

Summary
単一分子レベルでのDNAの部位特異的切断を測定するための高度に平行な方法が記載されている。このプロトコルは、制限エンドヌクレアーゼNdeIを用いた手法を示す。この方法は、部位特異的なDNA切断をもたらす任意のプロセスを研究するために容易に改変することができる。
Abstract
部位特異的DNA切断(SSDC)は、多くの細胞プロセスにおける重要なステップであり、遺伝子編集にとって極めて重要です。本研究は、多くの単DNA分子で同時にSSDCを測定することができる運動アッセイを記述する。ビーズ・テザード基板DNAは、それぞれ標的配列の単一コピーを含み、マイクロ流体流路に調製される。外部磁石は、常磁性ビーズに弱い力を与えます。広視野、低倍率の目的を使用して暗視野イメージング下のマイクロビーズを視覚化することによって、最大1,000個の個々のDNAの完全性を監視することができます。制限エンドヌクレアーゼの注入は、NdeI、切断反応を開始する。ビデオ顕微鏡は、関連するビーズが目的の焦点面を上下に移動するフレームを観察することによって、各DNA切断の正確な瞬間を記録するために使用される。フレームごとのビーズカウントは反応を定量化し、指数適合によって反応速度が決定されます。この方法は単一の実験で単一分子SSDC反応に関する定量的および統計的に有意なデータの収集を可能にする。
Introduction
部位特異的DNA切断(SSDC)は、多くのゲノム取引における重要なステップである。例えば、細菌制限修飾(RM)11およびCRISPR2システムは、特定の配列で外来DNAを認識および切断することによって、ファージおよびプラスミドによる攻撃から細胞を保護する。II型RMでは、制限エンドヌクレアーゼ(R)は、タンパク質-核酸相互作用を介して短い4–8塩基対(bp)配列を認識する。CRISPR関連エンドヌクレアーゼは、Cas9などの、エンドヌクレアーゼ4に結合したcrRNAを有する標的部位のハイブリダイゼーションを介して部位に結合する。部位特異的二本鎖破断(DSB)の作成も、多くのDNA組換えイベント5の第一歩である。例えば、V(D)J組換えによって作成された抗原結合領域の多様性は、特異的標的部位6の認識および切断を必要とする。いくつかのトランスポソンは、特定のDNA配列を標的とすることが知られている、7.当然のことながら、Cas9のようなこれらのプロセスに関与する多くの部位特異的ヌクレアーゼは、遺伝子編集技術8の重要な構成要素である。また、新規部位特異的エンドヌクレアーゼ(すなわち、ジンクフィンガーヌクレアーゼ9およびTALENS10)もゲノムを編集するように設計されている。
核酸の部位特異的切断の運動を測定するために多くの方法が採用されている。これらには、ゲル分析、蛍光11、12、,12およびシーケンシングベースの方法13が含まれる。DNAの単一分子のDSBが鎖分離後のビーズの動きによって検出されることを可能にするマイクロビーズのテザリングで大きな進歩が達成されました。これらの方法では、異なるタイプの力が、ビード後切断のストランド分離および運動を確実にするために使用される。あるケースでは、光トラップがEcoRV14によるDNAの切断を測定するために使用されてきた。これらの実験では、ターゲット検索は調査の目的であり、サイト固有の結合がレート制限ステップになるように条件が最適化されています。光トラップの欠点の1つは、一度に1つのDNAしか観察できないということです。さらに、ストランド分離のテストには、周期的に大きな引き上げ力を適用する必要があります。
別の技術は、連続的な方法でビードを引っ張るために流れと弱い磁力の組み合わせを使用します15.このようにして、NdeIによる拡散制限された切断が測定される。この方法を用いると、同時に数百個のDNAを同時に測定することができ、1回の実験で統計的有意性を得ることができます。磁気ピンセットを用いた実験も行われている。そのような研究の1つにおいて、挿入オリゴヌクレオチド16にDSBを含めることによってレトロウイルスインテグラーゼを研究した。統合に成功すると、結合されたDNAにDSBが組み込まれ、付着したビーズが失われた。ATP依存性III型制限エンドヌクレアーゼEcoPIの同様の研究では、1回の実験17で数十のDNAが観察された。磁気ピンセットは、反応中に緊張を制御し、監視することができるという利点を有する。
