

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
L’expansion clonale est une caractéristique clé de la réponse des lymphocytes T spécifiques à l’antigène. Cependant, le cycle cellulaire des lymphocytes T répondant à l’antigène a été mal étudié, en partie à cause de limitations techniques. Nous décrivons une méthode de cytométrie en flux pour analyser les lymphocytes T CD8 spécifiques de l’antigène à expansion clonale dans la rate et les ganglions lymphatiques de souris vaccinées.
Abstract
Le cycle cellulaire des lymphocytes T spécifiques de l’antigène in vivo a été examiné à l’aide de quelques méthodes, qui présentent toutes certaines limites. La bromodésoxyuridine (BrdU) marque les cellules qui sont en phase S ou récemment terminées, et l’ester de carboxyfluorescéine succinimidyle (CFSE) détecte les cellules filles après la division. Cependant, ces colorants ne permettent pas d’identifier la phase du cycle cellulaire au moment de l’analyse. Une autre approche consiste à exploiter Ki67, un marqueur fortement exprimé par les cellules dans toutes les phases du cycle cellulaire à l’exception de la phase de repos G0. Malheureusement, Ki67 ne permet pas une différenciation plus poussée car il ne sépare pas les cellules en phase S qui sont engagées dans la mitose de celles de G1 qui peuvent rester dans cettephase, procéder au cycle, ou passer à G0.
Ici, nous décrivons une méthode cytométrique en flux pour capturer un « instantané » des cellules T dans différentes phases du cycle cellulaire dans les organes lymphoïdes secondaires de souris. La méthode combine ki67 et la coloration de l’ADN avec une coloration complexe d’histocompatibilité majeure (CMH)-peptide-multimère et une stratégie de contrôle innovante, nous permettant de différencier avec succès les lymphocytes T CD8 spécifiques de l’antigène dans les phases G0,G1 et S-G2/ M du cycle cellulaire dans la rate et les ganglions lymphatiques drainants de souris après la vaccination avec des vecteurs viraux portant le modèle antigène du virus de l’immunodéficience humaine (VIH)-1.
Les étapes critiques de la méthode étaient le choix du colorant d’ADN et la stratégie de contrôle pour augmenter la sensibilité du test et inclure des lymphocytes T spécifiques de l’antigène hautement activés / proliférants qui auraient été omis par les critères d’analyse actuels. Le colorant ADN, Hoechst 33342, nous a permis d’obtenir une discrimination de haute qualité des pics d’ADN G0/G1 etG2/M,tout en préservant la coloration membranaire et intracellulaire. La méthode a un grand potentiel pour accroître les connaissances sur la réponse des lymphocytes T in vivo et pour améliorer l’analyse de l’immunosurveillance.
Introduction
Les lymphocytes T naïfs subissent une expansion clonale et une différenciation lors de l’amorçage de l’antigène. Les lymphocytes T différenciés présentent des fonctions effectrices essentielles à la clairance de l’antigène et au maintien de la mémoire spécifique de l’antigène, ce qui est essentiel pour une protection durable. Au cours des premières étapes de la réponse primaire, l’interaction naïve des lymphocytes T avec les cellules présentatrices d’antigènes (APC) dans des niches spécialisées dans les organes lymphoïdes est essentielle pour induire l’énorme prolifération des lymphocytes T qui caractérise la phase d’expansion clonale1,2,3. L’interaction lymphocyte T-APC est finement régulée par la concentration et la persistance de l’antigène, des signaux co-stimulateurs et des facteurs solubles (cytokines et chimiokines) qui influencent la quantité et la qualité de la progéniture clonale des lymphocytes T4,5,6,7.
Malgré des études intensives sur l’expansion clonale des lymphocytes T, on ne sait toujours pas si les lymphocytes T amorcés par l’antigène terminent tout leur cycle cellulaire sur le site de reconnaissance de l’antigène, ou s’ils migrent vers d’autres organes au cours de la progression du cycle cellulaire. Ce manque de connaissances est dû aux propriétés des outils disponibles pour l’analyse du cycle cellulaire. Il s’agit notamment des anticorps monoclonaux (mAbs) spécifiques au marqueur nucléaire, Ki67, et des colorants cellulaires qui identifient les cellules qui ont subi la phase S du cycle cellulaire (par exemple, la bromodésoxyuridine (BrdU)) ou discriminent entre les cellules filles et leurs ancêtres (par exemple, l’ester de carboxyfluorescéine succinimidyle (CFSE)).
Cependant, les colorants de marquage cellulaire, tels que CFSE et BrdU, ne permettent pas de déterminer si les cellules trouvées dans un organe particulier ont proliféré localement ou plutôt migré vers ce site après la division8,9. De plus, la protéine intranucléaire, Ki67, n’est capable de distinguer que les cellules G0 (cellules Ki67-négatives) de celles de toute autre phase du cycle cellulaire (cellules Ki67-positives). Ainsi, l’analyse Ki67 ne distingue pas les cellules en prolifération active (c’est-à-dire en S,G2ou M) de celles en G1,qui peuvent soit progresser rapidement vers la division, soit rester pendant de longues périodes dans G1 ou revenir à la quiescence10,11.
Nous décrivons ici une nouvelle méthode de cytométrie en flux pour l’analyse du cycle cellulaire des lymphocytes T CD812 spécifiques de l’antigène provenant de la rate et des ganglions lymphatiques (LN) de souris vaccinées(Figure 1). La méthode exploite une combinaison de Ki67 et de coloration de l’ADN qui était précédemment utilisée pour analyser le cycle cellulaire des cellules hématopoïétiques de la moelle osseuse (BM) de souris13,14. Ici, nous avons appliqué avec succès la coloration de l’ADN Ki67 plus, ainsi que la stratégie de contrôle innovante12récemment publiée, à l’analyse de l’expansion clonale des lymphocytes T CD8. Nous avons pu faire une distinction claire entre les lymphocytes T CD8 spécifiques de l’antigène dans G0, dans G1et dans les phases S-G2/ M dans la rate et les LN drainants de souris vaccinées.
Protocol
Les souris ont été hébergées à Plaisant Animal Facility, et le travail a été effectué sous le numéro d’autorisation du ministère italien de la Santé 1065/2015-PR. Le protocole suivait les directives en matière de soins aux animaux conformément aux lois et politiques nationales et internationales (directive UE 2010/63/UE; Décret législatif italien 26/2014).
1. Préparation du milieu et de la solution de coloration
- Préparer un milieu complet : Milieu du Roswell Park Memorial Institute (RPMI) avec 2 mM de glutamine, 100 U/mL de pénicilline/streptomycine, 50 μM de bêta-mercaptoéthanol et 10 % volume/volume (v/v) de sérum fœtal bovin (FBS)
- Préparer le tampon de coloration : Solution saline tamponnée au phosphate sans Ca2+/ Mg2+ (PBS) avec 1% poids / volume (p / v) d’albumine sérique bovine (BSA) et 2 mM de sel disodique d’acide éthylènediaminetétraacétique (EDTA)
2. Traitement de la souris
- Souris Balb/c femelles de 7 à 8 semaines par injection intramusculaire (i.m.) dans les quadriceps du virus de l’immunodéficience humaine (VIH)-1-gag-exprimant-le-vecteur adénoviral du chimpanzé (ChAd3-gag) avec une dose de 10à 7 particules virales.
- À 1-4 mois après l’amorçage, stimuler une fois les souris par i.m. injection du virus modifié de la vaccine Ankara exprimant le VIH-1-gag (MVA-gag) avec une dose de 106 unités formant de la plaque.
- Au jour 3 après le boost, sacrifiez les souris boostées par luxation cervicale et analysez-les en parallèle avec des souris non traitées.
- Récoltez les LN en drainant les quadriceps (iliaques, poplités et inguinaux) et la rate de souris boostées et non traitées. De plus, prélever le BM des deux pattes postérieures de souris non traitées et utiliser ce BM pour les réglages du cytomètre en flux et comme témoin positif pour l’analyse du cycle cellulaire (Figure 2).
REMARQUE: Générez les vecteurs ChAd3-gag et MVA-gag comme décrit précédemment12,15,16,17.
