

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
La expansión clonal es una característica clave de la respuesta de las células T específicas del antígeno. Sin embargo, el ciclo celular de las células T que responden a los antígenos ha sido poco investigado, en parte debido a limitaciones técnicas. Describimos un método citométrico de flujo para analizar células T CD8 específicas de antígeno en expansión clonal en bazo y ganglios linfáticos de ratones vacunados.
Abstract
El ciclo celular de las células T específicas de antígeno in vivo se ha examinado mediante el uso de algunos métodos, todos los cuales poseen algunas limitaciones. La bromodeoxiuridina (BrdU) marca las células que están en fase S o recientemente completadas, y el éster de succinimidil carboxifluoresceína (CFSE) detecta las células hijas después de la división. Sin embargo, estos colorantes no permiten la identificación de la fase del ciclo celular en el momento del análisis. Un enfoque alternativo es explotar Ki67, un marcador que es altamente expresado por las células en todas las fases del ciclo celular excepto la fase quiescente G0. Desafortunadamente, Ki67 no permite una mayor diferenciación, ya que no separa las células en fase S que están comprometidas con la mitosis de las de G1 que pueden permanecer en estafase, proceder al ciclo o pasar a G0.
Aquí, describimos un método citométrico de flujo para capturar una "instantánea" de las células T en diferentes fases del ciclo celular en órganos linfoides secundarios de ratón. El método combina Ki67 y tinción de ADN con tinción de complejo mayor de histocompatibilidad (MHC)-péptido-multímero y una innovadora estrategia de gating, lo que nos permite diferenciar con éxito entre células T CD8 específicas de antígeno en G0,en G1 y en fases S-G2/M del ciclo celular en el bazo y drenaje de ganglios linfáticos de ratones después de la vacunación con vectores virales portadores del modelo de antígeno mordaza del virus de la inmunodeficiencia humana (VIH)-1.
Los pasos críticos del método fueron la elección del colorante de ADN y la estrategia de cierre para aumentar la sensibilidad del ensayo e incluir células T específicas de antígeno altamente activadas / proliferantes que se habrían perdido según los criterios de análisis actuales. El colorante de ADN, Hoechst 33342, nos permitió obtener una discriminación de alta calidad de los picos de ADN G0/ G1 y G2/ M, al tiempo que preserva la membrana y la tinción intracelular. El método tiene un gran potencial para aumentar el conocimiento sobre la respuesta de las células T in vivo y mejorar el análisis de inmunomonitorización.
Introduction
Las células T naïve experimentan expansión clonal y diferenciación tras el cebado de antígenos. Las células T diferenciadas muestran funciones efectoras que son esenciales para la eliminación de antígenos y para el mantenimiento de la memoria específica del antígeno, que es clave para una protección duradera. Durante los primeros pasos de la respuesta primaria, la interacción ingenua de las células T con las células presentadoras de antígenos (APC) dentro de nichos especializados en órganos linfoides es crítica para inducir la enorme proliferación de células T que caracteriza la fase de expansión clonal1,2,3. La interacción célula t-APC está finamente regulada por la concentración y persistencia de antígeno, señales coestimuladoras y factores solubles (citoquinas y quimiocinas) que influyen en la cantidad y calidad de la progenie clonal de células T4,5,6,7.
A pesar de los estudios intensivos de la expansión clonal de las células T, todavía no se sabe si las células T preparadas con antígenos completan todo su ciclo celular en el sitio de reconocimiento del antígeno, o si migran a otros órganos durante la progresión del ciclo celular. Esta falta de conocimiento se debe a las propiedades de las herramientas disponibles para el análisis del ciclo celular. Estos incluyen anticuerpos monoclonales (mAbs) específicos para el marcador nuclear, Ki67, y colorantes celulares que identifican células que han sufrido la fase S del ciclo celular (por ejemplo, bromodeoxiuridina (BrdU)) o discriminan entre las células hijas y sus antepasados (por ejemplo, éster de carboxifluoresceína succinimidil (CFSE)).
