капли передаются в агарозные скважины с покрытием после первоначального образования сфероида. Это обеспечивает больший объем среды культуры присутствует для поддержания постоянно растущего числа клеток в сфероидов. В результате, модель сфероида HepG2 остается более чем на 70% жизнеспособной после 10 дней в культуре и может быть использована для долгосрочной оценки опасности invitro.
В то время как модель сфероида HepG2 может поддерживать как острые, так и долгосрочные режимы воздействия, освежающий клеточный культурный средний период в течение длительных периодов культуры ограничен для этой модели, поскольку полная замена среды не рекомендуется из-за потенциальной потери сфероидов. Предполагается, что при воздействии ЭНМ наблюдается высокая тенденция к однородным дисперсиям ЭНМ на агломерат и осадочные отложения. Тем не менее, примечательно, что скорость, с которой отложения ENM могут варьироваться в зависимости от параметров частиц (например, размер, форма и плотность) и может быть определена теоретически с помощью модели осадочных отложений in vitro, диффузии и дозиметрии (ISDD), или ее последних производных, о которых часто говорят, когда в отношении ENM (подвеска) экспозицияприближается к 27,28. При этом ум, предполагается, что если только 50% клеточной культуры среды тщательно удалены с поверхности клеточной культуры, нарушение и последующее удаление дозы ЕММ теоретически должно быть минимальным. Тем не менее, с броурианского движения в игре, это не может быть строго так и дальнейшей работы в осаждения и осаждения каждого конкретного ЕМ для тестирования должны быть предприняты для обеспечения правильной дозиметрии сохраняется на протяжении долгосрочных режимоввоздействия 27. Главным образом это потенциальное ограничение, чтобы рассмотреть при выполнении повторных режимов досирования, поскольку это может иметь решающее значение для окончательной, накопленной концентрации. Химические воздействия, с другой стороны, в то время как не без своих собственных ограничений, чтобы рассмотреть, предлагают более упрощенный подход в том, что химические вещества, как правило, остаются в растворе и, таким образом, прямая замена первоначальной химической концентрации в дополнение к вновь добавленной концентрации гарантирует, что любое химическое вещество, потерянное во время освежения средств массовой информациизаменяется соответственно 29. Будущие применения будут включать оценку пригодности модели для режимов повторного воздействия в течение долгосрочных периодов культуры, поскольку неоднократные стратегии досирования имеют решающее значение для оценки способности конкретной органной системы к улучшению или преодолению неблагоприятных последствий, если таковые имеются, вызванных биоаккумуляцией ксенобиотического вещества.
В заключение я хотел бы сказать, что эта 3D-печеночная печеночная модель может быть использована для оценки целого ряда реалистичных сценариев воздействия, обеспечивая тем самым будущий подход in vitro для более эффективной поддержки как ENM, так и оценки химической опасности в обычном и легкодоступном виде.
Disclosures
Авторов нечего раскрывать.