ここでは、DAの大規模テザリングの最近の改善を利用したSSDCキネティクスを測定するための非常に平行な単一分子法を紹介します。この方法は、DNA複製18、DNA19の輪郭長、および、R15による切断を測定するために用いられる以前15の方法の改良および延長である。19この技術では、認識配列の単一のコピーを含む線状DNAは、一方の端にビオチン、他方でジゴキシゲニンで調製される。ビオチンは、常磁性マイクロビーズに共有結合するストレプトアビジンに結合する。DNAビーズ複合体は、抗ジゴキシゲニンFAB断片で機能化されたマイクロ流体チャネルに注入される。次いでDNAは、二ゴキシゲニンを吸着したFAB断片に結合して表面付着点にテザーを付ける。永久磁石で弱い磁力を加えた場合、ビードが表面に非特異的に貼り付けられるのを防ぎます。サンプルは、流路に迅速に注入することができる(<30 s)切断反応を活性化する。データ収集中にフローがオフになります。各DNAが切断されると、ビーズが目的の焦点計画を上下に移動するフレームを記録することによって切断の正確な時間を決定することができ、したがってビデオレコードから消える。残りのビーズのフレームごとのカウントは、反応の進行状況を定量化するために使用することができます。
以下に示す完全なプロトコルと、NdeIを使用して収集されたデータの例を示します。この手法を適用する方法の一例として、タンパク質濃度の範囲に対する切断速度は、必須の金属補因子であるマグネシウムの2つの異なる濃度で測定される。このプロトコルの適用はNdeIを使用するが、この方法はDNA基質設計を変えることによって任意の部位特異的ヌクレアーゼとの使用のために適合させることができる。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. フローセルの作成
- カバーリップを洗う
- エタノール(EtOH)で瓶と超音波処理にカバースリップを置き、1 M KOH(それぞれ30分間)を使用します。EtOHのKOH沈殿を避けるために、洗浄の間にddH2Oで十分に洗いす。
- EtOH と KOH 洗浄ステップ 1x の両方を、合計 4 回の洗浄回数 (2 回の EtOH と 2 つの KOH) に対して繰り返します。汚れた瓶の中にddH2Oでクリーニングされたカバーリップを保管してください。
- クリーンなカミソリを使用してローディングと出口チューブ(長さ8cm)をカットし、きれいなガラススライドの穴に挿入します。入口にはPE-20、出口にはPE-60を使用してください。エポキシは、チューブを確保し、余分なチューブをトリミングするために5分間。
注:ガラススライドは2'x 3'x 1 mmを測定します。穴は15のmmの間隔でペアで開く。各ペアは、1 つのチャネルの両端を形成します。小さい ID のチューブは、上流のデッド ボリュームを減らし、必要な混合時間を減らすため、入口に使用されます (図 1)。 - スライドの穴にチャンネルパターンを切り取ったプレカット両面テープを並べて適用します。プラスチック製の鉗子で滑らかにして、良好なシールを実現します。
メモ:この実験で使用した両面テープの厚さは120μmで、チャンネル幅は2mm、長さは15mmです。チャンネルはナイフプリンタ(材料表)を使用して切断されます。1つのカバースリップに4つのチャネルまで合わせることは可能である。フローセルの画像については、 図1 を参照してください。 - 裏地を剥がした後、テープの上にきれいなカバースリップ(圧縮空気で乾燥)を適用し、良好なシールのためのプラスチック製の鉗子で再び滑らかにします。
- フローセルを密封し、それを治すためにカバースリップからエッジをエポキシ。
テザリング用標識DNAの調製
- PCRチューブでは、0.02 U/μLの高忠実度DNAポリメラーゼ、200μM dNTP、0.5 μMフォワードプライマー、0.5 μMリバースプライマー、および250 ngのM13mp18ベクターDNAを含むPCR反応ミックスを50 μL調製します。
注:フォワードプライマー(ビオチン-CCAACTTAATCGCcTTGC)およびリバースプライマー(ジゴキシゲニン-TGACCATTAGATACATTCGC)は、M1338から107までの位置に及ぶ長さ約1,000 bpの領域を増幅するために選択されました。増幅領域の中央に単一のNdeIサイトがあります。フォワードプライマーは、5ʹ、カバースリップ上の抗ジゴキシゲニンを結合するジゴキシゲニンで標識される。逆プライマーは、ストレプトアビジンコーティングビーズを結合するビオチンで標識5ʹ。 - PCR チューブをサーモサイクラーに挿入し、 表 1に示すようにサイクルに従います。
- メーカーのプロトコルに従って PCR クリーンアップ キットを使用して PCR 製品を精製します。
注: 材料表に指定されたキットを使用して、典型的なDNA収量は〜2μgです。
3. DNAとビーズのテザリング
- バッファーA(1 Mトリス-HCl[pH = 7.5]、50 mM NaCl、2 mM MgCl2、1mg/mL βカゼイン、1 mg/mLプルロニックF-127)の10 mLを調製します。少なくとも1時間真空デシケーター中のドガ。