Sin embargo, los colorantes de marcado celular, como CFSE y BrdU, no permiten determinar si las células que se encuentran en un órgano en particular proliferaron localmente o más bien migraron a este sitio después de la división8,9. Además, la proteína intranuclear, Ki67, solo es capaz de distinguir las células en G0 (células Ki67-negativas) de las de cualquier otra fase del ciclo celular (células Ki67-positivas). Por lo tanto, el análisis de Ki67 no distingue las células en proliferación activa (es decir, en S, G2o M) de las de G1,que pueden progresar rápidamente a la división o permanecer durante largos períodos en G1 o volver a la inactividad10,11.
Aquí, describimos un nuevo método citométrico de flujo para el análisis del ciclo celular de las células T CD812 específicas del antígeno del bazo y los ganglios linfáticos (LN) de ratones vacunados(Figura 1). El método explota una combinación de Ki67 y tinción de ADN que se utilizó previamente para analizar el ciclo celular de las células hematopoyéticas de médula ósea de ratón(BM) 13,14. Aquí, aplicamos con éxito Ki67 más tinción de ADN, junto con la innovadora estrategia de gating12recientemente publicada, al análisis de la expansión clonal de células T CD8. Pudimos discriminar claramente entre las células T CD8 específicas del antígeno en G0,en G1y en las fases S-G2/ M en el bazo y las LN drenantes de ratones vacunados.
Protocol
Los ratones fueron alojados en Plaisant Animal Facility, y el trabajo se realizó bajo la autorización del Ministerio de Salud italiano número 1065/2015-PR. El protocolo siguió las directrices de cuidado de los animales de acuerdo con las leyes y políticas nacionales e internacionales (Directiva ue 2010/63/UE; Decreto Legislativo italiano 26/2014).
1. Preparación del medio y solución de tinción
- Prepare el medio completo: medio del Roswell Park Memorial Institute (RPMI) con 2 mM de glutamina, 100 U/mL de penicilina/estreptomicina, 50 μM de beta-mercaptoetanol y 10% de volumen/volumen (v/v) de suero fetal bovino (FBS)
- Prepare el tampón de tinción: Solución salina tamponada con fosfato sin Ca2+/ Mg2 + (PBS) con 1% de peso / volumen (p / v) albúmina sérica bovina (BSA) y 2 mM de sal disódica de ácido etilendiaminotetraacético (EDTA)
2. Tratamiento con ratones
- Ratones Balb/c hembra de 7-8 semanas de edad mediante inyección intramuscular (i.m.) en los cuádriceps del virus de la inmunodeficiencia humana (VIH)-1-gag-expressing-chimpanzee adenoviral vector (ChAd3-gag) con una dosis de 107 partículas virales.
- A los 1-4 meses después del cebado, aumente una vez a los ratones mediante i.m. inyección del virus de Ankara modificado que expresa la vaccinia que expresa la mordaza del VIH-1 (MVA-gag) con una dosis de 106 unidades formadoras de placa.
- En el día 3 después del impulso, sacrifique a los ratones estimulados por dislocación cervical y analícelos en paralelo con los ratones no tratados.
- Cosecha los LNs drenando los cuádriceps (ilíaco, poplíteo e inguinal) y los bazos de ratones potenciados y no tratados. Además, recoja el BM de las dos patas traseras de ratones no tratados y use este BM para la configuración del citómetro de flujo y como control positivo para el análisis del ciclo celular(Figura 2).
NOTA: Genere vectores ChAd3-gag y MVA-gag como se describió anteriormente12,15,16,17.
3. Aislamiento de células LN, bazo y BM drenantes
- Aislamiento del bazo y las células LN
- Coloque 5 ml de medio completo en cada uno de los dos tubos de 15 ml y manténgalos en hielo, listos para que se recolecten los órganos.
- Sacrificar un ratón adulto por luxación cervical.
- Coloque el ratón sobre su espalda y esterilice la superficie de la piel con etanol al 70% v/v.
- Para recolectar LN inguinales, haga una incisión longitudinal de ~ 1 cm en el abdomen con tijeras y estire la incisión con las pinzas.
- Visualice las LN inguinales en la superficie interna de la piel y recójalas con las pinzas. Coloque las LN inguinales en uno de los dos tubos de 15 ml preparados en el paso 3.1.1.