3. Isolement des cellules LN, rate et BM drainantes
- Isolement de la rate et des cellules LN
- Placez 5 mL de milieu complet dans chacun des deux tubes de 15 mL et conservez-les sur de la glace, prêts pour le prélèvement d’organes.
- Sacrifier une souris adulte par luxation cervicale.
- Placez la souris sur le dos et stérilisez la surface de la peau avec 70% v / v d’éthanol.
- Pour recueillir les LN inguinaux, faites une incision longitudinale d’environ 1 cm sur l’abdomen avec des ciseaux et étirez l’incision avec la pince.
- Visualisez les LN inguinaux sur la surface interne de la peau et récoltez-les avec la pince. Placer les LN inguinaux dans l’un des deux tubes de 15 mL préparés à l’étape 3.1.1.
- Pour recueillir la rate, faites une incision péritonéale avec des ciseaux et retirez la rate. Après avoir coupé le tissu conjonctif environnant, placez la rate dans le deuxième tube de 15 mL préparé à l’étape 3.1.1.
- Pour recueillir les LN iliaques, déplacez les intestins de côté et visualisez les LN iliaques près de la veine cave inférieure, puis collectez-les à l’aide de la pince. Placez les LN iliaques dans le même tube contenant les LN inguinaux.
REMARQUE: Pour obtenir suffisamment de cellules LN pour la coloration (voir rubrique 4), il est souvent nécessaire de regrouper les LN poplitées, inguinales et iliaques d’une souris. Ces LN drainent tous les quadriceps (le site de la vaccination i.m). Ce protocole n’utilise qu’un seul tube de 15 mL de LN groupés. - Pour recueillir les LN poplités, saisissez la peau des pattes postérieures et tirez-la doucement vers le bas pour découvrir les muscles. Ensuite, insérez les pinces entre les muscles sous l’articulation du genou et collectez les LN poplités. Placez les LN poplités dans le même tube contenant des LN inguinaux et iliaques.
NOTE: Voir la note après 3.1.7. - Placer la rate dans une passoire cellulaire de 70 μm dans une boîte de culture de 60 mm remplie de 5 mL de milieu complet. À l’aide d’un piston de seringue de 5 mL, écraser doucement l’organe jusqu’à sa désagrégation complète.
- Retirez la crépine et transférez la suspension cellulaire dans un tube propre de 15 mL.
- Ajouter 5 mL de milieu complet à la cuve de culture et laver soigneusement la vaisselle et la passoire pour s’assurer que toutes les cellules ont été récupérées. Piscine avec le reste de la suspension de la cellule de rate dans le tube de 15 mL.
- Pour les LN inguinaux, iliaques et poplités regroupés, préparer une suspension unicellulaire en suivant une procédure similaire à celle utilisée aux étapes 3.1.9 à 3.1.11 pour la rate.
- Cellules centrifuges à 400 × g pendant 10 min à 4 °C. Jetez le surnageant et remettez en suspension les pastilles de cellules dans PBS.
- Comptez les cellules avec une chambre de Neubauer en utilisant un tampon de lyse des globules rouges et du bleu de trypan à 0,04% v / v dans le PBS.
- Isolement des cellules BM
- Placez le milieu complet de 5 mL dans un tube de 15 mL et conservez-le sur la glace, prêt pour la collecte des pattes postérieures.
- Sacrifier une souris adulte par luxation cervicale.
- Stériliser la surface de la peau avec 70% v/v d’éthanol.
- Faites une incision transversale d’environ 1 cm sur la peau ventrale avec des ciseaux, saisissez fermement la peau des deux côtés de la coupe et tirez doucement vers le bas pour découvrir les muscles des pattes postérieures.
- Pour éliminer la peau de l’arrière des pattes postérieures, maintenez la souris en position couchée, placez la pince sous le genou et tirez vers le haut pour exposer les muscles.
- Coupez les os aux deux extrémités d’une patte arrière: l’articulation pelvienne / hanche et la cheville.
- Transférer les deux pattes postérieures dans le tube de 15 ml préparé à l’étape 3.2.1. Gardez le tube sur la glace.
- Prenez les pattes postérieures du tube de 15 ml et transférez-les sur du papier de soie. Coupez les pattes postérieures juste en dessous de l’articulation du genou pour enlever le tibia. Disséquez le fémur et le tibia des muscles environnants, enlevez l’excès de tissu à l’aide de ciseaux et mouillez le papier de soie.
- Coupez les extrémités osseuses avec des ciseaux pour exposer la tige de moelle interne. Insérer le tibia et le fémur dans le tube d’extraction BM (voir préparation au 3.2.9.1-3.2.9.218), avec l’extrémité la plus large en bas.
- Coupez une pointe de pipette de 200 μL à la ligne juste au-dessus de l’extrémité de la pointe et à la ligne de 100 μL.
- Placez la partie centrale dans la partie supérieure, plus grande de la pointe, et placez-la dans un tube de microfuge de 1,5 mL.
- Faire tourner le tube d’extraction BM à 800 × g pendant 1 min.
- Jeter l’os et remettre vigoureusement la pastille dans 1 mL de milieu complet pour éliminer les grappes. Filtrer la suspension cellulaire à travers un filtre de 70 μm placé sur le dessus d’un tube de 15 mL.
- Lavez le tube d’extraction BM deux fois avec 1 mL de milieu complet à chaque fois. Filtrer à travers un filtre de 70 μm et regrouper le volume avec le reste de la suspension cellulaire obtenue à l’étape 3.2.11.
REMARQUE: Un seul tube de 15 mL contiendra des cellules des deux pattes postérieures d’une souris. - Cellules centrifuges à 400 × g pendant 10 min à 4 °C. Jetez le surnageant et remettez en suspension la pastille de cellule dans PBS.
- Comptez les cellules avec une chambre de Neubauer en utilisant un tampon de lyse des globules rouges et du bleu de trypan à 0,04% v / v dans le PBS.
4. Coloration des cellules de la rate, de la LN et de la BM
- Diviser les échantillons cellulaires à colorier en 3 sous-groupes : les échantillons cellulaires pour compensation,y compris les cellules BM de souris non traitées à teindre uniquement avec Hoechst 33342 (ci-après dénommé Hoechst) et les cellules de rate de souris non traitées à utiliser pour préparer un mélange de cellules mortes/vivantes pour la compensation de colorants de cellules mortes ; contrôle positif pour l’analysedu cycle cellulaire , consistant en un échantillon de BM provenant de souris non traitées; et des échantillons expérimentaux contenant des échantillons de rate et de LN provenant de souris non traitées et vaccinées.
REMARQUE: S’assurer qu’il y a suffisamment de cellules de rate et de LN pour l’analyse d’un nombre suffisant de lymphocytes T CD8 spécifiques au bâillon. Il est souvent nécessaire d’utiliser des cellules de rate regroupées et des cellules LN groupées de 3 souris vaccinées et de colorer deux ou plusieurs échantillons identiques de cellules regroupées, chacune contenant 3 × 106 cellules. Fusionner des échantillons identiques à l’étape de la coloration Hoechst. De même, les cellules de rate et les cellules LN de 3 souris non traitées se sont regroupées et fusionnent des échantillons identiques à la fin. Réserver un échantillon non coloré de cellules de rate provenant d’une souris non traitée à utiliser pour la configuration de l’instrument et de la compensation. - Préparez un mélange de cellules mortes / vivantes pour la compensation des colorants de cellules mortes (ce mélange de cellules ne sera coloré qu’avec le colorant de cellules mortes).
- Chauffer un bain-marie à 65 °C.
- Prenez une aliquote de cellules de la rate (~3 × 106).
- Transférer la suspension cellulaire dans un tube de microfuge, la placer au bain-marie à 65 °C pendant 5 min, puis la placer immédiatement sur de la glace pendant 10 min.
- Mélanger les cellules tuées par la chaleur avec des cellules vivantes de la rate (~3 ×10 6) dans un rapport de 1:1, et transférer la moitié du mélange dans une plaque inférieure bien ronde de 96 (~3 × 106 cellules / puits pour le contrôle de la coloration des cellules mortes).