- フローセルを機能化するには、25 μL の抗ジゴキシゲニン FAB フラグメント(20 μg/mL)をPBSに流路に注入します。PE-60チューブに収まるようにゲルローディングチップを使用してください。室温(RT)で30分間インキュベートします。
- インキュベーション後、0.5 mLのバッファAをスポイトを使用してチャネルを通して流路をフラッシュする。チャネルに空気を導入しないように注意してください。
- 機能化後、逆顕微鏡にフローセルを取り付けます。出口管をシリンジポンプに引き上げ、注入口チューブをバッファーAを含むマイクロ遠心分離管に入れます。
- 手動でシステムをフラッシュし、ポンプをプライミングするために、バッファーAの少なくとも0.5 mLを引っ張ります。ポンプを10 μL/minで少なくとも5分間稼働させ、システムを平衡化します。
- ビーズ(材料表)を調製するために、10 mg/mLストックビーズの1.6μLのビーズとピペットのストックボトルを50 μLのバッファーAに、次に渦を作ります。
- 磁気セパレータを使用して、バッファーをピペットアウトし、バッファーAの50 μLで再中断し、次に渦を再中断します。
- 3回の打ち上がりに対して、ステップ3.7 2xを繰り返します。最後の洗浄では、バッファAと渦の100 μLで再中断して、160 μg/mLの最終濃度を達成します。
- DNAとビーズを複雑化させるために、まず、0.5 pM標識DNA基質の480μLをバッファーAに調製する。次に、160 μg/mLビーズ懸濁液の20 μLのピペットを、ピペット処理の前に必ずビーズをボルテックスします。3分間、回転器の上に置きます。
- 3分後、15分間の流速10μL/minで、または十分なビーズテザリングが観察されるまで、すぐにチャネルにロードします。
注: ビーズは、サーフェス上で相互に相互作用するほど密集してはなりません (説明のセクションを参照してください)。 - すべてのフリービーズのチャネルを洗浄するには、インレットチューブをバッファーAの新鮮なチューブに切り替え、50 μL/minで少なくとも10分間、または緩いビーズが観察されないまで流れ込みます。
4. データ収集と分析
- データ収集の準備をするには、インレットチューブを、バッファAのNdeI(0.25~4.00 U/mL)の少なくとも100 μLを含むマイクロ遠心管に入管を入れ、永久磁石を流路の上に下げ、暗視野イメージング用の軸外に光源を配置します。
注:2つの環状希土類磁石は、一緒にエポキシスされ、データ収集中に片持ち光学ポストを使用して流路のアクティブ面の上に8mm保持されます。オフ軸ライトコースにはガチョウネックランプが使用されています。 - 市販の顕微鏡、ビデオカメラ、データ収集ソフトウェア(資料表)を使用します。 ソフトウェアで [露出] タブをクリックし、[露出時間] を 10 ミリ秒に設定します。[Timelapse]タブをクリックし、[イメージの数] を 600 に設定し、[期間] を 20 分に設定し、[間隔] を 2 秒に設定します。[実行] をクリックしてデータ収集を開始します。
- シリンジポンプで流量を150 μL/minに、注入量を80 μLに設定します。Run注入後、ポンプの電源を切り、データ収集中の流れを防ぐためにバルブを閉じます。
- データが収集されたら、画像解析ソフトウェア(表)を開きます。[ファイル] タブで、[インポート] をクリックします。 "画像シーケンス"ポップアップメニューで画像ファイルを見つけ、「開く」をクリックします。
- しきい値を設定するには、[イメージ] プルダウン メニューの [Thesholdを調整する] をクリックします。スライダ バーを使用して、イメージ内のビーズに対応する明るいスポットを識別するしきい値を設定します。
- [分析] プルダウン メニューの [パーティクルの分析] をクリックして、各フレームの明るいスポットをカウントします。[OK] をクリックし、すべてのイメージを処理するには [はい] をクリックします。結果ファイルを保存します。
注: これは、各録画ビデオ フレーム内のビーズの数を含むデータ ファイルを保存します。 - データ分析ソフトウェア (表) を開き、結果ファイルをインポートするには、[ファイル] プルダウン メニューの [テキスト ファイルからインポート] をクリックします。ビード数データと時間をプロットします。
- [分析] プルダウン メニューの[カーブ フィット] をクリックして、ビードの数と時間を合わせます。"自然指数" 方程式を選択し、[適合]をクリックします。 |"OK".