- Para recoger el bazo, haga una incisión peritoneal con tijeras y retire el bazo. Después de cortar el tejido conectivo circundante, coloque el bazo en el segundo tubo de 15 ml preparado en el paso 3.1.1.
- Para recolectar LN ilíacas, mueva los intestinos a un lado y visualice las LN ilíacas cerca de la vena cava inferior, y luego recójalas usando los fórceps. Coloque las LN ilíacas en el mismo tubo que contiene las LN inguinales.
NOTA: Para obtener suficientes células LN para la tinción (ver sección 4), a menudo es necesario agrupar LN poplíteas, inguinales e ilíacas de un ratón. Todos estos LN están drenando los cuádriceps (el sitio de la vacunación i.m. Este protocolo utiliza solo un tubo de 15 ml de LN agrupados. - Para recolectar LN poplíteas, agarre la piel de las patas traseras y tire suavemente de ella hacia abajo para descubrir los músculos. Luego, inserte los fórceps entre los músculos debajo de la articulación de la rodilla y recoja los LN poplíteos. Coloque los LN poplíteos en el mismo tubo que contiene LN inguinales e ilíacos.
NOTA: Véase la nota posterior al punto 3.1.7. - Coloque el bazo en un colador de células de 70 μm dentro de un plato de cultivo de 60 mm lleno de 5 ml de medio completo. Usando un émbolo de jeringa de 5 ml, triture suavemente el órgano hasta su desagregación completa.
- Retire el colador y transfiera la suspensión celular a un tubo limpio de 15 ml.
- Agregue 5 ml de medio completo al plato de cultivo y lave cuidadosamente el plato y el colador para asegurarse de que se hayan recuperado todas las células. Piscina con el resto de la suspensión de células del bazo en el tubo de 15 ml.
- Para las LN inguinales, ilíacas y poplíteas agrupadas, prepare una suspensión de una sola célula siguiendo un procedimiento similar al utilizado en los pasos 3.1.9 a 3.1.11 para el bazo.
- Células centrífugas a 400 × g durante 10 min a 4 °C. Deseche el sobrenadante y vuelva a suspender los gránulos celulares en PBS.
- Cuente las células con una cámara de Neubauer utilizando un tampón de lisis de glóbulos rojos y 0.04% v / v de azul de tripano en PBS.
- Aislamiento de células BM
- Coloque 5 ml de medio completo en un tubo de 15 ml y manténgalo en hielo, listo para la recolección de patas traseras.
- Sacrificar un ratón adulto por luxación cervical.
- Esterilizar la superficie de la piel con etanol al 70% v/v.
- Haga una incisión transversal de ~ 1 cm en la piel ventral con tijeras, agarre firmemente la piel a ambos lados del corte y tire suavemente hacia abajo para descubrir los músculos de las patas traseras.
- Para eliminar la piel de la parte posterior de las patas traseras, manteniendo al ratón en posición supina, coloque la abrazadera debajo de la rodilla y tire hacia arriba para exponer los músculos.
- Corte los huesos en las dos extremidades de una pierna trasera: la articulación pélvica/ cadera y el tobillo.
- Transfiera ambas patas traseras al tubo de 15 ml preparado en el paso 3.2.1. Mantenga el tubo sobre hielo.
- Tome las patas traseras del tubo de 15 ml y transfiéralas al papel de seda. Corte las patas traseras justo debajo de la articulación de la rodilla para eliminar la tibia. Diseccionar el fémur y la tibia de los músculos circundantes, eliminar el exceso de tejido con tijeras y mojar el papel de seda.
- Corte los extremos óseos con tijeras para exponer el eje interior de la médula. Inserte la tibia y el fémur en el tubo de extracción BM (ver preparación en 3.2.9.1-3.2.9.218), con el extremo más ancho en la parte inferior.
- Corte una punta de pipeta de 200 μL en la línea justo encima del final de la punta y en la línea de 100 μL.
- Coloque la parte media en la sección superior más grande de la punta y colóquela en un tubo de microfuge de 1,5 ml.
- Gire el tubo de extracción BM a 800 × g durante 1 min.
- Deseche el hueso y resuspenda vigorosamente el pellet en 1 ml de medio completo para eliminar cualquier racimo. Filtre la suspensión celular a través de un filtro de 70 μm colocado en la parte superior de un tubo de 15 ml.