- Coloration des cellules mortes des échantillons expérimentaux, contrôle positif pour l’analyse du cycle cellulaire et mélange de cellules mortes /vivantes
- Transférer la rate, la LN, les cellules BM (3 × 106 cellules/puits) et le mélange de cellules mortes/vivantes (section 4.2) dans une plaque à fond rond de 96 puits, selon le schéma de coloration (étape 4.1), et centrifuger à 400 × g pendant 3 min à 4 °C.
- Remettre en suspension chaque pastille de cellule dans 50 μL de colorant de cellules mortes dilué dans du PBS, et remettre en suspension en pipetant de haut en bas 3 fois immédiatement.
- Incuber pendant 30 min à 4 °C, à l’abri de la lumière.
- Laver les cellules 2 fois avec un tampon de coloration; la première fois avec 200 μL et la deuxième fois avec 250 μL. Pour chaque centrifugeuse de lavage, la plaque à 400 × g pendant 3 min à 4 °C.
- Jetez le surnageant et remettez en suspension la pastille de cellule dans 20 μL de PBS.
- Coloration cellulaire membranaire avec complexe majeur d’histocompatibilité (CMH)-multimères peptidiques et mAbs.
- En tenant compte des volumes nécessaires selon le schéma de coloration (réglages du cytomètre en flux, tableau 1),préparez les réactifs suivants:
- Diluer mAb 2.4G2 dans le tampon de coloration en fonction de la dilution appropriée (voir tableau des matériaux); pour chaque échantillon à colorier, utiliser 10 μL de cette dilution.
REMARQUE: 2,4G2 mAb bloque la liaison non spécifique de l’antigène des immunoglobulines aux récepteurs FcγIII et FcγII. - Diluer le tétramère marqué À l’allophycocyanine (Tetr-gag) H-2k(d) AMQMLKETI (APC) dans le tampon de coloration pour obtenir la dilution appropriée (voir tableau des matériaux); pour chaque échantillon à colorer, utiliser 20 μL de cette dilution.
- Préparer le mélange d’anticorps en diluant les mAbs dans le tampon de coloration en fonction de la dilution appropriée (voir tableau des matériaux)qui a été préalablement déterminée lors d’expériences de titrage; pour chaque échantillon à colorier, utiliser 20 μL de ce mélange d’anticorps.
REMARQUE: Ici, la protéine chlorophylle de périditonine anti-CD3e (PerCP-Cy5.5) (clone 145-2C11), l’ultraviolet brillant anti-CD8a (BUV805) (clone 53-6.7) et la cyanine phycoérythrine anti-CD62L (PECy7) (clone MEL-14) ont été utilisés.
- Diluer mAb 2.4G2 dans le tampon de coloration en fonction de la dilution appropriée (voir tableau des matériaux); pour chaque échantillon à colorier, utiliser 10 μL de cette dilution.
- Ajouter 10 μL du 2,4G2 mAb préalablement dilué (étape 4.4.1.1), et incuber pendant 10 min à 4 °C, à l’abri de la lumière.
- Ajouter 20 μL de l’APC Tetr-gag précédemment dilué (étape 4.4.1.2) et 10 μL de pentamère H-2k(d) AMQMLKETI phycoérythrine (PE) (pent-gag). Incuber pendant 15 min à 4 °C, à l’abri de la lumière.
- Ajouter 20 μL du mélange d’anticorps préalablement préparé (étape 4.4.1.3) et incuber 15 min à 4 °C, à l’abri de la lumière.
REMARQUE : Par conséquent, le volume final est de 80 μL par puits (étape 4.3.5, étapes 4.4.2 à 4.4.4). - Lavez les cellules avec 200 μL de tampon de coloration. Centrifuger à 400 × g pendant 5 min à 4 °C.
- Remettez en suspension la pastille de cellule dans 250 μL de tampon de coloration et transférez la suspension cellulaire dans des tubes de 5 mL. Ajouter 1 mL de tampon de coloration dans le tube et centrifuger à 400 × g pendant 5 min à 4 °C.
- Prélever l’aliquote des cellules BM (3 × 106 cellules) (voir la liste des échantillons de cellules, rubrique 4.1) à utiliser pour compenser le canal hoechst (Hoechst 33342 est excité par un laser ultraviolet (réglages du cytomètre en flux (tableau 2)),et transférer la suspension cellulaire dans un tube de 5 mL. Ajouter 1 mL de tampon de coloration au tube et centrifuger 400 × g pendant 5 min à 4 °C.
- En tenant compte des volumes nécessaires selon le schéma de coloration (réglages du cytomètre en flux, tableau 1),préparez les réactifs suivants:
5. Fixation/perméabilisation
- Préparer un tampon de fixation/perméabilisation frais en diluant 1 partie de concentré de fixation/perméabilisation avec 3 parties de diluant de fixation/perméabilisation, conformément aux instructions du fabricant.
- Jetez le surnageant et le vortex d’impulsions des échantillons pour désagréger complètement la pastille.
- Ajouter 1 mL du tampon de fixation/perméabilisation fraîchement préparé à chaque tube, y compris un tube avec des cellules de rate non colorées (3 x 106, voir la liste des échantillons de cellules, rubrique 4.1), et un vortex.
- Incuber pendant 16 h à 4 °C.
REMARQUE: Le protocole peut être mis en pause ici.
6. Coloration intracellulaire
- Coloration Ki67
- Préparer un tampon de perméabilisation frais 1x en diluant le tampon de perméabilisation 10x avec de l’eau distillée, conformément aux instructions du fabricant. Avant utilisation, le tampon de perméabilisation 1x doit être filtré à travers un filtre de 0,45 μm pour éliminer les agrégats.
- Diluer l’isothiocyanate de fluorescéine mAb Ki67 (FITC) (clone SolA15) dans un tampon de perméabilisation 1x (voir Tableau des matériaux),tel que déterminé précédemment dans des expériences de titrage (volume final de 100 μL par échantillon).
- Ajouter 3 mL de tampon de perméabilisation 1x à chaque tube, et centrifuger à 400 × g pendant 5 min à température ambiante (RT).
- Jetez le surnageant et répétez l’étape 6.1.3.
- Jeter le surnageant et remettre en suspension la pastille de cellule dans 100 μL de mAb Ki67 FITC préalablement dilué (étape 6.1.2).
- Incuber pendant 30 min à RT, à l’abri de la lumière.
- Lavez les cellules 2 fois avec 4 mL de tampon de perméabilisation 1x. Pour chaque centrifugeuse de lavage à 400 × g pendant 5 min à RT.
- Remettre en suspension la pastille cellulaire dans le PBS en tenant compte des volumes suivants : 350 μL de PBS pour les échantillons à acquérir directement au cytomètre en flux ; 250 μL de PBS pour les échantillons à incuber avec Hoechst peu avant la cytométrie en flux (rubrique 6.2).
- Coloration de l’ADN
- Ajouter 250 μL de 4 μg/mL de Hoechst dans du PBS à chaque échantillon (la concentration finale de Hoechst est de 2 μg/mL).
REMARQUE: Dans le cas où deux ou plusieurs échantillons identiques de 250 μL dans pbS ont été préparés, les fusionner à cette étape et ajouter un volume égal de 4 μg / mL de solution de Hoechst dans PBS (la concentration finale de Hoechst est de 2 μg / mL). Le nombre de cellules influence grandement l’étape de coloration de l’ADN. Utilisez le même numéro de cellule dans chaque échantillon. Sachez que même un nombre de cellules légèrement réduit (par exemple, en raison de la perte de cellules lors des étapes de lavage précédentes) entraîne une liaison Hoechst plus élevée à l’ADN et une intensité Hoechst plus élevée. - Incuber pendant 15 min à RT, à l’abri de la lumière.
- Centrifuger les échantillons à 400 × g pendant 5 min à RT.
- Remettre en suspension la pastille de cellule dans 350 μL de PBS.
- Ajouter 250 μL de 4 μg/mL de Hoechst dans du PBS à chaque échantillon (la concentration finale de Hoechst est de 2 μg/mL).
7. Préparation des échantillons de perles de compensation
- Préparer 5 μL de l’anticorps en diluant correctement le mAb dans le tampon de coloration.
REMARQUE: Pour chaque mAb conjugué au fluorochrome utilisé dans l’expérience, préparer son échantillon de billes de compensation correspondant. - Vortex Negative Control et Anti-Rat/Hamster Ig,κ Comp Perles avant utilisation.