注: サンプルの注入後に記録されたデータのみを継ぎ手領域に含める必要があります。フィッティング関数の指数の適合パラメータは、切断率になります。
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
この技術を用いて、2つの異なる濃度のマグネシウム(2 mMおよび4 mM)で、さまざまなタンパク質濃度(0.25~4.00 U/mL)について、NdeIのSSDC速度を測定した。これらの条件のそれぞれは、実験ごとに数百から1,000のテザリングされたDNAで、少なくとも2回複製された。 図2 は実験計画を説明する。 図 3 は、データ収集と分析の詳細の例を示しています。 図4 は、マグネシウムの2つの濃度におけるタンパク質濃度にいかに依存するかを示す。十分に低いタンパク質濃度では、その速度は、タンパク質に比例し、マグネシウムから独立であることが観察することができる。十分に高いタンパク質濃度の場合、速度はマグネシウムに依存しますが、タンパク質濃度に依存しません。
| ステップ | 説明 | 温度(°C) | 時間 (複数可) |
| 1 | 変性 | 98 | 30 |
| 2 | 溶融 | 98 | 10 |
| 3 | アニール | 60 | 30 |
| 4 | 拡張 | 72 | 30 |
| 5 | 最終拡張 | 72 | 120 |
表1:PCRパラメータ 図は、プロトコルのステップ 2.2 で使用されるサーモサイサー プログラムのステップの温度と継続時間です。溶融、アニール、及び延長ステップ(ステップ2、3、および4)は30倍繰り返される。
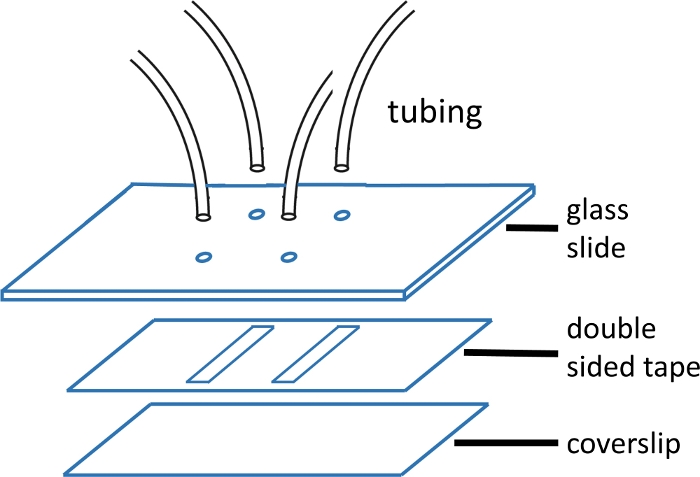
図1:マイクロ流体フローセル構造
上部のガラススライド(2''x 3'、厚さ1mm)は、チャンネルパターンに一致する穴で事前に掘削されています。入口および出口の管は穴に挿入され、テープおよびカバーガラスを取り付ける前にエポキシで固定される。両面テープは、チャンネルパターンで事前にカットされています。下部スライド(#1または#1.5カバーガラス)は、メインテキストに記載されているプロトコルを使用して、以前にクリーニングされています。一度組み立てると、カバーガラスの端部はエポキシで密封される。 この図の大きなバージョンを表示するには、ここをクリックしてください。

図2:実験計画
(A)DNAテザリングの方法。5ʹ末端にジゴキシゲニンで標識された二本鎖DNA(1キロビット)は、抗ジゴキシゲニン・ジゴキシゲニン相互作用を介してフロー細胞の表面に結合する。ビオチンで標識されたDNAの3ʹ末端は、ストレプトアビジンとビオチンの相互作用を介してマイクロビーズに結合する。NdeI切断部位は、DNAの中心に位置する。(B) 磁石と客観的な位置を示すデータ収集中の実験セットアップ。永久磁石は、切断反応中にビーズに弱い上向きの力を維持します。この図の大きなバージョンを表示するには、ここをクリックしてください。
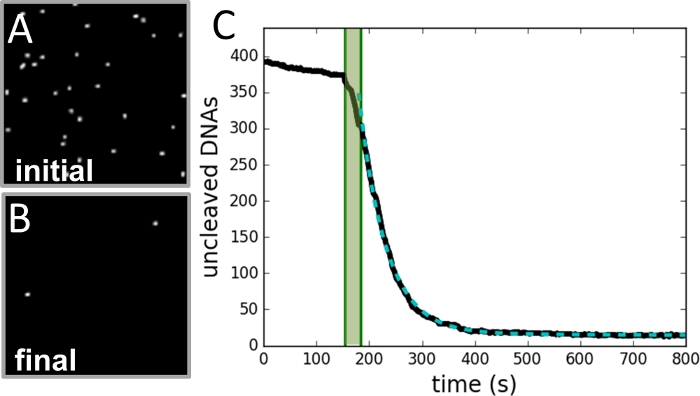
図3:データ収集と分析の例
(A)切断反応が開始される前に採取したビーズの領域の画像。(B)反応終了後の同領域の画像。(C) ビデオレコードの各フレームから決定されるビーズ数と時間(黒い曲線)のプロット。緑のシェーディングエリアは酵素の注入期間を示し、フィット感には含まれていません。データは、単一の指数曲線(緑の破線曲線)に適合し、減衰定数は反応速度と等しくなります。この図の大きなバージョンを表示するには、ここをクリックしてください。

図4:NdeI切断は、タンパク質およびマグネシウムの濃度に依存する。
2つの異なる濃度のマグネシウムにおけるタンパク質濃度の範囲に対するNdeIの測定されたSSDC率のプロット:2 mM(青い円)および4 mM(緑の正方形)。エラー バーは SEM を表します。破線曲線はトレンドラインであり、理論に適合することを表しません。 この図の大きなバージョンを表示するには、ここをクリックしてください。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
