

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Die klonale Expansion ist ein Schlüsselmerkmal der antigenspezifischen T-Zell-Antwort. Der Zellzyklus von Antigen-reagierenden T-Zellen wurde jedoch schlecht untersucht, teilweise aufgrund technischer Einschränkungen. Wir beschreiben eine durchflusszytometrische Methode zur Analyse klonal expandierender Antigen-spezifischer CD8-T-Zellen in Milz und Lymphknoten geimpfter Mäuse.
Abstract
Der Zellzyklus von Antigen-spezifischen T-Zellen in vivo wurde mit einigen Methoden untersucht, die alle einige Einschränkungen aufweisen. Bromodeoxyuridin (BrdU) markiert Zellen, die sich in oder kürzlich abgeschlossener S-Phase befinden, und Carboxyfluorescein-Succinimidylester (CFSE) erkennt Tochterzellen nach der Teilung. Diese Farbstoffe erlauben jedoch keine Identifizierung der Zellzyklusphase zum Zeitpunkt der Analyse. Ein alternativer Ansatz ist die Nutzung von Ki67, einem Marker, der von Zellen in allen Phasen des Zellzyklus mit Ausnahme der Ruhephase G0stark exprimiert wird. Leider erlaubt Ki67 keine weitere Differenzierung, da es Zellen in der S-Phase, die der Mitose verschrieben sind, nicht von denen in G1 trennt, die in dieser Phasebleiben, in den Zyklus übergehen oder sich in G0bewegen können.
Hier beschreiben wir eine durchflusszytometrische Methode zur Erfassung eines "Schnappschusses" von T-Zellen in verschiedenen Zellzyklusphasen in sekundären lymphatischen Organen der Maus. Die Methode kombiniert Ki67- und DNA-Färbung mit Major Histocompatibility Complex (MHC)-Peptid-Multimer-Färbung und einer innovativen Gating-Strategie, die es uns ermöglicht, erfolgreich zwischen antigenspezifischen CD8-T-Zellen in G0, in G1 und in S-G2/ M-Phasen des Zellzyklus in der Milz und drainierenden Lymphknoten von Mäusen nach der Impfung mit viralen Vektoren zu unterscheiden, die den Modell-Antigen-Gag des humanen Immunschwächevirus (HIV)-1 tragen.
Kritische Schritte der Methode waren die Wahl des DNA-Farbstoffs und die Gating-Strategie, um die Assay-Sensitivität zu erhöhen und hochaktivierte/proliferierende antigenspezifische T-Zellen einzubeziehen, die nach aktuellen Analysekriterien übersehen worden wären. Der DNA-Farbstoff Hoechst 33342 ermöglichte es uns, eine qualitativ hochwertige Unterscheidung der DNA-Peaks G0/G1 und G2/Mzu erhalten und gleichzeitig die Membran- und intrazelluläre Färbung zu erhalten. Die Methode hat ein großes Potenzial, das Wissen über die T-Zell-Antwort in vivo zu erweitern und die Immunmonitoring-Analyse zu verbessern.
Introduction
Naive T-Zellen durchlaufen eine klonale Expansion und Differenzierung beim Antigen-Priming. Differenzierte T-Zellen zeigen Effektorfunktionen, die für die Antigen-Clearance und für die Aufrechterhaltung des Antigen-spezifischen Gedächtnisses unerlässlich sind, was für einen lang anhaltenden Schutz entscheidend ist. Während der ersten Schritte der primären Antwort ist die naive T-Zell-Interaktion mit Antigen-präsentierenden Zellen (APCs) in spezialisierten Nischen in lymphatischen Organen entscheidend, um die riesige T-Zell-Proliferation zu induzieren, die die klonale Expansionsphasecharakterisiert 1,2,3. Die T-Zell-APC-Interaktion wird durch die Konzentration und Persistenz von Antigen, kostimulatorischen Signalen und löslichen Faktoren (Zytokine und Chemokine), die die Quantität und Qualität der klonalen T-Zell-Nachkommen4,5 ,6,7beeinflussen,fein reguliert.
Trotz intensiver Studien zur klonalen T-Zellexpansion ist noch nicht bekannt, ob Antigen-präparierte T-Zellen ihren gesamten Zellzyklus am Ort der Antigenerkennung abschließen oder ob sie während des Zellzyklusverlaufs in andere Organe wandern. Dieser Mangel an Wissen ist auf die Eigenschaften der verfügbaren Werkzeuge für die Zellzyklusanalyse zurückzuführen. Dazu gehören monoklonale Antikörper (mAbs), die spezifisch für den Kernmarker Ki67 sind, und Zellfarbstoffe, die entweder Zellen identifizieren, die die S-Phase des Zellzyklus durchlaufen haben (z. B. Bromodeoxyuridin (BrdU)) oder zwischen Tochterzellen und ihren Vorfahren diskriminieren (z. B. Carboxyfluorescein-Succinimidylester (CFSE)).
Zellmarkierungsfarbstoffe wie CFSE und BrdU erlauben jedoch keine Bestimmung darüber, ob sich zellen in einem bestimmten Organ gefundene Zellen lokal vermehrt haben oder eher nach derTeilung 8,9an diesen Ort gewandert sind. Darüber hinaus ist das intranukleäre Protein Ki67 nur in der Lage, Zellen in G0 (Ki67-negative Zellen) von denen in jeder anderen Zellzyklusphase (Ki67-positive Zellen) zu unterscheiden. Daher unterscheidet die Ki67-Analyse Zellen in aktiver Proliferation (d.h. in S,G2oder M) nicht von denen in G 1 , die entweder schnell zurTeilungfortschreiten oder für längere Zeit in G1 bleiben oder zur Ruhe zurückkehrenkönnen 10,11.
Hier beschreiben wir eine neue durchflusszytometrische Methode zur Zellzyklusanalyse von antigenspezifischen CD8-T-Zellen12 aus milz und lymphknoten (LNs) geimpfter Mäuse (Abbildung 1). Die Methode nutzt eine Kombination aus Ki67 und DNA-Färbung, die zuvor zur Analyse des Zellzyklus von hämatopoetischen Zellen des Mausknochenmarks (BM) verwendet wurde13,14. Hier haben wir Ki67 plus DNA-Färbung zusammen mit der kürzlich veröffentlichten innovativen Gating-Strategie12erfolgreich auf die Analyse der klonalen Expansion von CD8-T-Zellen angewendet. Wir konnten eindeutig zwischen antigenspezifischen CD8-T-Zellen in G0, in G1und in S-G2/ M-Phasen in der Milz und drainierenden LNs von geimpften Mäusen unterscheiden.
Protocol
Mäuse wurden in der Plaisant Animal Facility untergebracht, und die Arbeit wurde unter der Genehmigungsnummer 1065/2015-PR des italienischen Gesundheitsministeriums durchgeführt. Das Protokoll folgte den Tierpflegerichtlinien gemäß den nationalen und internationalen Gesetzen und Richtlinien (EU-Richtlinie 2010/63/EU; Italienisches Gesetzesdekret 26/2014).
1. Herstellung von Medium und Färbelösung
- Bereiten Sie ein komplettes Medium vor: Roswell Park Memorial Institute (RPMI) Medium mit 2 mM Glutamin, 100 U / ml Penicillin / Streptomycin, 50 μM Beta-Mercaptoethanol und 10% Volumen / Volumen (v / v) fötalem Rinderserum (FBS)
- Färbepuffer vorbereiten: Phosphatgepufferte Kochsalzlösung ohne Ca2+/Mg2+ (PBS) mit 1% Gewicht/Volumen (w/v) Rinderserumalbumin (BSA) und 2 mM Ethylendiamintetraessigsäure-Dinatriumsalz (EDTA)
2. Behandlung mit der Maus
- Prime 7-8 Wochen alte, weibliche Balb/c-Mäuse durch intramuskuläre (i.m.) Injektion in den Quadrizeps des humanen Immundefizienz-Virus (HIV)-1-gag-exprimierenden-Schimpansen-Adenoviralvektors (ChAd3-gag) mit einer Dosis von 107 Viruspartikeln.
- 1-4 Monate nach der Grundierung verstärken die Mäuse einmal durch i.m. Injektion des HIV-1-Gag-exprimierenden modifizierten Vaccinia Ankara Virus (MVA-Gag) mit einer Dosis von 106 plaquebildenden Einheiten.
- Am Tag 3 nach dem Boost opfern Sie die verstärkten Mäuse durch zervikale Luxation und analysieren Sie sie parallel zu unbehandelten Mäusen.
- Ernten Sie die LNs, die den Quadrizeps (Becken, Kniekehle und Leisten) und die Milz von verstärkten und unbehandelten Mäusen entwässern. Sammeln Sie außerdem die BM von den beiden Hinterbeinen von unbehandelten Mäusen und verwenden Sie diese BM für Durchflusszytometereinstellungen und als Positivkontrolle für die Zellzyklusanalyse (Abbildung 2).