- Lave el tubo de extracción BM dos veces con 1 ml de medio completo cada vez. Filtrar a través de un filtro de 70 μm, y agrupar el volumen con el resto de la suspensión celular obtenida en el paso 3.2.11.
NOTA: Un solo tubo de 15 ml contendrá células de ambas patas traseras de un ratón. - Células centrífugas a 400 × g durante 10 min a 4 °C. Deseche el sobrenadante y vuelva a suspender el pellet celular en PBS.
- Cuente las células con una cámara de Neubauer utilizando un tampón de lisis de glóbulos rojos y 0.04% v / v de azul de tripano en PBS.
4. Tinción de células del bazo, LN y BM
- Divida las muestras de células que se teñirán en 3 subgrupos: muestras de células para compensación,incluidas las células BM de ratones no tratados que se tiñen solo con Hoechst 33342 (en adelante, Hoechst) y células de bazo de ratones no tratados que se utilizarán para preparar una mezcla de células muertas / vivas para la compensación de colorantes de células muertas; control positivo para el análisis del ciclo celular,que consiste en una muestra de BM de ratones no tratados; y muestras experimentales que contienen muestras de bazo y LN de ratones no tratados y vacunados.
NOTA: Asegúrese de que haya suficientes células del bazo y LN para el análisis de un número suficiente de células T CD8 específicas de gag. A menudo es necesario utilizar células del bazo agrupadas y células LN agrupadas de 3 ratones vacunados y teñir dos o más muestras idénticas de células agrupadas, cada una de las cuales contiene 3 × 106 células. Combine muestras idénticas en el paso de tinción de Hoechst. Del mismo modo, tiñe las células del bazo agrupadas y las células LN de 3 ratones no tratados, y fusiona muestras idénticas al final. Apartar una muestra no teñida de células del bazo de un ratón no tratado para utilizarla para la configuración del instrumento y la compensación. - Prepare la mezcla de células muertas / vivas para la compensación de colorantes de células muertas (esta mezcla de células se teñirá solo con el tinte de células muertas).
- Calentar un baño de agua a 65 °C.
- Tome una alícuota de células del bazo (~ 3 × 106).
- Transfiera la suspensión celular a un tubo de microfuge, colóquela en el baño de agua a 65 ° C durante 5 minutos y luego colóquela inmediatamente en hielo durante 10 minutos.
- Mezcle las células muertas por calor con células vivas del bazo (~ 3 × 106) en una proporción de 1: 1, y transfiera la mitad de la mezcla a una placa inferior de 96 bien redonda (~ 3 × 106 células / pozo para el control de tinción de células muertas).
- Tinción de células muertas de muestras experimentales, control positivo para el análisis del ciclo celular y mezcla de células muertas / vivas
- Transfiera el bazo, LN, células BM (3 × 106 células/pozo) y la mezcla de células muertas/vivas (sección 4.2) a una placa de fondo redondo de 96 pocillos, de acuerdo con el esquema de tinción (paso 4.1), y centrífuga a 400 × g durante 3 min a 4 °C.
- Resuspenda cada gránulo celular en 50 μL de colorante de células muertas diluido en PBS, y resuspenda mediante pipeteo hacia arriba y hacia abajo 3 veces inmediatamente.
- Incubar durante 30 min a 4 °C, protegido de la luz.
- Lave las células 2 veces con tampón de tinción; la primera vez con 200 μL y la segunda vez con 250 μL. Para cada centrifugadora de lavado, la placa a 400 × g durante 3 min a 4 °C.
- Deseche el sobrenadante y vuelva a suspender el gránulo celular en 20 μL de PBS.
- Tinción celular de membrana con complejos principales de histocompatibilidad (MHC)-péptidos multímeros y mAbs.
- Teniendo en cuenta los volúmenes necesarios de acuerdo con el esquema de tinción (Configuración del citómetro de flujo, Tabla 1),prepare los siguientes reactivos:
- Diluir mAb 2.4G2 en el tampón de tinción de acuerdo con la dilución apropiada (ver Tabla de Materiales); para cada muestra a teñir, utilizar 10 μL de esta dilución.