- Pour chaque échantillon, introduisez une goutte (~20 μL) de CompBeads de contrôle négatif et une goutte de CompBeads Ig,k Anti-Rat/Hamster.
- Ajouter 5 μL de l’anticorps prédilué (étape 7.1) au tube et pipeter de haut en bas.
- Incuber pendant 15 min à 4 °C, à l’abri de la lumière.
- Laver les échantillons avec 2 mL de tampon de coloration. Centrifuger à 400 × g pendant 5 min à 4 °C.
- Jetez le surnageant et remettez en suspension la pastille en ajoutant 500 μL de PBS à chaque tube et vortex.
8. Configuration de l’instrument et de la compensation et acquisition expérimentale d’échantillons au cytomètre en flux
REMARQUE : Reportez-vous aux paramètres du cytomètre en flux (Tableau 2) pour la configuration du cytomètre.
- Configuration générale de l’instrument et de la compensation
- Ouvrez le logiciel pour l’acquisition d’échantillons (voir Tableau des matériaux)et créez une nouvelle expérience en cliquant sur Nouvelle expérience dans la section du ruban de l’espace de travail et en sélectionnant Nouvelle expérience vide.
- Double-cliquez sur l’expérience créée pour l’ouvrir.
- Dans la fenêtre Paramètres du cytomètre, cliquez sur Paramètres et sélectionnez tous les canaux (par exemple, PE, APC, etc.) utilisés dans le panneau de coloration, y compris les paramètres de diffusion directe (FSC) et de dispersion latérale (SSC).
- Sélectionnez l’échelle linéaire comme paramètre Hoechst en décochant l’échelle logarithmique et vérifiez la largeur (W) de l’impulsion de tension pour FCS, SSC et Hoechst.
REMARQUE : Tous les paramètres sont affichés par défaut à l’échelle logarithmique (log), à l’exception de FSC et SSC qui sont à l’échelle linéaire. Tous les paramètres sont analysés par la zone (A) et la hauteur (H) de l’impulsion de tension. - Dans la feuille de calcul globale, créez un diagramme à points avec FSC-A sur l’axe des x et SSC-A sur l’axe des y.
- Exécutez l’exemple de rate non colorée en cliquant sur Acquérir des données dans le tableau de bord d’acquisition.
- Définissez les paramètres FSC et SSC appropriés pour visualiser les cellules en modifiant les valeurs de tension dans la section Paramètres et créez une porte pour sélectionner toutes les cellules affichées dans le diagramme à points FSC-A/SSC-A en cliquant sur Polygon Gate dans la barre d’outils de l’espace de travail de la feuille de calculglobale .
- Affichez les cellules fermées dans des histogrammes avec chaque paramètre de fluorescence sur l’axe des x.
- Exécutez des échantillons de rate non colorés et entièrement colorés pour ajuster le détecteur de fluorescence (PMT) afin d’avoir une séparation claire entre les signaux négatifs et positifs des cellules colorées pour chaque paramètre de fluorescence.
- Pour effectuer la configuration de la compensation, cliquez sur Expérimenter dans le ruban de l’espace de travail et sous la section Configuration de la compensation, sélectionnez Créer des contrôles de compensation. Décochez inclure le tube de contrôle non coloré / puits et cliquez sur OK.
REMARQUE : Cette opération entraînera la création d’un spécimen nommé Contrôles de compensation et d’une feuille de calcul normale contenant plusieurs feuilles correspondant à chaque paramètre sélectionné. - Exécuter un échantillon de perles de compensation (voir la section 7); définissez les paramètres FSC et SSC appropriés pour visualiser les billes en modifiant les valeurs de tension et le seuil d’acquisition de 5 000 sur les paramètres FSC dans la fenêtre Cytomètre.
- Ajustez la porte P1 sur la population de billes et vérifiez que les pics positifs et négatifs sont tous deux visibles sur l’axe des x. Répétez cette opération pour chaque échantillon de perles de compensation, puis enregistrez chaque fichier d’échantillon en cliquant sur Données d’enregistrement dans le tableau de bord d’acquisition (enregistrez au moins 5 000 événements pour chaque échantillon).
- Pour chaque échantillon de billes enregistré, placez les portes P2 et P3 sur les pics positifs et négatifs, respectivement.
- Exécutez les échantillons de cellules pour la compensation (voir les étapes 4.2 et 4.4.7 et les sections 5 et 6). Modifiez les tensions FSC et SSC et la valeur de seuil pour visualiser les cellules, ajuster la porte P1 et enfin enregistrer chaque fichier d’échantillon (enregistrer au moins 10 000 événements). Réglez les portes P2 et P3 sur les pics positifs et négatifs, respectivement.
REMARQUE: Pour la compensation du canal de Hoechst, utilisez le G0/ G1 comme pic négatif (P3) et le G2/ M comme positif (P2). - Cliquez sur Expérimenter dans la section du ruban de l’espace de travail et dans la section Configuration de la compensation, sélectionnez Calculer la compensation.
- Nommez le paramètre de compensation créé, liez-le et enregistrez-le dans l’expérience en cours.
- Acquisition expérimentale d’échantillons
- Ouvrez un spécimen en cliquant sur Nouveau spécimen dans la barre d’outils du navigateur et créez la stratégie de contrôle dans la feuille de calcul globale.
REMARQUE : La stratégie de contrôle de l’acquisition d’échantillons est semblable à celle de l’analyse d’échantillons, décrite à la figure 3 et à la section 9. - Afficher toute la population d’événements dans un histogramme avec CD3-A sur l’axe des x. Créez une porte d’intervalle pour sélectionner uniquement les cellulesCD3 +.
- Dans le tableau de bord d’acquisition, sélectionnez La porte de stockage comme Tous les événements pour les échantillons LN et Tous les événements ou les cellules CD3+ pour les échantillons de rate.
- Exécutez les échantillons expérimentaux à basse vitesse et enregistrez enfin tous les fichiers en vous assurant de collecter au moins 100 à 200 lymphocytes T CD8 spécifiques de l’antigène pour chaque échantillon des souris vaccinées.
REMARQUE: La taille du fichier des échantillons expérimentaux est généralement grande (30-120 Mo), en particulier lorsque la fréquence des lymphocytes T CD8 spécifiques à l’antigène est faible. Par conséquent, un nombre élevé d’événements (> 1 × 106) doivent être collectés pour enregistrer au moins 100 à 200 lymphocytes T CD8 spécifiques de l’antigène. Les fichiers volumineux peuvent ralentir le processus d’analyse des données ultérieur. L’acquisition de cellules CD3+ uniquement dans des échantillons de rate (voir l’étape 8.2.2 ci-dessus) est utile pour réduire la taille du fichier. - Exécutez et enregistrez le témoin positif pour l’analyse du cycle cellulaire, c’est-à-dire l’échantillon de BM de souris non traitées.
- Ouvrez un spécimen en cliquant sur Nouveau spécimen dans la barre d’outils du navigateur et créez la stratégie de contrôle dans la feuille de calcul globale.
9. Analyse des données
- Ouvrez le logiciel (voir Tableau des matériaux)et créez différents groupes correspondant aux différents organes à analyser en cliquant sur Créer un groupe dans la section du ruban de l’espace de travail (c’est-à-dire créer un groupe « a-LNs »; « b-rate »; « c-BM »).
REMARQUE: Les groupes nouvellement créés apparaîtront dans la liste des groupes, tandis que le groupe « Compensation » est automatiquement généré par le logiciel. - Ouvrez la fenêtre Modifier le groupe en double-cliquant sur le nom du groupe et vérifiez que les groupes nouvellement créés sont synchronisés. Si ce n’est pas le cas, cochez la fonction Synchronisée.
- Faites glisser chaque fichier .fcs dans son groupe correspondant.
- Créez la stratégie de contrôle en commençant par le groupe « a-LNs ».
- Double-cliquez sur l’échantillon entièrement coloré dans le groupe pour ouvrir la fenêtre graphique; Les axes x et y sont étiquetés comme dans les fichiers fcs (voir paramètres du cytomètre en flux, tableau 2).