HINWEIS: Generieren Sie ChAd3-Gag- und MVA-Gag-Vektoren wie zuvor beschrieben12,15,16,17.
3. Isolierung von drainierenden LN-, Milz- und BM-Zellen
- Isolierung von Milz- und LN-Zellen
- Legen Sie 5 ml komplettes Medium in jedes der beiden 15-ml-Röhrchen und bewahren Sie sie auf Eis auf, bereit für die Entnahme von Organen.
- Opfern Sie eine erwachsene Maus durch zervikale Luxation.
- Legen Sie die Maus auf den Rücken und sterilisieren Sie die Hautoberfläche mit 70% v / v Ethanol.
- Um Leisten-LNs zu sammeln, machen Sie einen ~ 1 cm langen Schnitt am Bauch mit einer Schere und dehnen Sie den Schnitt mit der Pinzette.
- Visualisieren Sie Leisten-LNs auf der inneren Oberfläche der Haut und ernten Sie sie mit der Pinzette. Legen Sie die Leisten-LNs in eines der beiden in Schritt 3.1.1 vorbereiteten 15-ml-Röhrchen.
- Um die Milz zu sammeln, machen Sie einen Peritonealschnitt mit einer Schere und entfernen Sie die Milz. Nach dem Schneiden des umgebenden Bindegewebes wird die Milz in das zweite 15-ml-Röhrchen gegeben, das in Schritt 3.1.1 vorbereitet wurde.
- Um Becken-LNs zu sammeln, bewegen Sie den Darm zur Seite und visualisieren Sie Becken-LNs in der Nähe der unteren Hohlvene und sammeln Sie sie dann mit der Pinzette. Legen Sie die Becken-LNs in dasselbe Röhrchen, das die Leisten-LNs enthält.
HINWEIS: Um genügend LN-Zellen für die Färbung zu erhalten (siehe Abschnitt 4), ist es häufig notwendig, Popliteal-, Leisten- und Becken-LNs einer Maus zu bündeln. Diese LNs entwässern alle den Quadrizeps (die Stelle der i.m. Impfung). Dieses Protokoll verwendet nur eine 15-ml-Röhre aus gepoolten LNs. - Um Kniekehlen-LNs zu sammeln, greifen Sie die Haut der Hinterbeine und ziehen Sie sie sanft nach unten, um die Muskeln freizulegen. Führen Sie dann die Pinzette zwischen den Muskeln unter dem Kniegelenk ein und sammeln Sie die Kniekehlen-LNs. Legen Sie die Kniekehlen-LNs in die gleiche Röhre, die Leisten- und Becken-LNs enthält.
ANMERKUNG: Siehe Anmerkung nach 3.1.7. - Legen Sie die Milz in ein 70 μm Zellsieb in eine 60 mm Kulturschale, die mit 5 ml komplettem Medium gefüllt ist. Mit einem 5-ml-Spritzenkolben das Organ vorsichtig zerdrücken, bis es vollständig disaggregiert ist.
- Entfernen Sie das Sieb und geben Sie die Zellsuspension in ein sauberes 15-ml-Röhrchen.
- Geben Sie 5 ml komplettes Medium in die Kulturschale und waschen Sie die Schale und das Sieb sorgfältig ab, um sicherzustellen, dass alle Zellen wiederhergestellt wurden. Mit dem Rest der Milzzellensuspension in das 15-ml-Röhrchen einfließen lassen.
- Für die gepoolten Leisten-, Becken- und Popliteal-LNs wird eine Einzelzellsuspension nach einem ähnlichen Verfahren wie in den Schritten 3.1.9 bis 3.1.11 für die Milz hergestellt.
- Zentrifugenzellen bei 400 × g für 10 min bei 4 °C. Verwerfen Sie den Überstand und resuspendieren Sie die Zellpellets in PBS.
- Zählen Sie die Zellen mit einer Neubauer-Kammer unter Verwendung eines Lysepuffers für rote Blutkörperchen und 0,04% v / v Trypanblau in PBS.
- Isolierung von BM-Zellen
- Legen Sie 5 ml komplettes Medium in ein 15 ml Rohr und bewahren Sie es auf Eis auf, bereit für die Sammlung von Hinterbeinen.
- Opfern Sie eine erwachsene Maus durch zervikale Luxation.
- Sterilisieren Sie die Hautoberfläche mit 70% v/v Ethanol.
- Machen Sie mit einer Schere einen ~ 1 cm langen Querschnitt auf der ventralen Haut, greifen Sie die Haut auf beiden Seiten des Schnitts fest und ziehen Sie ihn vorsichtig nach unten, um die Muskeln der Hinterbeine freizulegen.
- Um die Haut von der Rückseite der Hinterbeine zu entfernen, halten Sie die Maus in Rückenlage, legen Sie die Klemme unter das Knie und ziehen Sie nach oben, um die Muskeln freizulegen.
- Schneiden Sie die Knochen an den beiden Extremitäten eines Hinterbeins ab: dem Becken- / Hüftgelenk und dem Knöchel.
- Beide Hinterbeine werden in das in Schritt 3.2.1 vorbereitete 15-ml-Röhrchen überführt. Halten Sie die Röhre auf Eis.
- Nehmen Sie die Hinterbeine aus dem 15-ml-Röhrchen und übertragen Sie sie auf Seidenpapier. Schneiden Sie die Hinterbeine direkt unter dem Kniegelenk ab, um die Tibia zu entfernen. Sezieren Sie den Femur und die Tibia von den umliegenden Muskeln, entfernen Sie überschüssiges Gewebe mit einer Schere und befeuchten Sie das Seidenpapier.
- Schneiden Sie die Knochenenden mit einer Schere ab, um den inneren Markschaft freizulegen. Tibia und Femur in das BM-Extraktionsröhrchen einführen (siehe Vorbereitung in 3.2.9.1-3.2.9.218), wobei das breiteste Ende unten ist.
- Schneiden Sie eine 200-μL-Pipettenspitze an der Linie knapp über dem Ende der Spitze und an der 100-μL-Linie.
- Legen Sie den mittleren Teil in den oberen, größeren Teil der Spitze und legen Sie diesen in ein 1,5 ml Mikrofugenrohr.
- Das BM-Extraktionsrohr bei 800 × g für 1 min drehen.
- Entsorgen Sie den Knochen und resuspen sie das Pellet kräftig in 1 ml vollständigem Medium, um alle Cluster zu entfernen. Filtrieren Sie die Zellsuspension durch einen 70-μm-Filter, der auf der Oberseite eines 15-ml-Röhrchens angebracht ist.
- Waschen Sie das BM-Extraktionsrohr zweimal mit jeweils 1 ml vollständigem Medium. Durch einen 70-μm-Filter filtriert und das Volumen mit dem Rest der in Schritt 3.2.11 erhaltenen Zellsuspension zusammenfasst.
HINWEIS: Ein einzelnes 15-ml-Röhrchen enthält Zellen aus beiden Hinterbeinen einer Maus. - Zentrifugenzellen bei 400 × g für 10 min bei 4 °C. Verwerfen Sie den Überstand und resuspendieren Sie das Zellpellet in PBS.
- Zählen Sie die Zellen mit einer Neubauer-Kammer unter Verwendung eines Lysepuffers für rote Blutkörperchen und 0,04% v / v Trypanblau in PBS.
4. Färbung von Milz-, LN- und BM-Zellen
- Teilen Sie die zu färbenden Zellproben in 3 Untergruppen auf: Zellproben zur Kompensation,einschließlich BM-Zellen von unbehandelten Mäusen, die nur mit Hoechst 33342 (im Folgenden als Hoechst bezeichnet) gefärbt werden sollen, und Milzzellen von unbehandelten Mäusen, die zur Herstellung einer Tot-/Lebendzellmischung für die Kompensation von Totzellenfarbstoffen verwendet werden; Positivkontrolle für die Zellzyklusanalyse, bestehend aus einer BM-Probe von unbehandelten Mäusen; und experimentelle Proben, die Milz- und LN-Proben von unbehandelten und geimpften Mäusen enthalten.
HINWEIS: Stellen Sie sicher, dass genügend Milz- und LN-Zellen für die Analyse einer ausreichenden Anzahl von Gag-spezifischen CD8-T-Zellen vorhanden sind. Es ist oft notwendig, gepoolte Milzzellen und gepoolte LN-Zellen von 3 geimpften Mäusen zu verwenden und zwei oder mehr identische Proben von gepoolten Zellen zu färben, die jeweils 3 × 10 6 Zellenenthalten. Identische Proben beim Hoechst-Färben zusammenführen. In ähnlicher Weise färben Sie gepoolte Milzzellen und LN-Zellen von 3 unbehandelten Mäusen und verschmelzen identische Proben am Ende. Legen Sie eine nicht gefärbte Probe von Milzzellen von einer unbehandelten Maus beiseite, um sie für die Einrichtung von Instrumenten und Kompensationen zu verwenden. - Bereiten Sie die Mischung aus toten / lebenden Zellen für die Kompensation von Totzellfarbstoffen vor (diese Zellmischung wird nur mit dem Totzellfarbstoff angefärbt).