Acknowledgments
Авторы хотели бы отметить, что это исследование получило финансирование от научно-исследовательской и инновационной программы Европейского союза Horizon 2020 для проекта PATROLS в соответствии с грантовое соглашение No760813
Materials
| Name | Company | Catalog Number | Comments |
| Aflotoxin B1 | Sigma Aldrich, UK | A6636-5MG | |
| Agarose | Sigma Aldrich, UK | A9539-50G | |
| Autoclave Tape | |||
| BCG Albumin Assay | Sigma Aldrich, UK | MAK124 | |
| Bovine Serum Albumin Powder | Sigma Aldrich, UK | A9418 | |
| Cell Freezing Aid | Thermo Fisher Scientific, UK | 5100-0001 - Mr Frosty | |
| Centrifuge | Eppendorf | 5810 R | |
| Cytochalasin B | Merck, UK | 250233 | |
| Cytology Metal Clips | |||
| Cytospin 4 Centrifuge | ThermoFisher Scientific, UK | CM00730202 | |
| DMEM with 4.5g/L D-Glucose, L-Glutamine | GIBCO, Paisley, UK | 41965-039 | |
| DMEM, phenol-red free with 4.5g/L D-Glucose, L-Glutamine with Hepes | GIBCO, Paisley, UK | 21063-029 | |
| DPX Mounting Medium | FisherScientific, UK | D/5330/05 | |
| Ethanol | FisherScientific, UK | 10048291 | |
| FBS | GIBCO, Paisley, UK | 10270-106 | |
| Filter Cards for Shandon Cytospin | FisherScientific, UK | 15995742 | |
| Frosted Glass Slides | ThermoFisher Scientific, UK | ||
| Giemsa's Stain Improved R66 Solution, Gurr | VWR Chemicals, UK | MFCD00081642 | |
| Glass Coverslips (24 x 60) | Deckglaser, VWR | ECN631-1575 | |
| Haemocytometer and Coverslip | |||
| Immersion Oil for Microscope | Zeiss, UK | 518F, ISO8034 | |
| Laminar Class II Tissue Culture Hood | Scanlaf Mars | ||
| Light Microscope | Zeiss, UK | Axiovert 40C | |
| Liquid Nitrogen | |||
| Methanol | FisherScientific, UK | 10284580 | |
| Microwave | |||
| Non-Filtered, Sterile 200µl and 1000µl Pipette tips | Greiner-Bio-One, UK | ||
| NuncMicroWell 96-Well Microplates | ThermoFisher Scientific, Denmark | 167008 | |
| P1000 and P200 micropipettes | |||
| P300 and P50 multi-channel pipettes | |||
| PBS pH 7.4 1X, MgCl2 and CaCl2 Free | GIBCO, Paisley, UK | 14190-094 | |
| Pen/Strep | GIBCO, Paisley, UK | 15140-122, Penicillin/Strepmyocin 100X or 10,000U/ml | |
| Phosphatase Buffer Tablets | GIBCO, Paisley, UK | 10582-013 | |
| Pipette Boy | |||
| Simport Scientific CytoSep Funnels for Shandon Cytospin 4 Centrifuges | FisherScientific, UK | 11690581 | |
| Sonifier SFX 550 240V CE 1/2" - Probe | Branson, USA | 101-063-971R | |
| T-25 and T-75 Tissue Culture Flask | Greiner-Bio-One, UK | T-25 (690175) and T-75 (660175) | |
| Trypan Blue Solution | Sigma Aldrich, UK | T8154-100mL | |
| Urea Assay Kit | Sigma Aldrich, UK | MAK006 | |
| Virkon Disinfectant | DuPont, UK | Rely+On Virkon | |
| Water Bath (37?C) | Grant JBNova 18 | ||
| Weighing Balance | |||
| Xylene | FisherScientific, UK | 10588070 | |
| 0.05% Trypsin-EDTA | GIBCO, Paisley, UK | 5300-054 | |
| 0.2mL and 1.0mL Eppendorf Tubes | Greiner-Bio-One, UK | ||
| 0.45µm Filter Unit | Millex HA, MF-Millipore, UK | SLHA033SS | |
| 1.0mL Syringe | BD Plastipak, FisherScientific, UK | 300185 | |
| 20mL LS Scintillation Glass Vials, 22-400 Foil Lined PP Caps | DWK Life Sciences GmbH, Germany | WHEA986581 | |
| 37?C and 5% CO2 ISO Class 5 Hepa Filter Incubator | NUAIRE DHD Autoflow | ||
| 3mL Pasteur Pipette | Greiner-Bio-One, UK | ||
| 50mL Conical Falcon Tubes | Greiner-Bio-One, UK | ||
| 50mL or 100mL Glass Bottles | |||
| 50mL Skirted Falcon Tubes | Greiner-Bio-One, UK | ||
| 5mL, 10mL and 25mL Pipettes | Greiner-Bio-One, UK | ||
| 9.4cm Square, Petri Dish | Greiner-Bio-One, UK | 688161 |
References
- Geiser, M., Kreyling, W. G. Deposition and biokinetics of inhaled nanoparticles. Particle and Fibre Toxicology. 7, 2 (2010).