- Affichez le nombre total d’événements acquis pour cet échantillon dans un diagramme à points avec l’ADN-A sur l’axe des x et l’ADN-W sur l’axe des y.
- Sélectionnez uniquement la population d’une seule cellule en cliquant sur Rectangle dans la section de l’outil de contrôle de la fenêtre graphique.
REMARQUE: Les cellules individuelles ont des valeurs d’ADN-A comme suit: 2N (faible): entre 2N et 4N (intermédiaire), ou égales à 4N (élevées), tandis que les valeurs d’ADN-W sont identiques pour toutes (étape 1 de la figure 3). - Double-cliquez au centre de la porte rectangulaire pour afficher des cellules simples dans un diagramme à points avec le paramètre FSC-A sur l’axe des x et le colorant de cellules mortes sur l’axe des y.
- Sélectionnez uniquement la population de cellules vivantes en cliquant sur Polygone dans la section de l’outil de contrôle de la fenêtre graphique. Les cellules vivantes sont négatives pour le colorant cellulaire mort (étape 2 de la figure 3).
- Double-cliquez au centre de la porte polygonale pour afficher les cellules d’un diagramme à points avec le paramètre FSC-A sur l’axe des x et le paramètre SSC-A sur l’axe des y.
- Cliquez sur Rectangle, et créez une porte « détendue » pour inclure toutes les cellules vivantes uniques dans ce graphique12 (étape 3 de la figure 3).
- Double-cliquez au centre de la porte « détendue » pour afficher les cellules dans un diagramme à points avec CD3 sur l’axe des x et CD8 sur l’axe des y.
- Sélectionnez les cellules CD3+CD8+ en cliquant sur Polygone (étape 4 de la figure 3).
- Double-cliquez au centre de la porte CD3+CD8+ pour afficher les cellules dans un diagramme à points avec Tetr-gag sur l’axe des x et Pent-gag sur l’axe des y.
- Sélectionnez les lymphocytes T CD8 spécifiques de l’antigène (positifs pour Tetr-gag et Pent-gag) en cliquant sur Polygone (étape 5 de la figure 3).
- Double-cliquez au centre de la porte spécifique au bâillon pour afficher les cellules dans un diagramme à points avec l’ADN-A sur l’axe des x et Ki67 sur l’axe des y(Figure 4).
- Sélectionnez les cellules dans les différentes phases du cycle cellulaire en cliquant sur Quad dans la section de l’outil de contrôle de la fenêtre graphique.
REMARQUE: Les cellules en phase G0 sont des cellules basses à ADN Ki67neg (quadrant inférieur gauche); les cellules de G1 sont Ki67pos-ADN faible (quadrant supérieur gauche); les cellules de S-G2/M sont Ki67pos-DNA intermédiaire/élevé (quadrant supérieur droit) (Figure 4). - Copiez la stratégie de contrôle créée dans un exemple dans le groupe correspondant pour appliquer les portes à tous les échantillons du groupe.
- Répétez les étapes 9.5 à 9.18 pour le « groupe a-LN ».
- Vérifiez que toutes les portes sont appropriées pour chaque échantillon du groupe « b-rate ». Pour analyser le cycle cellulaire parmi les cellules BM (contrôle positif), cliquez au centre de la porte « détendue » pour afficher les cellules dans un diagramme à points avec l’ADN-A sur l’axe des x et Ki67 sur l’axe des y.
- Vérifiez que toutes les portes sont appropriées pour chaque échantillon des 3 groupes (c.-à-d. pour les cellules de la rate, LN et BM).
REMARQUE: La porte de population unicellulaire (étape 9.7) et la porte quadruple pour le cycle cellulaire (étape 9.17) peuvent avoir des coordonnées de porte différentes dans différents échantillons, principalement en raison des légères différences possibles de l’intensité du colorant Hoechst entre les échantillons (section 6.2). Pour cette raison, il peut être nécessaire de modifier la porte de population unicellulaire et les portes Quad pour le cycle cellulaire dans chaque échantillon. Cela se fera comme suit: double-cliquez sur le nom du groupe et supprimez la synchronisation des propriétés du groupe. Cette opération permet de modifier les portes dans un échantillon sans modifier les mêmes portes dans tous les autres échantillons du groupe. Après la suppression de la synchronisation, modifiez les portes si nécessaire. - Pour visualiser les résultats obtenus par cette analyse, cliquez sur Éditeur de mise en page dans la section du ruban de l’espace de travail pour l’ouvrir. Faites glisser chaque porte de la stratégie de contrôle dans le volet d’exemple vers l’éditeur de mise en page et placez les tracés en fonction de la séquence de la stratégie de contrôle. Si nécessaire, modifiez le type de graphique en double-cliquant sur le tracé correspondant dans la mise en page et en sélectionnant le type approprié dans la fenêtre Définition du graphique.
- Cliquez sur le groupe et itérez par fonctions sur le ruban de mise en page pour visualiser les résultats obtenus dans chaque organe, et comparer différents échantillons.
Representative Results
Les phases du cycle cellulaire des cellules de la rate, des LN et des BM de souris Balb/c ont été analysées à l’aide du colorant d’ADN fluorescent, Hoechst, et d’un anti-Ki67 mAb, selon le protocole résumé à la figure 1. Cette coloration a permis la différenciation des cellules dans les phases suivantes du cycle cellulaire : G0 (Ki67neg, avec 2N d’ADN défini comme DNAlow), G1 (Ki67pos, DNAlow), et S-G2/M (Ki67pos, avec une teneur en ADN comprise entre 2N et 4N, ou égale à 4N d’ADN définie comme DNAintermediate/high).
Nous avons d’abord effectué une analyse du cycle cellulaire des cellules BM pour reproduire les résultatsprécédemment publiés 13,14, puis analysé les cellules d’intérêt, c’est-à-dire les cellules T CD8. La figure 2 montre un exemple typique d’analyse du cycle cellulaire des cellules BM (Figure 2A). Le protocole a donné un faible coefficient de variation (CV) des pics d’ADN G0/ G1 et G2/ M, indiquant l’excellente qualité de la coloration de l’ADN (Figure 2B, montrant un exemple avec CV < 2.5; CV a toujours été < 5 dans toutes les expériences).
Nous avons ensuite appliqué le même protocole aux lymphocytes T CD8 spécifiques de l’antigène provenant de souris vaccinées. Les souris BALB/c ont été vaccinées contre le bâillon antigénique du VIH-1 en utilisant Chad3-gag pour l’amorçage et MVA-gag pour le renforcement, tous deux conçus pour transporter le bâillon VIH-1. Au jour (d) 3 après le boost, nous avons analysé la fréquence des lymphocytes T CD8 spécifiques du bâillon provenant de la rate et des LN drainants. Nous avons tiré parti de la stratégie de contrôle nouvellement définie pour les lymphocytes T dans la phase précoce de la réponse immunitaire, qui, contrairement à la stratégie conventionnelle, est appropriée pour détecter les lymphocytes T CD8 hautement activés répondant à l’antigène12. Nous avons exécuté la nouvelle stratégie en cinq étapes suivantes. À l’étape 1, nous avons exclu les doublets ou les agrégats par porte ADN-A/-W, et à l’étape 2, nous avons identifié les cellules vivantes par exclusion des marqueurs de cellules mortes. À l’étape 3, nous avons identifié la population d’intérêt à l’aide d’une porte FSC-A/SSC-A « détendue » non conventionnelle(figure 3A)au lieu de la porte lymphocytaire étroite canonique12. Après avoir râpé sur des cellules CD3+CD8+ (étape 4 de la figure 3A), nous avons identifié des lymphocytes T CD8 spécifiques au bâillon en utilisant deux multimères MHC différents, à savoir Pent-gag et Tetr-gag (étape 5 de la figure 3A). Nous avons utilisé deux multimères au lieu d’un pour améliorer la sensibilité de la détection des lymphocytes T CD8 spécifiques au bâillon chez les souris vaccinées, sans augmenter le fond de coloration chez les souris non traitées(Figure 3B et C,étape 5). Ainsi, nous avons réussi à distinguer des souris non traitées (0,00 % et 0,00 % de lymphocytes T CD8 spécifiques de l’antigène dans les LN et la rate, respectivement) des souris vaccinées (0,46 % et 0,29 % de lymphocytes T CD8 spécifiques de l’antigène dans les LN et la rate, respectivement, Figure 3B et C).