- Ein Wasserbad bei 65 °C erhitzen.
- Nehmen Sie ein Aliquot von Milzzellen (~ 3 × 106).
- Übertragen Sie die Zellsuspension in ein Mikrofugenröhrchen, legen Sie sie für 5 min in das Wasserbad bei 65 °C und legen Sie sie dann sofort für 10 min auf Eis.
- Mischen Sie die hitzeabgetöteten Zellen mit lebenden Milzzellen (~ 3 × 10 6 ) ineinemVerhältnis von 1: 1 und übertragen Sie die Hälfte der Mischung auf eine 96 well-runde Bodenplatte (~ 3 × 106 Zellen / Well für die Totzellfärbungskontrolle).
- Totzellfärbung experimenteller Proben, Positivkontrolle für Zellzyklusanalyse und Tot-/Lebendzellmischung
- Milz- und LN-, BM-Zellen (3 × 106 Zellen/Well) und das Tot-/Lebendzellgemisch (Abschnitt 4.2) gemäß dem Färbeschema (Schritt 4.1) in die 96-Well-Rundbodenplatte überführen und bei 400 × g für 3 min bei 4 °C zentrifugieren.
- Resuspend jedes Zellpellet in 50 μL toter Zellfarbstoff, verdünnt in PBS, und resuspend durch pipettieren Sie sofort 3 Mal auf und ab.
- 30 min bei 4 °C vor Licht geschützt inkubieren.
- Zellen 2 mal mit Färbepuffer waschen; das erste Mal mit 200 μL und das zweite Mal mit 250 μL. Für jede Waschzentrifuge die Platte bei 400 × g für 3 min bei 4 °C.
- Verwerfen Sie den Überstand und resuspenieren Sie das Zellpellet in 20 μL PBS.
- Membranzellfärbung mit Major Histocompatibility Complex (MHC)-Peptid-Multimeren und mAbs.
- Unter Berücksichtigung der erforderlichen Volumina gemäß dem Färbeschema (Durchflusszytometereinstellungen, Tabelle 1) stellen Sie die folgenden Reagenzien her:
- Verdünnen Sie mAb 2.4G2 im Färbepuffer entsprechend der entsprechenden Verdünnung (siehe Tabelle der Materialien); für jede zu färbende Probe 10 μL dieser Verdünnung verwenden.
HINWEIS: 2,4G2 mAb blockiert die nicht-antigenspezifische Bindung von Immunglobulinen an die FcγIII- und FcγII-Rezeptoren. - Verdünnen Sie das H-2k(d) AMQMLKETI Allophycocyanin (APC)-markierte Tetramer (Tetr-Gag) im Färbepuffer, um die entsprechende Verdünnung zu erhalten (siehe Tabelle der Materialien); für jede zu färbende Probe 20 μL dieser Verdünnung verwenden.
- Die Antikörpermischung wird durch Verdünnen von mAbs im Färbepuffer entsprechend der entsprechenden Verdünnung (siehe Materialtabelle),die zuvor in Titrationsexperimenten bestimmt wurde, hergestellt; für jede zu färbende Probe 20 μL dieser Antikörpermischung verwenden.
HINWEIS: Hier wurden Anti-CD3e-Peridinin-Chlorophyllprotein (PerCP-Cy5.5) (Klon 145-2C11), Anti-CD8a-Brillant-Ultraviolett (BUV805) (Klon 53-6.7) und Anti-CD62L-Phycoerythrin Cyanin7 (PECy7) (Klon MEL-14) verwendet.
- Verdünnen Sie mAb 2.4G2 im Färbepuffer entsprechend der entsprechenden Verdünnung (siehe Tabelle der Materialien); für jede zu färbende Probe 10 μL dieser Verdünnung verwenden.
- 10 μL der zuvor verdünnten 2,4G2 mAb (Schritt 4.4.1.1) zugeben und 10 min bei 4 °C lichtgeschützt inkubieren.
- Fügen Sie 20 μL des zuvor verdünnten Tetr-Gag APC (Schritt 4.4.1.2) und 10 μL H-2k(d) AMQMLKETI Phycoerythrin (PE) Pentamer (Pent-Gag) hinzu. 15 min bei 4 °C vor Licht geschützt inkubieren.
- 20 μL der zuvor hergestellten Antikörpermischung (Schritt 4.4.1.3) zugeben und 15 min bei 4 °C lichtgeschützt inkubieren.
HINWEIS: Daher beträgt das Endvolumen 80 μL pro Vertiefung (Schritt 4.3.5, Schritte 4.4.2 bis 4.4.4). - Zellen mit 200 μL Färbepuffer waschen. Zentrifuge bei 400 × g für 5 min bei 4 °C.
- Resuspendieren Sie das Zellpellet in 250 μL Färbepuffer und übertragen Sie die Zellsuspension auf 5 ml Röhrchen. 1 ml Färbepuffer in das Röhrchen geben und bei 400 × g für 5 min bei 4 °C zentrifugieren.
- Nehmen Sie das Aliquot von BM-Zellen (3 × 106 Zellen) (siehe Liste der Zellproben, Abschnitt 4.1), das zur Kompensation des Hoechst-Kanals verwendet werden soll (Hoechst 33342 wird durch einen ultravioletten Laser angeregt (Durchflusszytometereinstellungen (Tabelle 2)), und übertragen Sie die Zellsuspension in ein 5-ml-Röhrchen. 1 ml Färbepuffer in das Röhrchen geben und 400 × g für 5 min bei 4 °C zentrifugieren.
- Unter Berücksichtigung der erforderlichen Volumina gemäß dem Färbeschema (Durchflusszytometereinstellungen, Tabelle 1) stellen Sie die folgenden Reagenzien her:
5. Fixierung/Permeabilisierung
- Bereiten Sie einen frischen Fixierungs- / Permeabilisierungspuffer vor, indem Sie 1 Teil Fixierungs- / Permeabilisierungskonzentrat mit 3 Teilen Fixierungs- / Permeabilisierungsverdünnungsmittel gemäß den Anweisungen des Herstellers verdünnen.
- Verwerfen Sie den Überstand und pulsieren Sie die Proben, um das Pellet vollständig zu disaggregieren.
- Jedem Röhrchen, einschließlich eines Rohrs mit ungefärbten Milzzellen (3 x 106,siehe Liste der Zellproben, Abschnitt 4.1) und Wirbel, wird 1 ml des frisch zubereiteten Fixations-/Permeabilisierungspuffers gegeben.
- 16 h bei 4 °C inkubieren.
HINWEIS: Das Protokoll kann hier angehalten werden.
6. Intrazelluläre Färbung
- Ki67 Färbung
- Bereiten Sie frischen Permeabilisierungspuffer 1x vor, indem Sie den Permeabilisierungspuffer 10x mit destilliertem Wasser gemäß den Anweisungen des Herstellers verdünnen. Vor der Verwendung muss der Permeabilisierungspuffer 1x durch einen 0,45 μm-Filter gefiltert werden, um Aggregate zu eliminieren.
- Verdünntes mAb Ki67 Fluoresceinisothiocyanat (FITC) (Klon SolA15) in Permeabilisierungspuffer 1x (siehe Materialtabelle),wie zuvor in Titrationsexperimenten bestimmt (Endvolumen von 100 μL pro Probe).
- 3 ml Permeabilisierungspuffer 1x in jedes Röhrchen geben und bei 400 × g für 5 min bei Raumtemperatur (RT) zentrifugieren.
- Verwerfen Sie den Überstand und wiederholen Sie Schritt 6.1.3.
- Verwerfen Sie den Überstand und resuspenieren Sie das Zellpellet in 100 μL zuvor verdünntem mAb Ki67 FITC (Schritt 6.1.2).
- 30 min bei RT inkubieren, vor Licht geschützt.
- Zellen 2 mal mit 4 ml Permeabilisierungspuffer 1x waschen. Für jede Waschzentrifuge bei 400 × g für 5 min bei RT.
- Resuspendieren Sie das Zellpellet in PBS unter Berücksichtigung der folgenden Volumina: 350 μL PBS für die Proben, die direkt am Durchflusszytometer entnommen werden sollen; 250 μL PBS für die mit Hoechst kurz vor der Durchflusszytometrie zu inkubierenden Proben (Abschnitt 6.2).
- DNA-Färbung
- Zu jeder Probe werden 250 μL 4 μg/ml Hoechst in PBS gegeben (die Endkonzentration von Hoechst beträgt 2 μg/ml).
HINWEIS: Falls zwei oder mehr identische Proben von 250 μL in PBS hergestellt wurden, verschmelzen Sie sie in diesem Schritt und fügen Sie das gleiche Volumen von 4 μg/ml Hoechst-Lösung in PBS hinzu (die Endkonzentration von Hoechst beträgt 2 μg/ml). Die Anzahl der Zellen beeinflusst den DNA-Färbeschritt stark. Verwenden Sie in jeder Stichprobe dieselbe Zellnummer. Beachten Sie, dass selbst eine leicht reduzierte Zellzahl (z.B. durch Zellverlust in früheren Waschschritten) zu einer höheren Hoechst-Bindung an DNA und einer höheren Hoechst-Intensität führt. - 15 min bei RT inkubieren, vor Licht geschützt.