NOTA: 2.4G2 mAb bloquea la unión no específica de antígenos de las inmunoglobulinas a los receptores FcγIII y FcγII. - Diluir el tetrámero marcado con aloficocianina (TEtr-gag) de Aloficocianina (APC) H-2k(d) en el tampón de tinción para obtener la dilución adecuada (ver Tabla de Materiales); para cada muestra a teñir, utilizar 20 μL de esta dilución.
- Preparar la mezcla de anticuerpos diluyendo mAbs en el tampón de tinción de acuerdo con la dilución apropiada (ver Tabla de Materiales)que se haya determinado previamente en experimentos de titulación; para cada muestra que se va a teñir, utilice 20 μL de esta mezcla de anticuerpos.
NOTA: Aquí, se utilizaron proteína de clorofila de peridinina anti-CD3e (PerCP-Cy5.5) (clon 145-2C11), ultravioleta brillante anti-CD8a (BUV805) (clon 53-6.7) y cianina de ficoeritrina anti-CD62L (PECy7) (clon MEL-14).
- Diluir mAb 2.4G2 en el tampón de tinción de acuerdo con la dilución apropiada (ver Tabla de Materiales); para cada muestra a teñir, utilizar 10 μL de esta dilución.
- Añadir 10 μL de los 2,4G2 mAb previamente diluidos (paso 4.4.1.1) e incubar durante 10 min a 4 °C, protegidos de la luz.
- Añadir 20 μL del APC tetr-gag previamente diluido (paso 4.4.1.2) y 10 μL de pentamero de ficoeritrina (PE) AMQMLKETI H-2k(d) (pent-gag). Incubar durante 15 min a 4 °C, protegido de la luz.
- Añadir 20 μL de la mezcla de anticuerpos preparada previamente (paso 4.4.1.3) e incubar 15 min a 4 °C, protegido de la luz.
NOTA: Por lo tanto, el volumen final es de 80 μL por pozo (paso 4.3.5, pasos 4.4.2 a 4.4.4). - Lavar las células con 200 μL de tampón de tinción. Centrifugar a 400 × g durante 5 min a 4 °C.
- Resuspenda el pellet celular en 250 μL de tampón de tinción y transfiera la suspensión celular a tubos de 5 ml. Añadir 1 ml de tampón de tinción al tubo y centrifugar a 400 × g durante 5 min a 4 °C.
- Tomar la alícuota de células BM (3 × 106 células) (ver lista de muestras celulares, sección 4.1) que se utilizará para compensar el canal de Hoechst (Hoechst 33342 se excita con un láser ultravioleta (ajustes del citómetro de flujo (Tabla 2)), y transferir la suspensión celular a un tubo de 5 ml. Añadir 1 ml de tampón de tinción al tubo y centrifugar 400 × g durante 5 min a 4 °C.
- Teniendo en cuenta los volúmenes necesarios de acuerdo con el esquema de tinción (Configuración del citómetro de flujo, Tabla 1),prepare los siguientes reactivos:
5. Fijación/permeabilización
- Prepare un tampón de fijación / permeabilización fresco diluyendo 1 parte del concentrado de fijación / permeabilización con 3 partes de diluyente de fijación / permeabilización, de acuerdo con las instrucciones del fabricante.
- Deseche el sobrenadante y el vórtice de pulso de las muestras para desagregar completamente el pellet.
- Añadir 1 ml del tampón de fijación/permeabilización recién preparado a cada tubo, incluido un tubo con células del bazo no teñidas (3 x 106, ver lista de muestras celulares, sección 4.1) y vórtice.
- Incubar durante 16 h a 4 °C.
NOTA: El protocolo se puede pausar aquí.
6. Tinción intracelular
- Tinción ki67
- Prepare el tampón de permeabilización fresco 1x diluyendo el tampón de permeabilización 10x con agua destilada, de acuerdo con las instrucciones del fabricante. Antes de su uso, el tampón de permeabilización 1x debe filtrarse a través de un filtro de 0,45 μm para eliminar los agregados.
- Diluir mAb Ki67 isotiocianato de fluoresceína (FITC) (clon SolA15) en tampón de permeabilización 1x (ver Tabla de Materiales),según lo determinado previamente en experimentos de titulación (volumen final de 100 μL por muestra).