Notamment, le protocole nous a permis d’avoir un fond extrêmement faible dans la porte des lymphocytes T CD8 spécifiques de l’antigène des LN et de la rate des souris non traitées (généralement 0,00% et au maximum 0,02%). La comparaison des diagrammes FSC-A / SSC-A spécifiques au bâillon et non spécifiques au bâillon a montré que les cellules spécifiques au bâillon avaient un taux élevé de SSC-A et de FSC-A(figure 3D),confirmant la nécessité d’utiliser une porte FSC-A / SSC-A « détendue » pour capturer ces cellules. Nous avons ensuite évalué les pourcentages de lymphocytes T CD8 spécifiques au bâillon dans différentes phases du cycle cellulaire(Figure 4A). Nous avons constaté que les lymphocytes T CD8 spécifiques du bâillon dans la rate et encore plus dans les LN drainants contenaient une forte proportion de cellules dans les phases S-G2/ M au jour 3 après le boost (18,60% et 33,52%, respectivement).
De plus, nous avons constaté que les lymphocytes T CD8 spécifiques au bâillon dans les phases S-G2/M présentaient un TAUX ÉLEVÉ de FSC-A et de SSC-A, lorsqu’ils étaient superposés sur le total des lymphocytes T CD8 du même organe(Figure 4B). L’expression de CD62L par les lymphocytes T CD8 spécifiques au bâillon était faible, comme prévu pour les lymphocytes T activés, à l’exception de quelques cellules dans G0 dans les LN(Figure 4C). Dans l’ensemble, ces résultats ont confirmé que la porte « détendue » (étape 3 des figures 3A, B et C)devait inclure tous les lymphocytes T CD8 spécifiques de l’antigène proliférant12. Le protocole a été extrêmement utile pour une évaluation « instantanée » des phases du cycle cellulaire des lymphocytes T CD8 spécifiques à l’antigène au moment de l’analyse et de l’expression de CD62L par les cellules dans différentes phases du cycle cellulaire.

Figure 1: Schéma du protocole d’analyse du cycle cellulaire des lymphocytes T CD8 spécifiques de l’antigène. Veuillez cliquer ici pour voir une version agrandie de cette figure.

Figure 2: Analyse du cycle cellulaire des cellules BM. Les cellules BM de souris Balb/c non traitées ont été colorées et analysées par cytométrie en flux. (A) Exemple de stratégie de contrôle. Nous avons fermé sur des cellules individuelles dans le diagramme DNA-A / -W (à gauche) et par la suite sur des cellules vivantes par exclusion de colorant de cellules mortes (au milieu). Ensuite, une porte FSC-A/SSC-A « détendue » a été utilisée pour toutes les cellules BM (à droite). (B) Exemple d’analyse du cycle cellulaire des cellules BM (à gauche). Nous avons utilisé une combinaison de Ki67 et de coloration de l’ADN pour identifier les cellules dans les phases suivantes du cycle cellulaire : G0 (quadrant inférieur gauche, cellules Ki67neg-DNAlow), G1 (quadrant supérieur gauche, Ki67pos-DNAlow), S-G2/M (quadrant supérieur droit, Ki67pos-DNAintermediate/high). Le contrôle de la fluorescence moins un (FMO) du Ki67 mAb (au milieu) et de l’histogramme de l’ADN (à droite) est montré. Dans le diagramme d’histogramme d’ADN, les portes gauche et droite correspondent respectivement au pic d’ADN G0/ G1 et au pic d’ADN G2/ M, et les nombres représentent les coefficients de variation (CV) de chaque pic. Dans tous les autres graphiques, les nombres représentent les pourcentages de cellules dans les portes indiquées. La figure montre 1 expérience représentative sur 5. Dans chaque expérience, nous avons analysé des cellules BM regroupées de 3 souris. Veuillez cliquer ici pour voir une version agrandie de cette figure.

Figure 3: Analyse des lymphocytes T CD8 spécifiques de l’antigène à partir de LN et de rate. Les souris Balb/c ont été amorcées par voie intramusculaire (i.m.) avec Chad3-gag et boostées i.m. avec MVA-gag. Au jour 3 après le boost, les cellules LN et rate drainantes de souris témoins vaccinées et non traitées ont été colorées et analysées par cytométrie en flux. (A) Schéma de la stratégie de contrôle en cinq étapes pour identifier les cellules individuelles (étape 1); cellules vivantes (étape 2); lymphocytes (étape 3); lymphocytes T CD8 (étape 4); et les cellules spécifiques au bâillon (étape 5). (B-C) Exemple de graphiques : analyse de cellules de LN (B) et de rate (C) de souris non traitées (en haut) et vaccinées (en bas). Nous avons identifié des cellules uniques sur le diagramme ADN-A/-W à l’étape 1. Ensuite, à l’étape 2, nous avons sélectionné des cellules vivantes par exclusion de colorant de cellules mortes. À l’étape 3, nous avons utilisé une porte « détendue » non canonique pour les lymphocytes. À l’étape 4, nous avons identifié les lymphocytes T CD8 par leur double expression de CD3 et CD8. Nous avons ensuite identifié des cellules spécifiques au bâillon et non spécifiques au bâillon à l’étape 5, en fonction de leur capacité à lier le pentamère H-2kd-gag-Pentamer (Pent-gag) et le tétramère H-2kd-gag marqué par fluorochrome (Tetr-gag), ou non, respectivement. D) Profils FSC-A/SSC-A de cellules spécifiques au bâillon (bleu) et non spécifiques au bâillon (gris) après le contrôle décrit ci-dessus. Les nombres représentent les pourcentages de cellules dans les portes indiquées. La figure montre 1 expérience représentative sur 5. Dans chaque expérience, nous avons analysé la rate groupée et les cellules LN groupées de 3 souris vaccinées et de 3 souris non traitées. Veuillez cliquer ici pour voir une version agrandie de cette figure.

Figure 4: Analyse du cycle cellulaire des lymphocytes T CD8 spécifiques de l’antigène. Les souris ont été vaccinées comme sur la figure 3 et l’analyse du cycle cellulaire des cellules spécifiques du bâillon a été effectuée au jour 3 après la relance, après avoir été contrôlée en 5 étapes comme dans la figure 3. (A)Exemple d’analyse du cycle cellulaire des lymphocytes T CD8 spécifiques du bâillon provenant de LN (en haut) et de rate (en bas) de souris vaccinées. Les phases du cycle cellulaire ont été identifiées comme à la figure 2B. Les panneaux représentent les cellules en G0, en G1, et en S-G2/ M (à gauche) et fluorescence moins un (FMO) contrôle de Ki67 mAb (à droite). Les nombres représentent les pourcentages de cellules dans les portes indiquées. (B) Diagrammes à points FSC-A/SSC-A montrant des lymphocytes T CD8 spécifiques au bâillon en phases S-G2/M(en rouge) superposés sur le total des lymphocytes T CD3+CD8+ (en gris) provenant des LN (en haut) et de la rate (en bas) de souris vaccinées. (C) Histogrammes décalés montrant l’expression de CD62L par les lymphocytes T CD8 spécifiques du bâillon dans G0 (vert), dans G1 (bleu) et dans S-G2/M (rouge) à partir de LN (en haut) et de rate (en bas) de souris vaccinées. Les axes y indiquent le nombre normalisé d’événements. La figure montre 1 exemple représentatif sur 5 expériences indépendantes avec un total de 15 souris. Veuillez cliquer ici pour voir une version agrandie de cette figure.
Matériel supplémentaire: Réglages du cytomètre en flux. Veuillez cliquer ici pour télécharger ce fichier.