- Zentrifugieren Sie die Proben bei 400 × g für 5 min bei RT.
- Resuspendieren Sie das Zellpellet in 350 μL PBS.
- Zu jeder Probe werden 250 μL 4 μg/ml Hoechst in PBS gegeben (die Endkonzentration von Hoechst beträgt 2 μg/ml).
7. Vorbereitung von Kompensationsperlenproben
- Bereiten Sie 5 μL des Antikörpers vor, indem Sie mAb im Färbepuffer entsprechend verdünnen.
ANMERKUNG: Bereiten Sie für jedes fluorchromkonjugierte mAb, das im Experiment verwendet wird, die entsprechende Kompensationsperlenprobe vor. - Vortex Negative Control und Anti-Rat/Hamster Ig,κ Comp Beads vor Gebrauch.
- Geben Sie für jede Probe einen Tropfen (~ 20 μL) Negativkontroll-CompBeads und einen Tropfen Anti-Ratten/Hamster-Ig,k CompBeads ein.
- 5 μL des vorverdünnten Antikörpers (Schritt 7.1) in das Röhrchen geben und auf und ab pipettieren.
- 15 min bei 4 °C vor Licht geschützt inkubieren.
- Proben mit 2 mL Färbepuffer waschen. Zentrifuge bei 400 × g für 5 min bei 4 °C.
- Verwerfen Sie den Überstand und resuspendieren Sie das Pellet, indem Sie 500 μL PBS zu jedem Rohr und Wirbel hinzufügen.
8. Instrumenten- und Kompensationsaufbau und experimentelle Probenaufnahme am Durchflusszytometer
HINWEIS: Die Konfiguration des Durchflusszytometers finden Sie unter Einstellungen des Durchflusszytometers (Tabelle 2).
- Allgemeiner Instrumenten- und Kompensationsaufbau
- Öffnen Sie die Software zur Probenerfassung (siehe Materialtabelle),und erstellen Sie ein neues Experiment, indem Sie im Menübandbereich des Arbeitsbereichs auf Neues Experiment klicken und Neues leeres Experimentauswählen.
- Doppelklicken Sie auf das erstellte Experiment, um es zu öffnen.
- Klicken Sie im Fenster Zytometereinstellungen auf Parameter und wählen Sie alle Kanäle (z. B. PE, APC usw.) aus, die im Färbefenster verwendet werden, einschließlich der Parameter Vorwärtsstreuung (FSC) und Seitenstreuung (SSC).
- Wählen Sie lineare Skalierung als Hoechst-Parameter aus, indem Sie die Log-Skala deaktivieren, und überprüfen Sie die Breite (W) des Spannungsimpulses für FCS, SSC und Hoechst.
HINWEIS: Alle Parameter werden standardmäßig in logarithmischer (logarithmischer) Skala angezeigt, mit Ausnahme von FSC und SSC, die im linearen Maßstab vorliegen. Alle Parameter werden durch die Fläche (A) und die Höhe (H) des Spannungsimpulses analysiert. - Erstellen Sie auf dem globalen Arbeitsblattein Punktdiagramm mit FSC-A auf der x-Achse und SSC-A auf der y-Achse.
- Führen Sie die nicht gebeizte Milzprobe aus, indem Sie im Erfassungsdashboard auf Daten erfassenklicken.
- Legen Sie die entsprechenden FSC- und SSC-Einstellungen fest, um die Zellen zu visualisieren, indem Sie die Spannungswerte im Abschnitt Parameter ändern, und erstellen Sie ein Gate, um alle im FSC-A/SSC-A-Punktdiagramm angezeigten Zellen auszuwählen, indem Sie in der Workspace-Symbolleiste des globalen Arbeitsblattsauf Polygon-Gate klicken.
- Zeigen Sie die gated Zellen in Histogrammen mit jedem Fluoreszenzparameter auf der x-Achse an.
- Führen Sie ungefärbte und vollständig gefärbte Milzproben aus, um den Fluoreszenzdetektor (PMT) so einzustellen, dass er für jeden Fluoreszenzparameter eine klare Trennung zwischen negativen und positiven Signalen der gefärbten Zellen aufweist.
- Um die Vergütungseinrichtung durchzuführen, klicken Sie im Arbeitsbereichsmenüband auf Experiment und wählen Sie im Abschnitt Vergütungseinrichtung die Option Vergütungssteuerung erstellenaus. Deaktivieren Sie Ungefärbtes Steuerrohr/Well einschließen und klicken Sie auf OK.
HINWEIS: Dieser Vorgang führt zur Erzeugung einer Probe mit dem Namen Compensation Controls und eines normalen Arbeitsblatts, das mehrere Blätter enthält, die jedem ausgewählten Parameter entsprechen. - Führen Sie eine Probe von Kompensationsperlen aus (siehe Abschnitt 7); Legen Sie die entsprechenden FSC- und SSC-Einstellungen fest, um die Kügelchen zu visualisieren, indem Sie die Spannungswerte und den Erfassungsschwellenwert von 5.000 für FSC-Parameter im Zytometerfenster ändern.
- Passen Sie das P1-Tor auf der Perlenpopulation an und überprüfen Sie, ob die positiven und negativen Spitzen beide auf der x-Achse sichtbar sind. Wiederholen Sie diesen Vorgang für jede Kompensationsperlenprobe und zeichnen Sie schließlich jede Probendatei auf, indem Sie im Erfassungs-Dashboard auf Daten aufzeichnen klicken (zeichnen Sie mindestens 5.000 Ereignisse für jede Probe auf).
- Stellen Sie für jede aufgezeichnete Perlenprobe die P2- und P3-Gatter auf die positiven bzw. negativen Peaks ein.
- Führen Sie die Zellproben zur Kompensation aus (siehe Schritte 4.2 und 4.4.7 sowie Abschnitte 5 und 6). Ändern Sie die FSC- und SSC-Spannungen und den Schwellenwert, um die Zellen zu visualisieren, passen Sie das P1-Gate an und zeichnen Sie schließlich jede Sample-Datei auf (zeichnen Sie mindestens 10.000 Ereignisse auf). Stellen Sie die P2- und P3-Gatter auf die positiven bzw. negativen Peaks ein.
HINWEIS: Für die Kompensation des Hoechst-Kanals verwenden Sie G0/ G1 als negativen Peak (P3) und G2/ M als positiven (P2). - Klicken Sie im Menübandbereich des Arbeitsbereichs auf Experiment und wählen Sie im Abschnitt Vergütungseinrichtung die Option Vergütung berechnenaus.
- Benennen Sie die erstellte Vergütungseinstellung, verknüpfen Sie sie, und speichern Sie sie im aktuellen Test.
- Experimentelle Probenentnahme
- Öffnen Sie ein Muster, indem Sie in der Symbolleiste des Browsers auf Neues Muster klicken, und erstellen Sie die Gating-Strategie im globalen Arbeitsblatt.
ANMERKUNG: Die Gating-Strategie der Probenentnahme ähnelt der der Probenanalyse, die in Abbildung 3 und Abschnitt 9 beschrieben ist. - Zeigen Sie alle Ereignispopulationen in einem Histogramm mit CD3-A auf der x-Achse an. Erstellen Sie ein Intervall-Gate, um nur die CD3+ -Zellen auszuwählen.
- Wählen Sie im ErfassungsdashboardStorage Gate als Alle Ereignisse für LN-Samples und entweder Alle Ereignisse oder CD3+ Zellen für Milzproben aus.
- Führen Sie die experimentellen Proben mit niedriger Geschwindigkeit aus und nehmen Sie schließlich alle Dateien auf, um sicherzustellen, dass mindestens 100-200 antigenspezifische CD8-T-Zellen für jede Probe von den geimpften Mäusen gesammelt werden.
HINWEIS: Die Dateigröße experimenteller Proben ist in der Regel groß (30-120 MB), insbesondere wenn die Häufigkeit von antigenspezifischen CD8-T-Zellen niedrig ist. Daher müssen eine hohe Anzahl von Ereignissen (> 1 × 106) gesammelt werden, um mindestens 100-200 Antigen-spezifische CD8-T-Zellen zu erfassen. Große Dateien können den nachfolgenden Datenanalyseprozess verlangsamen. Die Erfassung von nur CD3+ Zellen in Milzproben (siehe Schritt 8.2.2 oben) ist hilfreich, um die Dateigröße kleiner zu halten. - Führen Sie die Positivkontrolle für die Zellzyklusanalyse durch und zeichnen Sie sie auf, d. H. BM-Probe von unbehandelten Mäusen.
- Öffnen Sie ein Muster, indem Sie in der Symbolleiste des Browsers auf Neues Muster klicken, und erstellen Sie die Gating-Strategie im globalen Arbeitsblatt.