- Modrzynska, J. Toxicological effects of nanoparticle deposition in the liver. Kgs. Lyngby, Denmark: Technical University of Denmark. , (2018).
- Elje, E., et al. The comet assay applied to HepG2 liver spheroids. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 845, 403033 (2019).
- Breslin, S., O'Driscoll, L. Three-dimensional cell culture: the missing link in drug discovery. Drug Discovery Today. 18, 240-249 (2013).
- Shah, U. -K., et al. A three-dimensional in vitro HepG2 cells liver spheroid model for genotoxicity studies. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 825, 51-58 (2018).
- Lauschke, V. M., Hendriks, D. F. G., Bell, C. C., Andersson, T. B., Ingelman-Sundberg, M. Novel 3D Culture Systems for Studies of Human Liver Function and Assessments of the Hepatotoxicity of Drugs and Drug Candidates. Chemical Research in Toxicology. 29, 1936-1955 (2016).
- van Grunsven, L. A. 3D in vitro models of liver fibrosis. Advanced Drug Delivery Reviews. 121, 133-146 (2017).
- Corvi, R., Madia, F. In vitro genotoxicity testing - can the performance be enhanced. Food and Chemical Toxicology. 106, 600-608 (2017).
- Doak, S. H., Manshian, B., Jenkins, G. J. S., Singh, N. In vitro genotoxicity testing strategy for nanomaterials and the adaptation of current OECD guidelines. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 745, 104-111 (2012).
- Fenech, M. Cytokinesis-block micronucleus cytome assay. Nature Protocols. 2, 1084-1104 (2007).
- OECD. OECD Guidelines. Test 489: In vivo Mammalian Alkaline Comet Assay. , (2016).
- Gerets, H. H. J., et al. Characterization of primary human hepatocytes, HepG2 cells, and HepaRG cells at the mRNA level and CYP activity in response to inducers and their predictivity for the detection of human hepatotoxins. Cell Biology and Toxicology. 28, 69-87 (2012).
- Sison-Young, R. L., et al. A multicenter assessment of single-cell models aligned to standard measures of cell health for prediction of acute hepatotoxicity. Archives of Toxicology. 91, 1385-1400 (2017).
- European Guidelines 2019. European Agency for Safety and Health at Work. , Available from: https://osha.europa.eu/en/safety-and-health-legislation/european-guidelines (2019).
- Jensen, K. A. The NANOGENOTOX Dispersion Protocol for NANoREG. European Union Grant Agreement n° 2009. 21, 01 (2014).
- Marchese, S., et al. Aflatoxin B1 and M1: Biological Properties and Their Involvement in Cancer Development. Toxins. 10, 214 (2018).
- Rushing, B. R., Selim, M. I. Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food and Chemical Toxicology. 124, 81-100 (2019).
- Kermanizadeh, A., Brown, D. M., Moritz, W., Stone, V. The importance of inter-individual Kupffer cell variability in the governance of hepatic toxicity in a 3D primary human liver microtissue model. Scientific Reports. 9, 7295 (2019).
- Berger, B., et al. Comparison of Liver Cell Models Using the Basel Phenotyping Cocktail. Frontiers in Pharmacology. 7, 443 (2016).
- Ramaiahgari, S. C., et al. A 3D in vitro model of differentiated HepG2 cell spheroids with improved liver-like properties for repeated dose high-throughput toxicity studies. Archives of Toxicology. , (2014).
- Li, Y., et al. Factors affecting the in vitro micronucleus assay for evaluation of nanomaterials. Mutagenesis. 32 (1), 151-159 (2016).
- Kirkland, D., Reeve, L., Gatehouse, D., Vanparys, P. A core in vitro genotoxicity battery comprising the Ames test plus the in vitro micronucleus test is sufficient to detect rodent carcinogens and in vivo genotoxins. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. 721 (1), 27-73 (2011).
- Curcio, E., et al. Mass transfer and metabolic reactions in hepatocyte spheroids cultured in rotating wall gas-permeable membrane system. Biomaterials. 28, 5487-5497 (2007).