Discussion
Bien que l’expansion clonale des lymphocytes T ait été intensivement étudiée, certains aspects restent inconnus, principalement parce que les outils disponibles pour l’étudier sont peu nombreux et ont leurs propres inconvénients. De ce point de vue, nous avons mis en place une méthode cytométrique en flux très sensible pour analyser le cycle cellulaire des lymphocytes T CD8 spécifiques de l’antigène à un stade précoce après la vaccination dans un modèle murin. Le protocole est basé sur une combinaison de Ki67 et de coloration de l’ADN, qui était précédemment utilisée pour analyser le cycle cellulaire des cellules hématopoïétiques BM chez les souris13,14. Pour adapter le protocole aux lymphocytes T CD8 spécifiques à l’antigène, nous avons dû tenir compte de quelques questions critiques, notamment le choix du colorant ADN, les conditions appropriées pour obtenir une coloration comparable de l’ADN sur différents échantillons et la stratégie de contrôle pour l’analyse des données.
De nombreux colorants sont disponibles pour la coloration de l’ADN, y compris l’iodure de propidium et la 7-aminoactinomycine D; nous avons choisi Hoechst parce qu’il était compatible avec la coloration membranaire et le protocole de fixation / perméabilisation légère requis pour la coloration Ki67. Dans le même temps, la coloration avec Hoechst nous a permis d’obtenir un histogramme d’ADN d’excellente qualité, c’est-à-dire que les pics d’ADN G0/ G1 et G2/ M avaient un coefficient de variation (CV) beaucoup plus faible que les pics d’ADN généralement obtenus avec d’autres colorants ADN, par exemple, DRAQ519. En effet, Hoechst peut colorer l’ADN même dans les cellules vivantes20.
Certaines stratégies ont été utilisées pour éviter la fluctuation de l’intensité de Hoechst dans différents échantillons de la même expérience. La coloration de Hoechst a été effectuée juste avant l’acquisition de l’échantillon au cytomètre en flux afin de minimiser la baisse de l’intensité du colorant pendant le temps. Pour ceux qui souhaitent reproduire le protocole dans de grandes expériences avec de nombreux échantillons, nous recommandons d’effectuer une coloration Hoechst sur quelques échantillons à la fois. Un autre inconvénient est que l’intensité de Hoechst peut être fortement influencée par le nombre de cellules pendant l’incubation avec le colorant. Pour cette raison, nous recommandons fortement de toujours utiliser le même nombre de cellules et le même volume par échantillon pour la coloration de l’ADN. Si un nombre élevé de cellules est nécessaire pour l’acquisition au cytomètre en flux, nous recommandons de préparer deux ou plusieurs échantillons identiques, puis de les fusionner juste avant l’étape de coloration hoechst.
Un point clé du protocole est la stratégie de contrôle pour l’analyse des données. Nous avons récemment publié une nouvelle stratégie pour l’analyse des lymphocytes T aux premiers moments de la réponse immunitaire, ce qui nous a permis d’augmenter la sensibilité de détection des lymphocytes T spécifiques de l’antigène12. Nous avons appliqué cette stratégie aux données présentées ici comme suit. Tout d’abord, nous avons exclu les agrégats cellulaires dans le diagramme ADN-A/W. Deuxièmement, après avoir éliminé les cellules mortes, nous avons utilisé une porte lymphocytaire assez grande dans le diagramme FSC / SSC (« porte détendue »). Grâce à cette stratégie, nous avons pu inclure des lymphocytes T CD8 spécifiques de l’antigène hautement activés dans S-G2/ M qui sont généralement manqués par les stratégies de contrôle actuelles, car ces cellules ont un FSC-A et un SSC-A élevés. En résumé, l’analyse des données représente une partie essentielle de la méthode, qui est essentielle pour obtenir une détection sensible des lymphocytes T activés/proliférants spécifiques de l’antigène.
La méthode empêche la possibilité de manquer des données critiques sur les lymphocytes T aux premières phases de la réponse immunitaire et ouvre de nouvelles perspectives pour l’immunosurveillance des lymphocytes T. Une amélioration future pourrait être d’inclure la coloration pour la phospho-histone 3 qui permettrait la différenciation entreG2 et M21. Une limitation actuelle est que les cellules doivent être fixées et perméabilisées pour tacher le marqueur nucléaire, Ki67. Ainsi, les cellules ne peuvent pas être utilisées pour d’autres types d’analyse tels que le tri et l’analyse fonctionnelle ultérieure. De plus, les colorants d’ADN, y compris Hoechst, interfèrent généralement avec l’analyse génomique de l’ADN et ne conviennent pas à ce type d’évaluation. L’identification de marqueurs membranaires qui sont en corrélation avec différentes phases du cycle cellulaire et qui peuvent être colorés sur des cellules vivantes pourrait surmonter cette limitation. En conclusion, la méthode a un grand potentiel pour l’évaluation des lymphocytes T activés/proliférants dans plusieurs contextes tels que la vaccination, l’infection, les maladies à médiation immunologique et l’immunothérapie.
Disclosures
A. Folgori et S. Capone sont des employés de Reithera Srl. A. Nicosia est nommé inventeur sur la demande de brevet WO 2005071093 (A3) « Chimpanzee adenovirus vaccine carriers ». Les autres auteurs n’ont rien à divulguer.
Acknowledgments
Ce travail a été soutenu par Reithera, par le projet MIUR 2017K55HLC_006, et par 5 × subvention de 1000 de l’Associazione Italiana Ricerca sul Cancro (AIRC). Le tétramère suivant a été obtenu par l’intermédiaire de l’installation de tétramères des NIH : H-2K (d) homonyme du VIH conjugué APC 197-205 AMQMLKETI.
Materials
| Name | Company | Catalog Number | Comments |
| 1-200 μL universal fit bulk packed pipet tips | Corning | CLS4866-1000EA | |
| 2.4G2 anti-FcγR mAb | BD | 553141 | 10 μg/ml final concentration |
| 5 ml syringe plunger | BD Emerald | 307733 | |
| 15 ml conical tubes | MercK Millipore | SBHA025SB | |
| 60 mm TC-treated Cell Culture Dish | Falcon | 353002 | |
| 70 μm cell strainer | Falcon | 352097 | |
| 96-well Clear Round Bottom TC-treated Culture Microplate | Falcon | 353077 | |
| Anti-Rat/Hamster Ig,k/Negative Control Compensation Particles | BD- Bioscience | 552845 | |
| Beta-mercaptoethanol | Sigma | M3148 | |
| Bovine Serum Albumin | Sigma | A07030 | |
| BUV805 Rat Anti-Mouse CD8a | BD- Bioscience | 564920 | 4 μg/ml final concentration |
| Dulbecco's Phosphate Buffer Saline w/o Calcium w/o Magnesium | Euroclone | ECB4004L | |
| Eppendorf Safe-Lock Tubes, 1.5 mL | Eppendorf | 30120159 | |
| Ethanol | Sigma | 34852-1L-M | |
| Ethylenediaminetetraacetic Acid Disodium Salt solution (EDTA) | Sigma | E7889 | |
| Fetal Bovine Serum | Corning | 35-079-CV | |
| Filcon, Sterile, Syringe-Type 70 μm | Falcon | 352350 | |
| Fixable Viability Dye eFluor 780 | eBioscience | 65-0865-14 | 1:1000 final concentration |
| Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | 00-5523-00 | This Set contains fixation/permeabilization concentrate and diluent, and permeabilization buffer 10x |
| H-2k(d) AMQMLKETI allophycocyanin (APC)-labelled tetramer | provided by NIH Tetramer Core Facility | 6 μg/ml final concentration | |
| H-2k(d) AMQMLKETI phycoerythrine (PE) labelled pentamer | Proimmune | F176-2A-E - 176 | 10 μL / sample |
| Hoechst 33342, Trihydrochloride, Trihydrate - 10 mg/mL Solution in Water | ThermoFisher | H3570 | |
| Ki-67 Monoclonal Antibody (SolA15), FITC | eBioscience | 11-5698-82 | 5 μg/ml final concentration |
| L-Glutamine 100X (200 mM) | Euroclone | ECB3000D | |
| Millex-HA Filters 0,45 µm | BD | 340606 | |
| Penicillin/Streptomycin 100X | Euroclone | ECB3001D | |
| PE/Cyanine7 anti-mouse CD62L Antibody | Biolegend | 104418 | 0.2 μg/ml final concentration |
| PerCP-Cy™5.5 Hamster Anti-Mouse CD3e | BD- Bioscience | 551163 | 4.4 μg/ml final concentration |
| Red Blood Cell Lysis Buffer | Sigma | R7757 | |
| Round-Bottom Polystyrene Tubes, 5 mL | Falcon | 352058 | |
| RPMI 1640 Medium without L-Glutamine with Phenol Red | Euroclone | ECB9006L | |
| Software package for analyzing flow cytometry data | FlowJo | v.10 | |
| Software for acquisition of samples at flowcytometer | BD FACSDiva | v 6.2 | |
| Trypan Blue Solution | Euroclone | ECM0990D |
References
- Castellino, F., et al. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature. 440 (7086), 890-895 (2006).