- Agregue 3 ml de tampón de permeabilización 1x a cada tubo y centrífuga a 400 × g durante 5 minutos a temperatura ambiente (RT).
- Deseche el sobrenadante y repita el paso 6.1.3.
- Deseche el sobrenadante y vuelva a suspender el pellet celular en 100 μL de mAb Ki67 FITC previamente diluido (paso 6.1.2).
- Incubar durante 30 min en RT, protegido de la luz.
- Lavar las células 2 veces con 4 ml de tampón de permeabilización 1x. Para cada centrífuga de lavado a 400 × g durante 5 min en RT.
- Resuspendir el pellet celular en PBS considerando los siguientes volúmenes: 350 μL de PBS para que las muestras se adquieran directamente en el citómetro de flujo; 250 μL de PBS para las muestras que se incubarán con Hoechst poco antes de la citometría de flujo (sección 6.2).
- Tinción de ADN
- Añadir 250 μL de 4 μg/mL hoechst en PBS a cada muestra (la concentración final de Hoechst es de 2 μg/mL).
NOTA: En caso de que se prepararan dos o más muestras idénticas de 250 μL en PBS, combínelas en este paso y agregue un volumen igual de 4 μg/ml de solución de Hoechst en PBS (la concentración final de Hoechst es de 2 μg/mL). El número de células influye en gran medida en el paso de tinción del ADN. Utilice el mismo número de celda en cada muestra. Tenga en cuenta que incluso un número de células ligeramente reducido (por ejemplo, debido a la pérdida de células en los pasos de lavado anteriores) da como resultado una mayor unión de Hoechst al ADN y una mayor intensidad de Hoechst. - Incubar durante 15 min en RT, protegido de la luz.
- Centrifugar las muestras a 400 × g durante 5 min a RT.
- Resuspend el pellet celular en 350 μL de PBS.
- Añadir 250 μL de 4 μg/mL hoechst en PBS a cada muestra (la concentración final de Hoechst es de 2 μg/mL).
7. Preparación de muestras de cuentas de compensación
- Preparar 5 μL del anticuerpo diluyendo mAb en el tampón de tinción adecuadamente.
NOTA: Para cada mAb conjugado con fluorocromo utilizado en el experimento, prepare su correspondiente muestra de perla de compensación. - Vortex Negative Control y Anti-Rat/Hamster Ig,κ Comp Beads antes de su uso.
- Para cada muestra, introduzca una gota (~20 μL) de CompBeads de Control Negativo y una gota de Anti-Rat/Hamster Ig,k CompBeads.
- Añadir 5 μL del anticuerpo prediluido (paso 7.1) al tubo y canalizar hacia arriba y hacia abajo.
- Incubar durante 15 min a 4 °C, protegido de la luz.
- Lavar las muestras con 2 ml de tampón de tinción. Centrifugar a 400 × g durante 5 min a 4 °C.
- Deseche el sobrenadante y vuelva a suspender el pellet agregando 500 μL de PBS a cada tubo y vórtice.
8. Configuración del instrumento y la compensación y adquisición experimental de muestras en el citómetro de flujo
NOTA: Consulte la configuración del citómetro de flujo (Tabla 2) para la configuración del citómetro.
- Instrumento general y configuración de compensación
- Abra el software para la adquisición de muestras (consulte Tabla de materiales)y cree un nuevo experimento haciendo clic en Nuevo experimento en la sección de la cinta de opciones del espacio de trabajo y seleccionando Nuevo experimento en blanco.
- Haga doble clic en el experimento creado para abrirlo.
- En la ventana Configuración del citómetro, haga clic en Parámetros y seleccione todos los canales (por ejemplo, PE, APC, etc.) utilizados en el panel de tinción, incluidos los parámetros de dispersión directa (FSC) y dispersión lateral (SSC).
- Seleccione la escala lineal como parámetro hoechst desmarcando la escala logarítmica y compruebe el ancho (W) del pulso de voltaje para FCS, SSC y Hoechst.