9. Datenanalyse
- Öffnen Sie die Software (siehe Tabelle der Materialien) und erstellen Sie verschiedene Gruppen, die den verschiedenen zu analysierenden Organen entsprechen, indem Sie im Menübandbereich des Arbeitsbereichs auf Gruppe erstellen klicken (d. h. Gruppe "a-LNs erstellen"; "b-Milz"; "c-BM").
HINWEIS: Neu erstellte Gruppen werden in der Gruppenliste angezeigt, während die Gruppe "Vergütung" automatisch von der Software generiert wird. - Öffnen Sie das Fenster Gruppe ändern, indem Sie auf den Gruppennamen doppelklicken, und überprüfen Sie, ob die neu erstellten Gruppen synchronisiert sind. Wenn nicht, setzen Sie ein Häkchen in die Funktion Synchronisiert.
- Ziehen Sie jede FCS-Datei in die entsprechende Gruppe.
- Erstellen Sie die Gating-Strategie beginnend mit der Gruppe "a-LNs".
- Doppelklicken Sie auf die vollständig gebeizte Probe in der Gruppe, um das Diagrammfenster zu öffnen. x- und y-Achse sind wie in den fcs-Dateien gekennzeichnet (siehe Durchflusszytometereinstellungen, Tabelle 2).
- Zeigen Sie die für diese Probe erfassten Gesamtereignisse in einem Punktdiagramm mit DNA-A auf der x-Achse und DNA-W auf der y-Achse an.
- Wählen Sie nur die einzelne Zellpopulation aus, indem Sie im Abschnitt Gating-Werkzeug des Diagrammfensters auf Rechteck klicken.
HINWEIS: Einzelne Zellen haben DNA-A-Werte wie folgt: 2N (niedrig): zwischen 2N und 4N (intermediär) oder gleich 4N (hoch), während DNA-W-Werte für alle identisch sind (Schritt 1 von Abbildung 3). - Doppelklicken Sie in die Mitte des rechteckigen Tors, um einzelne Zellen in einem Punktdiagramm mit dem Parameter FSC-A auf der x-Achse und dem Farbstoff toter Zellen auf der y-Achse anzuzeigen.
- Wählen Sie nur die Lebende Zellpopulation aus, indem Sie im Abschnitt Gating-Werkzeug des Diagrammfensters auf Polygon klicken. Lebende Zellen sind negativ für den toten Zellfarbstoff (Schritt 2 von Abbildung 3).
- Doppelklicken Sie in die Mitte des polygonalen Gatters, um die Zellen in einem Punktplot mit dem Parameter FSC-A auf der x-Achse und dem Parameter SSC-A auf der y-Achse anzuzeigen.
- Klicken Sie auf Rechteck, und erstellen Sie ein "entspanntes" Tor, um alle einzelnen lebenden Zellen in diesem Diagramm12 einzuschließen (Schritt 3 in Abbildung 3).
- Doppelklicken Sie in die Mitte des "entspannten" Tors, um die Zellen in einem Punktdiagramm mit CD3 auf der x-Achse und CD8 auf der y-Achse anzuzeigen.
- Wählen Sie die Zellen CD3+CD8+ aus, indem Sie auf Polygon klicken (Schritt 4 in Abbildung 3).
- Doppelklicken Sie in die Mitte des CD3+CD8+ Gatters, um die Zellen in einem Punktdiagramm mit Tetr-Gag auf der x-Achse und Pent-Gag auf der y-Achse anzuzeigen.
- Wählen Sie die antigenspezifischen CD8-T-Zellen (positiv für Tetr-Gag und Pent-Gag) aus, indem Sie auf Polygon klicken (Schritt 5 von Abbildung 3).
- Doppelklicken Sie in die Mitte des Gag-spezifischen Tors, um die Zellen in einem Punktdiagramm mit DNA-A auf der x-Achse und Ki67 auf der y-Achse anzuzeigen (Abbildung 4).
- Markieren Sie die Zellen in den verschiedenen Zellzyklusphasen, indem Sie im Gating-Werkzeugabschnitt des Diagrammfensters auf Quad klicken.
HINWEIS: Zellen in der G0-Phase sind Ki67neg-DNA-niedrige Zellen (unterer linker Quadrant); Zellen in G1 sind Ki67pos-DNA niedrig (oberer linker Quadrant); Zellen in S-G2/M sind Ki67pos-DNA intermediate/high (oben rechts Quadrant) (Abbildung 4). - Kopieren Sie die in einem Beispiel erstellte Gating-Strategie in die entsprechende Gruppe, um die Gatter auf alle Stichproben der Gruppe anzuwenden.
- Wiederholen Sie die Schritte 9.5 bis 9.18 für die "a-LN-Gruppe".
- Überprüfen Sie, ob alle Tore für jede Probe der Gruppe "b-Milz" geeignet sind. Um den Zellzyklus zwischen den BM-Zellen zu analysieren (Positivkontrolle), klicken Sie in die Mitte des "entspannten" Tors, um die Zellen in einem Punktdiagramm mit DNA-A auf der x-Achse und Ki67 auf der y-Achse anzuzeigen.
- Überprüfen Sie, ob alle Gatter für jede Probe der 3 Gruppen geeignet sind (d. H. Für Zellen aus Milz, LN und BM).
ANMERKUNG: Das Einzelzellpopulations-Gate (Schritt 9.7) und das Quad-Gate für den Zellzyklus (Schritt 9.17) können in verschiedenen Proben unterschiedliche Gate-Koordinaten aufweisen, hauptsächlich aufgrund der möglichen geringfügigen Unterschiede der Hoechst-Farbintensität zwischen den Proben (Abschnitt 6.2). Aus diesem Grund kann es notwendig sein, das Single Cell Population Gate und die Quad Gates für den Zellzyklus in jeder Probe zu modifizieren. Dies geschieht wie folgt: Doppelklicken Sie auf den Gruppennamen und entfernen Sie die Synchronisation aus den Gruppeneigenschaften. Diese Operation ermöglicht die Modifikation der Gatter in einer Probe, ohne die gleichen Gatter in allen anderen Proben der Gruppe zu ändern. Ändern Sie nach dem Entfernen der Synchronisierung die Gates bei Bedarf. - Um die Ergebnisse dieser Analyse zu visualisieren, klicken Sie im Menübandbereich des Arbeitsbereichs auf Layout-Editor, um ihn zu öffnen. Ziehen Sie jedes Gate der Gating-Strategie im Beispielbereich in den Layout-Editor, und platzieren Sie die Plots entsprechend der Reihenfolge der Gating-Strategie. Ändern Sie bei Bedarf den Diagrammtyp, indem Sie auf das entsprechende Diagramm im Layout doppelklicken und im Fenster Diagrammdefinition den entsprechenden Typ auswählen.
- Klicken Sie auf die Gruppe und iterieren Sie nach Funktionen auf dem Layout-Menüband, um die in jedem Organ erzielten Ergebnisse zu visualisieren und verschiedene Proben zu vergleichen.
Representative Results
Die Zellzyklusphasen von Zellen aus Milz, LNs und BM von Balb /c-Mäusen wurden mit dem fluoreszierenden DNA-Farbstoff Hoechst und einem Anti-Ki67 mAb gemäß dem in Abbildung 1zusammengefassten Protokoll analysiert. Diese Färbung ermöglichte die Differenzierung von Zellen in den folgenden Phasen des Zellzyklus: G0 (Ki67neg, wobei 2N DNA als DNAlow definiert ist), G1 (Ki67pos, DNAlow) und S-G2/ M (Ki67pos, mit einem DNA-Gehalt zwischen 2N und 4N oder gleich 4N DNA, definiert als DNAintermedite / high).
Wir führten zunächst eine Zellzyklusanalyse von BM-Zellen durch, um zuvor veröffentlichte Ergebnisse13,14 zu reproduzieren, und analysierten dann die interessierenden Zellen, d.h. CD8-T-Zellen. Abbildung 2 zeigt ein typisches Beispiel für die Zellzyklusanalyse von BM-Zellen (Abbildung 2A). Das Protokoll ergab einen niedrigen Variationskoeffizienten (CV) von G0/G1 und G2/MDNA-Peaks, was auf die hervorragende Qualität der DNA-Färbung hinweist (Abbildung 2B, zeigt ein Beispiel mit CV < 2,5; CV war in allen Experimenten immer < 5).