このプロトコルは、実験中にストランド分離が観察される限り、任意のSSDCシステムの運動を測定するために使用することができる。切断の検出は、につながれたビーズの剥離を観察することによって影響を受け、したがって、ストランド分離の瞬間をマークします。すべての前のステップは、切断の検出前に発生します。したがって、合計通過時間のみが記録されます。
フローセルカバースリップは、クリーンガラスへの抗体タンパク質の非特異的吸着を介して機能化されます。十分に洗浄されたガラスは、抗体の結合に影響を与える可能性があります。テザリングでは、ビーズ密度が十分に低く、ビーズが相互作用しないようにする必要があります。取り付けポイントの表面密度は、官能化中の抗体の濃度によって制御することができる。ビーズの合計数は、視野のサイズによって異なります。この場合、数百から1,000個のつながれたビーズは良い統計のために十分であり、ビーズとビーズの相互作用を避けた。ビーズ注入中、表面テザリングはライブビデオを介して監視された。ビーズの推定数が500〜1,000ビーズの間であったときにビーズ注射を停止した。
正確に測定できる最速の切断速度は、フローセルの混合時間によって制限される。層流細胞における混合時間は、いくつかの要因の影響を受ける。表面への拡散は重要なステップです。したがって、混合時間は反応物の拡散係数に依存する。サンプル貯蔵所から流路にサンプルを輸送する入口管で発生する有意なせん断は、チャネル内の反応面で十分な混合を確保するために必要な時間を増加させることができます。上流のデッドボリュームを減らし、流量を増やすことで、混合時間を短縮できることが分かった。内径380μm、最大長8cm(流量150μL/min)を用いて、測定した切断速度に影響を与えることなく、射出時間を〜20秒に短縮できることが判明した。混合時間は反応物の拡散係数に依存するので、研究した酵素または切断活性化剤ごとに別々に決定されるべきである。
テザリング法は、ジゴキシゲニン抗体複合体の解離または表面からの抗体の放出のいずれかによる非特異的テザー破裂を可能にする。この結果、〜3 x10-4 s -1の酵素を注入する前に存在する再現性のあるバックグラウンドビーズの損失率が得られる。この系統的効果は、測定された切断率からバックグラウンドレートを差し引くか、フィッティング方程式の背景をモデル化することによって補正することができます。しかし、この下限より低い切断率は、より確実に測定される。
不完全な表面パッシベーションは、不適切なテザリングにつながる可能性があります。これは、実験中に消えない「立ち往生」ビーズの割合を増加theさせるか、または表面から非常にゆっくりと解離する不適切につながれたビーズにつながります。これにより、処理されたデータに、より高いベースラインが作成され、場合によっては傾斜するベースラインが作成されます。適切にクリーニングされたカバースリップと作りたてのβカゼインストックソリューションでは、これらの効果はほとんどのデータセットで最小限に抑えられることがわかりました。これを示す不定期のデータセットでは、フィッティング関数を変更して(傾斜ベースラインを含む)この効果を修正できます。
現在のプロトコルは、いくつかの方法で拡張できます。標的部位結合後の更なる機械化ステップの分離は、必須の補因子がない場合にタンパク質を注入する事前結合形式を用いて行うことができる。このアイデアは、マグネシウムの不在時にNdeIを注入することによってテストされます.これらの条件下では、タンパク質はその同結合部位に結合するが、DNAを切断しない。 この結合工程の後にマグネシウムを注入すると 、 切断が活性化し、急速なビーズ損失をもたらす。実験用セットアップでは、磁石の構成を変えたり流量を加えたりすることで、DNAの立体構造や張力を制御することもできます。これらの実験における低力の下で、DNAは部分的にコイル化される。力をわずかに変えることは、DNAの立体構造に劇的な影響を与える可能性があります。例えば、標的探索が速度制限される緩衝条件下では、DNAの立体構造を変化させることにより、標的探索に対するジャンプの効果を検査することができる。加水分解工程が制限速度である緩衝条件下では、力を変化させることにより、DNA張力がホスホジエステル結合加水分解に及ぼす影響をプローブすることができる。なお、使用される低倍率では、これらの立体構造変化は観察できない。結果として生じるビーズ位置の小さな動きは、DNAの立体構造を制御できることを確認するために、より高い倍率で追跡する必要があります。