NOTA: Todos los parámetros se muestran de forma predeterminada en escala logarítmica (log), excepto FSC y SSC que están en escala lineal. Todos los parámetros son analizados por el Área (A) y la Altura (H) del pulso de voltaje. - En la hoja de cálculo global,cree un diagrama de puntos con FSC-A en el eje x y SSC-A en el eje y.
- Ejecute el ejemplo de bazo sin manchas haciendo clic en Adquirir datos en el Panel de adquisición.
- Establezca la configuración FSC y SSC adecuada para visualizar las celdas modificando los valores de voltaje en la sección Parámetros, y cree una puerta para seleccionar todas las celdas que se muestran en el diagrama de puntos FSC-A/SSC-A haciendo clic en Puerta poligonal en la barra de herramientas del espacio de trabajo de la Hoja de cálculoglobal .
- Muestre las celdas cerradas en histogramas con cada parámetro de fluorescencia en el eje x.
- Ejecute muestras de bazo sin teñir y completamente teñidas para ajustar el detector de fluorescencia (PMT) para tener una separación clara entre las señales negativas y positivas de las células teñidas para cada parámetro de fluorescencia.
- Para realizar la configuración de compensación, haga clic en Experimentar en la cinta del espacio de trabajo y, en la sección Configuración de compensación, seleccione Crear controles de compensación. Desmarque Incluir tubo/pozo de control sin manchar y haga clic en Aceptar.
NOTA: Esta operación dará como resultado la creación de un espécimen denominado Controles de compensación y una hoja de cálculo normal que contiene varias hojas correspondientes a cada parámetro seleccionado. - Ejecutar una muestra de cuentas de compensación (ver sección 7); establezca la configuración FSC y SSC adecuada para visualizar las perlas modificando los valores de voltaje y el umbral de adquisición de 5.000 en los parámetros FSC en la ventana Citómetro.
- Ajuste la puerta P1 en la población de cuentas y verifique que los picos positivos y negativos sean visibles en el eje x. Repita esta operación para cada muestra de cuentas de compensación y, finalmente, registre cada archivo de muestra haciendo clic en Registrar datos en el Panel de adquisición (registre al menos 5.000 eventos para cada muestra).
- Para cada muestra de cuentas registrada, establezca las puertas P2 y P3 en los picos positivo y negativo, respectivamente.
- Ejecute las muestras de células para la compensación (consulte los pasos 4.2 y 4.4.7, y las secciones 5 y 6). Modifique los voltajes FSC y SSC y el valor umbral para visualizar las celdas, ajustar la puerta P1 y, finalmente, registrar cada archivo de muestra (registrar al menos 10,000 eventos). Establezca las puertas P2 y P3 en los picos positivo y negativo, respectivamente.
NOTA: Para la compensación del canal de Hoechst, utilice el G0/ G1 como pico negativo (P3) y el G2/ M como positivo (P2). - Haga clic en Experimentar en la sección de la cinta del espacio de trabajo y, en la sección Configuración de compensación, seleccione Calcular compensación.
- Asigne un nombre a la configuración de compensación creada, vincule y guárdela en el experimento actual.
- Adquisición experimental de muestras
- Abra un espécimen haciendo clic en Nuevo espécimen en la barra de herramientas del explorador y cree la estrategia de cierre en la hoja de cálculo global.
NOTA: La estrategia de cierre de la adquisición de muestras es similar a la del análisis de muestras, descrita en la Figura 3 y la sección 9. - Mostrar toda la población de eventos en un histograma con CD3-A en el eje x. Cree una puerta de intervalo para seleccionar solo las celdas CD3 +.
- En el Panel de adquisición, seleccione puerta de almacenamiento como Todos los eventos para muestras LN y Todos los eventos o celdas CD3+ para muestras de bazo.
- Ejecute las muestras experimentales a baja velocidad y, finalmente, grabe todos los archivos asegurándose de recolectar al menos 100-200 células T CD8 específicas de antígeno para cada muestra de los ratones vacunados.