Wir haben dann das gleiche Protokoll auf antigenspezifische CD8-T-Zellen von geimpften Mäusen angewendet. BALB / c-Mäuse wurden gegen den Antigen-Gag von HIV-1 geimpft, indem chad3-gag für Priming und MVA-Gag für Boosting verwendet wurden, beide entwickelt, um HIV-1-Gag zu tragen. Am Tag (d) 3 nach dem Boost analysierten wir die Häufigkeit von Gag-spezifischen CD8-T-Zellen aus der Milz und den drainierenden LNs. Wir nutzten die neu definierte Gating-Strategie für T-Zellen in der Frühphase der Immunantwort, die im Gegensatz zur konventionellen Strategie geeignet ist, hochaktivierte Antigen-reagierende CD8-T-Zellennachzuweisen 12. Wir haben die neuartige Strategie in fünf aufeinander folgenden Schritten umgesetzt. In Schritt 1 schlossen wir Dubletten oder Aggregate durch DNA-A/ -W-Gatter aus, und in Schritt 2 identifizierten wir lebende Zellen durch Ausschluss toter Zellmarker. In Schritt 3 identifizierten wir die interessierende Population unter Verwendung eines unkonventionellen "entspannten" FSC-A / SSC-A-Gates (Abbildung 3A) anstelle des kanonischen schmalen Lymphozyten-Gates12. Nach dem Gating auf CD3+CD8+ Zellen (Schritt 4 von Abbildung 3A) identifizierten wir Gag-spezifische CD8 T-Zellen unter Verwendung von zwei verschiedenen MHC-Multimeren, d.h. Pent-Gag und Tetr-Gag (Schritt 5 von Abbildung 3A). Wir verwendeten zwei Multimere anstelle von einem, um die Empfindlichkeit des Gag-spezifischen CD8-T-Zellnachweises bei geimpften Mäusen zu verbessern, ohne den Färbehintergrund bei unbehandelten Mäusen zu erhöhen(Abbildung 3B und C,Schritt 5). So haben wir erfolgreich unbehandelte Mäuse (0,00% bzw. 0,00% Antigen-spezifische CD8-T-Zellen in LNs bzw. Milz) von geimpften Mäusen (0,46% bzw. 0,29% Antigen-spezifische CD8-T-Zellen in LNs bzw. Milz) unterschieden, Abbildung 3B und C).
Bemerkenswerterweise erlaubte uns das Protokoll, einen extrem niedrigen Hintergrund im antigenspezifischen CD8-T-Zell-Gate von LNs und Milz von unbehandelten Mäusen zu haben (normalerweise 0,00% und maximal 0,02%). Der Vergleich von Gag-spezifischen und nicht Gag-spezifischen FSC-A / SSC-A-Plots zeigte, dass die Gag-spezifischen Zellen hohe SSC-A und FSC-A aufwiesen(Abbildung 3D),was die Notwendigkeit bestätigt, ein "entspanntes" FSC-A / SSC-A-Gate zu verwenden, um diese Zellen zu erfassen. Wir haben dann die Prozentsätze von Gag-spezifischen CD8-T-Zellen in verschiedenen Zellzyklusphasen ausgewertet (Abbildung 4A). Wir fanden heraus, dass Gag-spezifische CD8-T-Zellen in der Milz und noch mehr in den drainierenden LNs einen hohen Anteil an Zellen in S-G2/ M-Phasen am Tag 3 nach dem Boost enthielten (18,60% bzw. 33,52%).
Darüber hinaus fanden wir heraus, dass Gag-spezifische CD8-T-Zellen in S-G2/M-Phasen hohe FSC-A- und SSC-A-Zellen aufwiesen, wenn sie auf die gesamten CD8-T-Zellen desselben Organs überlagert wurden (Abbildung 4B). Die CD62L-Expression durch Gag-spezifische CD8-T-Zellen war niedrig, wie für aktivierte T-Zellen erwartet, mit Ausnahme einiger Zellen in G0 in den LNs (Abbildung 4C). Insgesamt bestätigten diese Ergebnisse, dass das "entspannte" Gate (Schritt 3 von Abbildung 3A, B und C)erforderlich war, um alle sich ausbreitenden antigenspezifischen CD8-T-Zelleneinzuschließen 12. Das Protokoll war äußerst wertvoll für eine "Momentaufnahme" der Zellzyklusphasen von Antigen-spezifischen CD8-T-Zellen zum Zeitpunkt der Analyse und der CD62L-Expression durch Zellen in verschiedenen Zellzyklusphasen.

Abbildung 1: Schema des Protokolls für die Zellzyklusanalyse von Antigen-spezifischen CD8-T-Zellen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 2: Zellzyklusanalyse von BM-Zellen. BM-Zellen von unbehandelten Balb/c-Mäusen wurden gefärbt und mittels Durchflusszytometrie analysiert. (A) Beispiel für eine Gating-Strategie. Wir haben einzelne Zellen im DNA-A/-W-Diagramm (links) und anschließend lebende Zellen durch Totzellfarbstoffausschluss (Mitte) abgegrenzt. Dann wurde ein "entspanntes" FSC-A/SSC-A-Gate für alle BM-Zellen verwendet (rechts). (B) Beispiel für die Zellzyklusanalyse von BM-Zellen (links). Wir verwendeten eine Kombination aus Ki67 und DNA-Färbung, um Zellen in den folgenden Phasen des Zellzyklus zu identifizieren: G0 (unterer linker Quadrant, Ki67neg-DNAlow-Zellen), G1 (oberer linker Quadrant, Ki67pos-DNAlow), S-G2/ M (oberer rechter Quadrant, Ki67pos-DNAintermedite / high). Fluoreszenz Minus Eins (FMO) Kontrolle von Ki67 mAb (Mitte) und DNA-Histogramm (rechts) werden gezeigt. Im DNA-Histogrammdiagramm entsprechen das linke und das rechte Gatter dem DNA-Peak G0/G1 bzw. dem DNA-PeakG2/M,und die Zahlen stellen die Variationskoeffizienten (CV) jedes Peaks dar. In allen anderen Diagrammen stellen die Zahlen Zellprozentsätze in den angegebenen Toren dar. Die Abbildung zeigt 1 repräsentatives Experiment von 5. In jedem Experiment analysierten wir gepoolte BM-Zellen von 3 Mäusen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 3: Analyse von antigenspezifischen CD8-T-Zellen aus LNs und Milz. Balb/c-Mäuse wurden intramuskulär (i.m.) mit Chad3-Gag und i.m. mit MVA-Gag präpariert. An Tag 3 nach dem Boost wurden abfließende LN- und Milzzellen von geimpften und unbehandelten Kontrollmäusen gefärbt und mittels Durchflusszytometrie analysiert. (A) Schema der Gating-Strategie in fünf Schritten zur Identifizierung einzelner Zellen (Schritt 1); lebende Zellen (Schritt 2); Lymphozyten (Schritt 3); CD8 T-Zellen (Schritt 4); und Gag-spezifische Zellen (Schritt 5). (B-C) Beispiel für Plots: Analyse von Zellen aus (B) LNs und (C) Milz von unbehandelten (oben) und geimpften (unten) Mäusen. Wir haben einzelne Zellen auf dem DNA-A/ -W-Diagramm in Schritt 1 identifiziert. In Schritt 2 haben wir dann lebende Zellen durch Ausschluss von Totzellfarbstoffen ausgewählt. In Schritt 3 haben wir ein nicht-kanonisches "entspanntes" Tor für Lymphozyten verwendet. In Schritt 4 identifizierten wir CD8-T-Zellen durch ihre doppelte Expression von CD3 und CD8. Wir identifizierten dann gag-spezifische Zellen und nicht gag-spezifische Zellen in Schritt 5, basierend auf ihrer Fähigkeit, fluorochrom-markiertes H-2kd-Gag-Pentamer (Pent-Gag) bzw. H-2kd-Gag-Tetramer (Tetr-Gag) zu binden oder nicht. (D) FSC-A/SSC-A-Profile von knebelspezifischen (blau) und nicht knebelspezifischen (grauen) Zellen nach dem Gating wie oben beschrieben. Zahlen stellen Zellprozentsätze in den angegebenen Gates dar. Die Abbildung zeigt 1 repräsentatives Experiment von 5. In jedem Experiment analysierten wir gepoolte Milz und gepoolte LN-Zellen von 3 geimpften Mäusen und 3 unbehandelten Mäusen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 4: Zellzyklusanalyse von Antigen-spezifischen CD8-T-Zellen. Die Mäuse wurden wie in Abbildung 3 geimpft und die Zellzyklusanalyse von Gag-spezifischen Zellen wurde an Tag 3 nach dem Boost durchgeführt, nach dem Gating in 5 Schritten wie in Abbildung 3. (A)Beispiel für eine Zellzyklusanalyse von Gag-spezifischen CD8-T-Zellen aus LNs (oben) und Milz (unten) geimpfter Mäuse. Zellzyklusphasen wurden wie in Abbildung 2Bidentifiziert. Die Panels repräsentieren Zellen in G0, in G1und in S-G2/M (links) und Fluoreszenz Minus Eins (FMO) Steuerung von Ki67 mAb (rechts). Zahlen stellen Zellprozentsätze in den angegebenen Gates dar. (B) FSC-A/SSC-A-Punktdiagramme, die Gag-spezifische CD8-T-Zellen in S-G2/M-Phasen (in rot) zeigen, überlagert auf insgesamt CD3+CD8+ T-Zellen (in grau) aus LNs (oben) und Milz (unten) geimpfter Mäuse. (C) Offset-Histogramme, die die CD62L-Expression durch Gag-spezifische CD8-T-Zellen in G0 (grün), in G1 (blau) und in S-G2/M(rot) aus LNs (oben) und Milz (unten) geimpfter Mäuse zeigen. Die y-Achsen zeigen die normalisierte Anzahl von Ereignissen an. Die Abbildung zeigt 1 repräsentatives Beispiel aus 5 unabhängigen Experimenten mit insgesamt 15 Mäusen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Ergänzendes Material: Durchflusszytometer-Einstellungen. Bitte klicken Sie hier, um diese Datei herunterzuladen.