データ分析は、さまざまな方法で拡張できます。この作業は単純なビーズカウント法に続いて、単一の指数関数の曲線フィッティングを適用します。滞留時間分析に基づく方法も20.個々のDNAの滞留時間の分布は、カーブフィッティングを介して、またはモーメント21の一般化された方法のようなより洗練された技術を使用して分析することができる。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
著者らは開示する利益相反を持っていません。
Acknowledgments
この研究は、国立科学財団がMCB-1715317を助成金によって支援されました。
Materials
| Name | Company | Catalog Number | Comments |
| 5 minute Epoxy | Devcon | 14250 | |
| anti-digoxigenin FAB fragments | Roche Diagnostics | 11214667001 | |
| camera and software | Jenoptik | GRYPHAX SUBRA | |
| data analysis software | Vernier Inc. | LP | |
| double sided tape | Grace Biolabs | SA-S-1L | |
| Dulbeccos Phosphate Buffered Saline | Corning | 21-031-CV | |
| ethanol 95% | VWR | 89370-082 | |
| forward primer: digoxigenin-CCAACTTAATCGCCTTGC | Integrated DNA Technologies | n/a | |
| image analysis software | National Institutes of Health | ImageJ | |
| inverted microscope | Nikon | TE2000 | |
| knife printer | Silhouette | ||
| M13mp18 DNA | New England Biolabs | N4040S | |
| MyOne streptavidin beads | Thermo Fisher Scientific | 65601 | |
| NdeI enzyme | New England Biolabs | R0111S | |
| PCR cleanup kit | Qiagen | 28104 | |
| pluronic F-127 | Anatrace | P305 | |
| polyethylene tubing PE-20 | BD Intramedic | 427406 | |
| polyethylene tubing PE-60 | BD Intramedic | 427416 | |
| Q5 Mastermix | New England Biolabs | M0492S | |
| rare earth magnet 0.5" OD 0.25" ID | National Imports | NSN0814 | |
| rare earth magnet 0.75" OD 0.5" ID | National Imports | NSN0615 | |
| reverse primer: biotin-TGACCATTAGATACATTTCGC | Integrated DNA Technologies | n/a | |
| syringe pump | Kent Scientific | Genie Plus | |
| β-Casein from bovine Milk | Sigma-Aldrich | C6905 |
References
- Tock, M. R., Dryden, D. T. The biology of restriction and anti-restriction. Current Opinion in Microbiology. 8 (4), 466-472 (2005).