- Glicklis, R., Merchuk, J. C., Cohen, S. Modeling mass transfer in hepatocyte spheroids via cell viability, spheroid size, and hepatocellular functions. Biotechnology and Bioengineering. 86, 672-680 (2004).
- Asthana, A., Kisaalita, W. S. Microtissue size and hypoxia in HTS with 3D cultures. Drug Discovery Today. 17, 810-817 (2012).
- Gaskell, H., et al. Characterization of a functional C3A liver spheroid model. Toxicology Research. 5, 1053-1065 (2016).
- Cho, E. C., Zhang, Q., Xia, Y. The effect of sedimentation and diffusion on cellular uptake of gold nanoparticles. Nature Nanotechnology. 6, 385-391 (2011).
- Hinderliter, P. M., et al. ISDD: A computational model of particle sedimentation, diffusion and target cell dosimetry for in vitro toxicity studies. Particle and Fiber Toxicology. 7, 36 (2010).
- Kramer, N. I., di Consiglio, E., Blaauboer, B. J., Testai, E. Biokinetics in repeated-dosing in vitro drug toxicity studies. Toxicology in vitro. 30, 217-224 (2015).
Tags
Биоинженерия выпуск 160 модели печени In Vitro наноматериалы оценка опасности долгосрочное воздействие нано(гено)токсикология повреждение ДНКErratum
Formal Correction: Erratum: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure
Posted by JoVE Editors on 01/26/2021.
Citeable Link.
An erratum was issued for: Advanced 3D Liver Models for In vitro Genotoxicity Testing Following Long-Term Nanomaterial Exposure. The Representative Results section was updated.
Figure 6 in the Representative Results section was updated from:
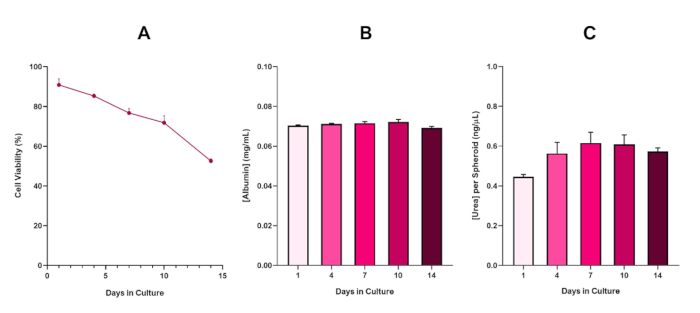
to:
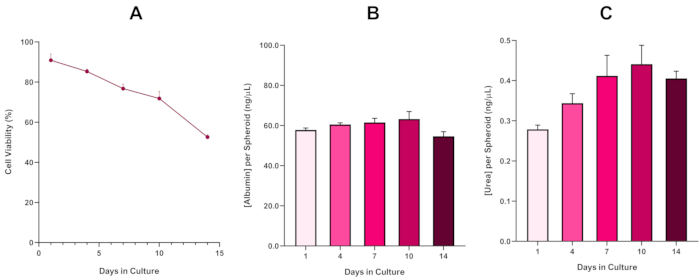
The fourth paragraph in the Representative Results section was updated from:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥0.06 mg/mL whilst urea production should be ≥0.4 ng/µL before conducting an in vitro toxicological assessment with this model.
to:
With the inevitable development of a necrotic core, a known limitation of 3D liver spheroid cultures, the viability of this HepG2 based model had to be established to demonstrate it was able to sustain long-term (5-10 day) exposure regimes whilst maintaining the proliferative capability required to support the micronucleus assay5. Indeed, this 3D liver spheroid model has been shown to retain >70% viability over 10 days in culture. Based on this and in conjunction with the sustained liver-like functionality observed over the ≥14 day culture period, this 3D liver spheroid model can thus support long-term, repeated ENM exposure regimes up to 10 days long (i.e., before viability of the spheroids drop below 70%). For reference, it is advised that albumin levels for HepG2 spheroids seeded at 4000 cells/spheroid should be ≥50.0 ng/μL whilst urea production should be ≥0.25 ng/µL before conducting an in vitro toxicological assessment with this model.