- Zhang, N., Bevan, M. J. CD8(+) T cells: foot soldiers of the immune system. Immunity. 35 (2), 161-168 (2011).
- Bajénoff, M., et al. Highways, byways and breadcrumbs: directing lymphocyte traffic in the lymph node. Trends Immunology. 28 (8), 346-352 (2007).
- Bevan, M. J., Fink, P. J. The CD8 response on autopilot. Nature Immunology. 2 (5), 381-382 (2001).
- Van Stipdonk, M. J., Lemmens, E. E., Schoenberger, S. P. Naïve CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nature Immunology. 2 (5), 423-429 (2001).
- Kaech, S. M., Wherry, E. J., Ahmed, R. Effector and memory T-cell differentiation: implications for vaccine development. Nature Review Immunology. 2 (4), 251-262 (2002).
- Beverley, P. C. Primer: making sense of T-cell memory. Nature Clinical Practice Rheumatology. 4 (1), 43-49 (2008).
- Parretta, E., et al. CD8 cell division maintaining cytotoxic memory occurs predominantly in the bone marrow. Journal of Immunology. 174 (12), 7654-7664 (2005).
- Di Rosa, F. Maintenance of memory T cells in the bone marrow: survival or homeostatic proliferation. Nature Review Immunology. 16 (4), 271 (2016).
- Di Rosa, F. Two niches in the bone marrow: a hypothesis on life-long T cell memory. Trends in Immunology. 37 (8), 503-512 (2016).
- Di Rosa, F. Commentary: Memory CD8(+) T cells colocalize with IL-7(+) stromal cells in bone marrow and rest in terms of proliferation and transcription. Frontiers in Immunology. 7, 102 (2016).
- Simonetti, S., et al. Antigen-specific CD8 T cells in cell cycle circulate in the blood after vaccination. Scandinavian Journal of Immunology. 89 (2), 12735 (2019).
- Wilson, A., et al. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes & Development. 18 (22), 2747-2763 (2004).
- Hirche, C., et al. Systemic virus infections differentially modulate cell cycle state and functionality of long-term hematopoietic stem cells in vivo. Cell Report. 19 (11), 2345-2356 (2017).
- Colloca, S., et al. Vaccine vectors derived from a large collection of simian adenoviruses induce potent cellular immunity across multiple species. Science Translational Medicine. 4 (115), (2012).
- Di Lullo, G., et al. Marker gene swapping facilitates recombinant Modified Vaccinia Virus Ankara production by host-range selection. Journal of Virological Methods. 156 (1-2), 37-43 (2009).
- Di Lullo, G., et al. The combination of marker gene swapping and fluorescence-activated cell sorting improves the efficiency of recombinant modified vaccinia virus Ankara vaccine production for human use. Journal of Virological Methods. 163 (2), 195-204 (2010).
- Mouse phenotype. , Available from: https://www.mousephenotype.org/data/secondaryproject/3i (2020).
- Yoon, H., Kim, T. S., Braciale, T. J. The cell cycle time of CD8+ T cells responding in vivo is controlled by the type of antigenic stimulus. PLoS One. 5 (11), 15423 (2010).
- Pauklin, S., Vallier, L. The cell-cycle state of stem cells determines cell fate propensity. Cell. 155 (1), 135-147 (2013).
- Vignon, C., et al. Flow cytometric quantification of all phases of the cell cycle and apoptosis in a two-color fluorescence plot. PLoS One. 8 (7), 68425 (2013).
Tags
Immunologie et infection numéro 167 lymphocytes T CD8 spécifiques à l’antigène cycle cellulaire Ki67 colorant ADN cytométrie en flux rate ganglions lymphatiques sourisErratum
Formal Correction: Erratum: A DNA/Ki67-Based Flow Cytometry Assay for Cell Cycle Analysis of Antigen-Specific CD8 T Cells in Vaccinated Mice
Posted by JoVE Editors on 11/03/2021.
Citeable Link.
An erratum was issued for: A DNA/Ki67-Based Flow Cytometry Assay for Cell Cycle Analysis of Antigen-Specific CD8 T Cells in Vaccinated Mice. The Authors section and a figure were updated.
The authors section was updated from:
Sonia Simonetti*1,2, Ambra Natalini*1,2, Giovanna Peruzzi3, Alfredo Nicosia4, Antonella Folgori5, Stefania Capone5, Angela Santoni2, Francesca Di Rosa1
1Institute of Molecular Biology and Pathology, National Research Council of Italy (CNR),
2Department of Molecular Medicine, University of Rome “Sapienza”,
3Center for Life Nano Science, Istituto Italiano di Tecnologia,
4Department of Molecular Medicine and Medical Biotechnology, University of Naples Federico II,
5Reithera Srl
* These authors contributed equally
To:
Sonia Simonetti*1,2, Ambra Natalini*1,2, Giovanna Peruzzi3, Alfredo Nicosia4, Antonella Folgori5, Stefania Capone5, Angela Santoni2,6, Francesca Di Rosa1
1Institute of Molecular Biology and Pathology, National Research Council of Italy (CNR),
2Department of Molecular Medicine, University of Rome “Sapienza”,
3Center for Life Nano Science, Istituto Italiano di Tecnologia,
4Department of Molecular Medicine and Medical Biotechnology, University of Naples Federico II,
5Reithera Srl
6IRCCS, Neuromed
* These authors contributed equally
Figure 1 was updated from:
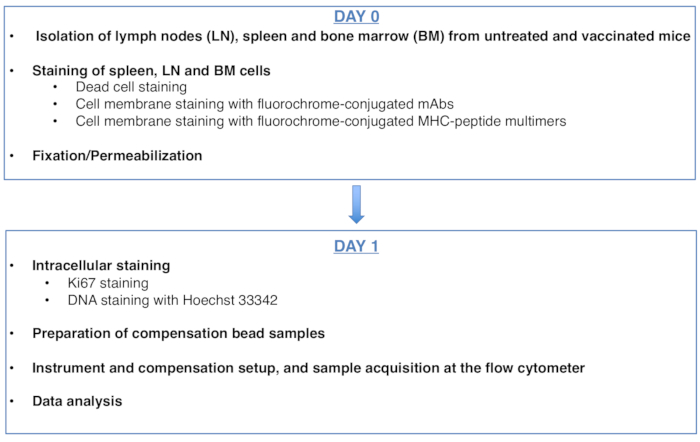
Figure 1: Scheme of the protocol for cell cycle analysis of antigen-specific CD8 T cells. Please click here to view a larger version of this figure.
To:
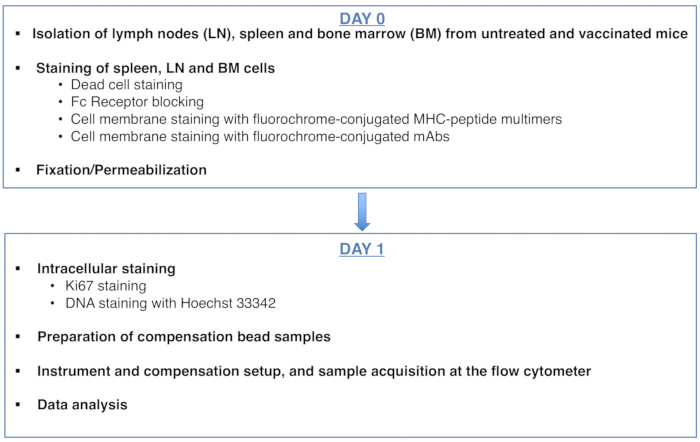
Figure 1: Scheme of the protocol for cell cycle analysis of antigen-specific CD8 T cells. Please click here to view a larger version of this figure.