- Garneau, J. E., et al. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 468 (7320), 67-71 (2010).
- Pingoud, A., Fuxreiter, M., Pingoud, V., Wende, W. Type II restriction endonucleases: structure and mechanism. Cellular and Molecular Life Sciences. 62 (6), 685-707 (2005).
- Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C., Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 507 (7490), 62-67 (2014).
- Sadowski, P. D. Site-specific genetic recombination: hops, flips, and flops. The FASEB Journal. 7 (9), 760-767 (1993).
- Schatz, D. G. Antigen receptor genes and the evolution of a recombinase. Seminars in Immunology. 16 (4), 245-256 (2004).
- Craig, N. L. Tn7: a target site-specific transposon. Molecular Microbiology. 5 (11), 2569-2573 (1991).
- Gori, J. L., et al. Delivery and Specificity of CRISPR/Cas9 Genome Editing Technologies for Human Gene Therapy. Human Gene Therapy. 26 (7), 443-451 (2015).
- Urnov, F. D., Rebar, E. J., Holmes, M. C., Zhang, H. S., Gregory, P. D. Genome editing with engineered zinc finger nucleases. Nature Reviews Genetics. 11 (9), 636-646 (2010).
- Joung, J. K., Sander, J. D. TALENs: a widely applicable technology for targeted genome editing. Nature Reviews Molecular Cell Biology. 14 (1), 49-55 (2013).
- Alves, J., Urbanke, C., Fliess, A., Maass, G., Pingoud, A. Fluorescence stopped-flow kinetics of the cleavage of synthetic oligodeoxynucleotides by the EcoRI restriction endonuclease. Biochemistry. 28 (19), 7879-7888 (1989).
- Deng, J., Jin, Y., Chen, G., Wang, L. Label-free fluorescent assay for real-time monitoring site-specific DNA cleavage by EcoRI endonuclease. Analyst. 137 (7), 1713-1717 (2012).
- Becker, W. R., et al. High-throughput analysis reveals rules for target RNA binding and cleavage by AGO2. Molecular Cell. 75 (4), 741-755 (2019).
- vanden Broek, B., Lomholt, M. A., Kalisch, S. M., Metzler, R., Wuite, G. J. How DNA coiling enhances target localization by proteins. Proceedings of the National Academy of Sciences. 105 (41), 15738-15742 (2008).
- Gambino, S., et al. A single molecule assay for measuring site-specific DNA cleavage. Analytical Biochemistry. 495, 3-5 (2016).
- Jones, N. D., et al. Retroviral intasomes search for a target DNA by 1D diffusion which rarely results in integration. Nature Communications. 7, 11409 (2016).
- van Aelst, K., et al. Type III restriction enzymes cleave DNA by long-range interaction between sites in both head-to-head and tail-to-tail inverted repeat. Proceedings of the National Academy of Sciences. 107 (20), 9123-9128 (2010).
- Williams, K., et al. A single molecule DNA flow stretching microscope for undergraduates. American Journal of Physics. 79 (11), 1112-1120 (2011).
- Song, D., et al. Tethered particle motion with single DNA molecules. American Journal of Physics. 83 (5), 418-426 (2015).
- Etson, C. M., Todorov, P., Walt, D. R. Elucidating Restriction Endonucleases Reaction Mechanisms via Dwell-Time Distribution Analysis. Biophys Journal. 106 (2), 22 (2014).
- Piatt, S., Price, A. C. Analyzing dwell times with the Generalized Method of Moments. PLoS One. 14 (1), 0197726 (2019).