Discussion
Obwohl die klonale Expansion von T-Zellen intensiv untersucht wurde, bleiben einige Aspekte unbekannt, vor allem, weil die verfügbaren Werkzeuge, um sie zu untersuchen, wenige sind und ihre eigenen Nachteile haben. Aus dieser Perspektive haben wir eine hochempfindliche durchflusszytometrische Methode aufgebaut, um den Zellzyklus von antigenspezifischen CD8-T-Zellen zu frühen Zeitpunkten nach der Impfung in einem Mausmodell zu analysieren. Das Protokoll basiert auf einer Kombination aus Ki67 und DNA-Färbung, die zuvor zur Analyse des Zellzyklus von BM-hämatopoetischen Zellen in Mäusen verwendet wurde13,14. Um das Protokoll an antigenspezifische CD8-T-Zellen anzupassen, mussten wir einige kritische Punkte berücksichtigen, darunter die Wahl des DNA-Farbstoffs, die geeigneten Bedingungen, um eine vergleichbare DNA-Färbung über verschiedene Proben hinweg zu erhalten, und die Gating-Strategie für die Datenanalyse.
Viele Farbstoffe sind für die DNA-Färbung verfügbar, einschließlich Propidiumiodid und 7-Aminoactinomycin D; Wir haben uns für Hoechst entschieden, weil es mit der Membranfärbung und dem für die Ki67-Färbung erforderlichen milden Fixations- / Permeabilisierungsprotokoll kompatibel war. Gleichzeitig ermöglichte uns die Färbung mit Hoechst ein DNA-Histogramm von ausgezeichneter Qualität, d.h. die DNA-Peaks G0/G1 und G2/M hatten einen viel niedrigeren Variationskoeffizienten (CV) als DNA-Peaks, die normalerweise mit anderen DNA-Farbstoffen, z.B. DRAQ519,erhalten werden. Tatsächlich kann Hoechst DNA sogar in lebenden Zellenfärben 20.
Einige Strategien wurden verwendet, um die Fluktuation der Hoechst-Intensität in verschiedenen Proben desselben Experiments zu vermeiden. Die Hoechst-Färbung wurde kurz vor der Probenentnahme am Durchflusszytometer durchgeführt, um den Rückgang der Farbstoffintensität im Laufe der Zeit zu minimieren. Für diejenigen, die daran interessiert sind, das Protokoll in großen Experimenten mit zahlreichen Proben zu reproduzieren, empfehlen wir, eine Hoechst-Färbung an einigen Proben gleichzeitig durchzuführen. Ein weiterer Nachteil ist, dass die Hoechst-Intensität während der Inkubation mit dem Farbstoff stark von der Zellzahl beeinflusst werden kann. Aus diesem Grund empfehlen wir dringend, für die DNA-Färbung immer die gleiche Anzahl von Zellen und das gleiche Volumen pro Probe zu verwenden. Wird für die Erfassung am Durchflusszytometer eine hohe Anzahl von Zellen benötigt, empfehlen wir, zwei oder mehr identische Proben vorzubereiten und diese dann kurz vor dem Hoechst-Färbeschritt zusammenzuführen.
Ein wichtiger Punkt des Protokolls ist die Gating-Strategie für die Datenanalyse. Wir haben kürzlich eine neuartige Strategie für die T-Zell-Analyse zu frühen Zeitpunkten der Immunantwort veröffentlicht, die es uns ermöglichte, die Empfindlichkeit des Nachweises von antigenspezifischen T-Zellen zu erhöhen12. Wir haben diese Strategie wie folgt auf die hier gezeigten Daten angewendet. Zuerst haben wir Zellaggregate im DNA-A/W-Diagramm ausgeschlossen. Zweitens, nachdem wir abgestorbene Zellen ausgesondert hatten, verwendeten wir ein ziemlich großes Lymphozyten-Gate im FSC / SSC-Diagramm ("entspanntes Gate"). Durch diese Strategie konnten wir hochaktivierte antigenspezifische CD8-T-Zellen in S-G2/M einschließen, die normalerweise von aktuellen Gating-Strategien übersehen werden, da diese Zellen einen hohen FSC-A und SSC-A aufweisen. Zusammenfassend stellt die Datenanalyse einen kritischen Teil der Methode dar, der essentiell ist, um einen sensitiven Nachweis von aktivierten/proliferierenden antigenspezifischen T-Zellen zu erhalten.
Die Methode verhindert die Möglichkeit, kritische T-Zell-Daten in frühen Phasen der Immunantwort zu verpassen und eröffnet neue Perspektiven für das T-Zell-Immun-Monitoring. Eine zukünftige Verbesserung könnte darin bestehen, eine Färbung für Phospho-Histon 3 aufzunehmen, die eine Unterscheidung zwischenG2 undM21ermöglichen würde. Eine aktuelle Einschränkung besteht darin, dass Zellen fixiert und permeabilisiert werden müssen, um für den Kernmarker Ki67 zu färben. Daher können Zellen nicht für andere Arten von Analysen wie Sortierung und anschließende Funktionsanalyse verwendet werden. Darüber hinaus stören DNA-Farbstoffe, einschließlich Hoechst, in der Regel die genomische DNA-Analyse und sind für diese Art der Bewertung nicht geeignet. Die Identifizierung von Membranmarkern, die mit verschiedenen Zellzyklusphasen korrelieren und auf lebenden Zellen gefärbt werden können, könnte diese Einschränkung überwinden. Zusammenfassend lässt sich sagen, dass die Methode ein großes Potenzial für die Bewertung von aktivierten /proliferierenden T-Zellen in verschiedenen Kontexten wie Impfung, Infektion, immunvermittelten Erkrankungen und Immuntherapie hat.
Disclosures
A. Folgori und S. Capone sind Mitarbeiter von Reithera Srl. A. Nicosia wird auf der Patentanmeldung WO 2005071093 (A3) "Schimpansen-Adenovirus-Impfstoffträger" zum Erfinder ernannt. Die anderen Autoren haben nichts zu verraten.
Acknowledgments
Diese Arbeit wurde von Reithera, vom MIUR-Projekt 2017K55HLC_006 und von 5 × 1000 Zuschuss der Associazione Italiana Ricerca sul Cancro (AIRC) unterstützt. Das folgende Tetramer wurde durch die NIH Tetramer Facility erhalten: APC-konjugiertes H-2K (d) HIV-Gag 197-205 AMQMLKETI.
Materials
| Name | Company | Catalog Number | Comments |
| 1-200 μL universal fit bulk packed pipet tips | Corning | CLS4866-1000EA | |
| 2.4G2 anti-FcγR mAb | BD | 553141 | 10 μg/ml final concentration |
| 5 ml syringe plunger | BD Emerald | 307733 | |
| 15 ml conical tubes | MercK Millipore | SBHA025SB | |
| 60 mm TC-treated Cell Culture Dish | Falcon | 353002 | |
| 70 μm cell strainer | Falcon | 352097 | |
| 96-well Clear Round Bottom TC-treated Culture Microplate | Falcon | 353077 | |
| Anti-Rat/Hamster Ig,k/Negative Control Compensation Particles | BD- Bioscience | 552845 | |
| Beta-mercaptoethanol | Sigma | M3148 | |
| Bovine Serum Albumin | Sigma | A07030 | |
| BUV805 Rat Anti-Mouse CD8a | BD- Bioscience | 564920 | 4 μg/ml final concentration |
| Dulbecco's Phosphate Buffer Saline w/o Calcium w/o Magnesium | Euroclone | ECB4004L | |
| Eppendorf Safe-Lock Tubes, 1.5 mL | Eppendorf | 30120159 | |
| Ethanol | Sigma | 34852-1L-M | |
| Ethylenediaminetetraacetic Acid Disodium Salt solution (EDTA) | Sigma | E7889 | |
| Fetal Bovine Serum | Corning | 35-079-CV | |
| Filcon, Sterile, Syringe-Type 70 μm | Falcon | 352350 | |
| Fixable Viability Dye eFluor 780 | eBioscience | 65-0865-14 | 1:1000 final concentration |
| Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | 00-5523-00 | This Set contains fixation/permeabilization concentrate and diluent, and permeabilization buffer 10x |
| H-2k(d) AMQMLKETI allophycocyanin (APC)-labelled tetramer | provided by NIH Tetramer Core Facility | 6 μg/ml final concentration | |
| H-2k(d) AMQMLKETI phycoerythrine (PE) labelled pentamer | Proimmune | F176-2A-E - 176 | 10 μL / sample |
| Hoechst 33342, Trihydrochloride, Trihydrate - 10 mg/mL Solution in Water | ThermoFisher | H3570 | |
| Ki-67 Monoclonal Antibody (SolA15), FITC | eBioscience | 11-5698-82 | 5 μg/ml final concentration |
| L-Glutamine 100X (200 mM) | Euroclone | ECB3000D | |
| Millex-HA Filters 0,45 µm | BD | 340606 | |
| Penicillin/Streptomycin 100X | Euroclone | ECB3001D | |
| PE/Cyanine7 anti-mouse CD62L Antibody | Biolegend | 104418 | 0.2 μg/ml final concentration |
| PerCP-Cy™5.5 Hamster Anti-Mouse CD3e | BD- Bioscience | 551163 | 4.4 μg/ml final concentration |
| Red Blood Cell Lysis Buffer | Sigma | R7757 | |
| Round-Bottom Polystyrene Tubes, 5 mL | Falcon | 352058 | |
| RPMI 1640 Medium without L-Glutamine with Phenol Red | Euroclone | ECB9006L | |
| Software package for analyzing flow cytometry data | FlowJo | v.10 | |
| Software for acquisition of samples at flowcytometer | BD FACSDiva | v 6.2 | |
| Trypan Blue Solution | Euroclone | ECM0990D |
References
- Castellino, F., et al. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature. 440 (7086), 890-895 (2006).
- Zhang, N., Bevan, M. J. CD8(+) T cells: foot soldiers of the immune system. Immunity. 35 (2), 161-168 (2011).
- Bajénoff, M., et al. Highways, byways and breadcrumbs: directing lymphocyte traffic in the lymph node. Trends Immunology. 28 (8), 346-352 (2007).
- Bevan, M. J., Fink, P. J. The CD8 response on autopilot. Nature Immunology. 2 (5), 381-382 (2001).
- Van Stipdonk, M. J., Lemmens, E. E., Schoenberger, S. P. Naïve CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nature Immunology. 2 (5), 423-429 (2001).
- Kaech, S. M., Wherry, E. J., Ahmed, R. Effector and memory T-cell differentiation: implications for vaccine development. Nature Review Immunology. 2 (4), 251-262 (2002).
- Beverley, P. C. Primer: making sense of T-cell memory. Nature Clinical Practice Rheumatology. 4 (1), 43-49 (2008).
- Parretta, E., et al. CD8 cell division maintaining cytotoxic memory occurs predominantly in the bone marrow. Journal of Immunology. 174 (12), 7654-7664 (2005).
- Di Rosa, F. Maintenance of memory T cells in the bone marrow: survival or homeostatic proliferation. Nature Review Immunology. 16 (4), 271 (2016).
- Di Rosa, F. Two niches in the bone marrow: a hypothesis on life-long T cell memory. Trends in Immunology. 37 (8), 503-512 (2016).
- Di Rosa, F. Commentary: Memory CD8(+) T cells colocalize with IL-7(+) stromal cells in bone marrow and rest in terms of proliferation and transcription. Frontiers in Immunology. 7, 102 (2016).
- Simonetti, S., et al. Antigen-specific CD8 T cells in cell cycle circulate in the blood after vaccination. Scandinavian Journal of Immunology. 89 (2), 12735 (2019).
- Wilson, A., et al. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes & Development. 18 (22), 2747-2763 (2004).
- Hirche, C., et al. Systemic virus infections differentially modulate cell cycle state and functionality of long-term hematopoietic stem cells in vivo. Cell Report. 19 (11), 2345-2356 (2017).
- Colloca, S., et al. Vaccine vectors derived from a large collection of simian adenoviruses induce potent cellular immunity across multiple species. Science Translational Medicine. 4 (115), (2012).
- Di Lullo, G., et al. Marker gene swapping facilitates recombinant Modified Vaccinia Virus Ankara production by host-range selection. Journal of Virological Methods. 156 (1-2), 37-43 (2009).
- Di Lullo, G., et al. The combination of marker gene swapping and fluorescence-activated cell sorting improves the efficiency of recombinant modified vaccinia virus Ankara vaccine production for human use. Journal of Virological Methods. 163 (2), 195-204 (2010).
- Mouse phenotype. , Available from: https://www.mousephenotype.org/data/secondaryproject/3i (2020).
- Yoon, H., Kim, T. S., Braciale, T. J. The cell cycle time of CD8+ T cells responding in vivo is controlled by the type of antigenic stimulus. PLoS One. 5 (11), 15423 (2010).
- Pauklin, S., Vallier, L. The cell-cycle state of stem cells determines cell fate propensity. Cell. 155 (1), 135-147 (2013).
- Vignon, C., et al. Flow cytometric quantification of all phases of the cell cycle and apoptosis in a two-color fluorescence plot. PLoS One. 8 (7), 68425 (2013).
Tags
Immunologie und Infektion Ausgabe 167 Antigen-spezifische CD8-T-Zellen Zellzyklus Ki67 DNA-Farbstoff Durchflusszytometrie Milz Lymphknoten MausErratum
Formal Correction: Erratum: A DNA/Ki67-Based Flow Cytometry Assay for Cell Cycle Analysis of Antigen-Specific CD8 T Cells in Vaccinated Mice
Posted by JoVE Editors on 11/03/2021.
Citeable Link.
An erratum was issued for: A DNA/Ki67-Based Flow Cytometry Assay for Cell Cycle Analysis of Antigen-Specific CD8 T Cells in Vaccinated Mice. The Authors section and a figure were updated.
The authors section was updated from:
Sonia Simonetti*1,2, Ambra Natalini*1,2, Giovanna Peruzzi3, Alfredo Nicosia4, Antonella Folgori5, Stefania Capone5, Angela Santoni2, Francesca Di Rosa1
1Institute of Molecular Biology and Pathology, National Research Council of Italy (CNR),
2Department of Molecular Medicine, University of Rome “Sapienza”,
3Center for Life Nano Science, Istituto Italiano di Tecnologia,
4Department of Molecular Medicine and Medical Biotechnology, University of Naples Federico II,
5Reithera Srl
* These authors contributed equally
To:
Sonia Simonetti*1,2, Ambra Natalini*1,2, Giovanna Peruzzi3, Alfredo Nicosia4, Antonella Folgori5, Stefania Capone5, Angela Santoni2,6, Francesca Di Rosa1
1Institute of Molecular Biology and Pathology, National Research Council of Italy (CNR),
2Department of Molecular Medicine, University of Rome “Sapienza”,
3Center for Life Nano Science, Istituto Italiano di Tecnologia,
4Department of Molecular Medicine and Medical Biotechnology, University of Naples Federico II,
5Reithera Srl
6IRCCS, Neuromed
* These authors contributed equally
Figure 1 was updated from:
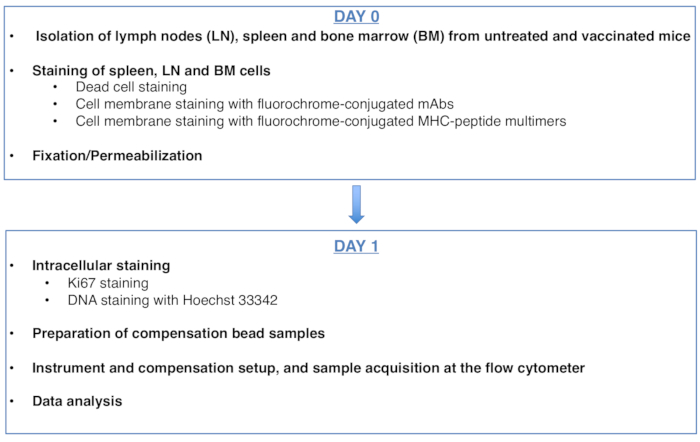
Figure 1: Scheme of the protocol for cell cycle analysis of antigen-specific CD8 T cells. Please click here to view a larger version of this figure.
To:
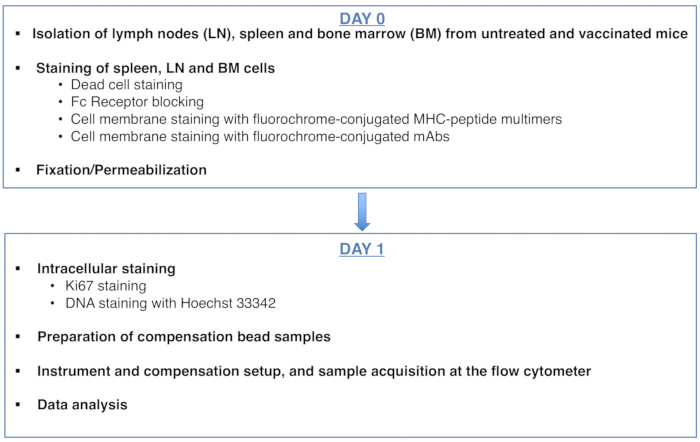
Figure 1: Scheme of the protocol for cell cycle analysis of antigen-specific CD8 T cells. Please click here to view a larger version of this figure.