NOTA: El tamaño del archivo de las muestras experimentales suele ser grande (30-120 MB), especialmente cuando la frecuencia de las células T CD8 específicas del antígeno es baja. Por lo tanto, se debe recolectar un gran número de eventos (> 1 ×10 6) para registrar al menos 100-200 células T CD8 específicas de antígenos. Los archivos grandes pueden ralentizar el proceso de análisis de datos posterior. La adquisición de solo células CD3+ en muestras de bazo (consulte el paso 8.2.2 anterior) es útil para mantener el tamaño del archivo más pequeño. - Ejecute y registre el control positivo para el análisis del ciclo celular, es decir, la muestra de BM de ratones no tratados.
- Abra un espécimen haciendo clic en Nuevo espécimen en la barra de herramientas del explorador y cree la estrategia de cierre en la hoja de cálculo global.
9. Análisis de datos
- Abra el software (consulte Tabla de materiales)y cree diferentes grupos correspondientes a los diferentes órganos que se analizarán haciendo clic en Crear grupo en la sección de la cinta del espacio de trabajo (es decir, crear grupo "a-LNs"; "bazo b"; "c-BM").
NOTA: Los grupos recién creados aparecerán en la lista de grupos, mientras que el grupo "Compensación" es generado automáticamente por el software. - Abra la ventana Modificar grupo haciendo doble clic en el nombre del grupo y compruebe que los grupos recién creados estén sincronizados. De lo contrario, inserte una marca de verificación en la función Sincronizado.
- Arrastre cada archivo .fcs en su grupo correspondiente.
- Cree la estrategia de cierre comenzando con el grupo "a-LNs".
- Haga doble clic en la muestra completamente teñida en el grupo para abrir la ventana del gráfico; Los ejes x e y están etiquetados como en los archivos fcs (consulte la configuración del citómetro de flujo, Tabla 2).
- Muestre el total de eventos adquiridos para esta muestra en un diagrama de puntos con ADN-A en el eje x y ADN-W en el eje y.
- Seleccione solo la población de una sola celda haciendo clic en Rectángulo en la sección de la herramienta de cierre de la ventana del gráfico.
NOTA: Las células individuales tienen valores de ADN-A de la siguiente manera: 2N (bajo): entre 2N y 4N (intermedio), o igual a 4N (alto), mientras que los valores de ADN-W son idénticos para todas ellas (paso 1 de la Figura 3). - Haga doble clic en el centro de la puerta rectangular para mostrar celdas individuales en un diagrama de puntos con el parámetro FSC-A en el eje X y el tinte de celda muerta en el eje y.
- Seleccione solo la población de células vivas haciendo clic en Polígono en la sección de herramientas de cierre de la ventana del gráfico. Las células vivas son negativas para el colorante de células muertas (paso 2 de la Figura 3).
- Haga doble clic en el centro de la puerta poligonal para mostrar las celdas en un diagrama de puntos con el parámetro FSC-A en el eje X y el parámetro SSC-A en el eje y.
- Haga clic en Rectánguloy cree una puerta "relajada" para incluir todas las celdas vivas individuales en ese gráfico12 (paso 3 de la Figura 3).
- Haga doble clic en el centro de la puerta "relajada" para mostrar las celdas en un diagrama de puntos con CD3 en el eje X y CD8 en el eje Y.
- Seleccione las celdas CD3+CD8+ haciendo clic en Polígono (paso 4 de la Figura 3).
- Haga doble clic en el centro de la puerta CD3+CD8+ para mostrar las celdas en un diagrama de puntos con Tetr-gag en el eje x y Pent-gag en el eje y.
- Seleccione las células T CD8 específicas del antígeno (positivas tanto para Tetr-gag como para Pent-gag) haciendo clic en Polígono (paso 5 de la Figura 3).
- Haga doble clic en el centro de la puerta específica de gag para mostrar las células en un diagrama de puntos con ADN-A en el eje x y Ki67 en el eje y(Figura 4).
- Seleccione las celdas en las diferentes fases del ciclo celular haciendo clic en Quad en la sección de la herramienta de cierre de la ventana del gráfico.
NOTA: Las células en fase G0 son células bajas Ki67neg-DNA (cuadrante inferior izquierdo); las células en G1 son Ki67pos-DNA bajas (cuadrante superior izquierdo); las células en S-G2/M son Ki67pos-DNA intermedio/alto (cuadrante superior derecho) (Figura 4). - Copie la estrategia de cierre creada en una muestra al grupo correspondiente para aplicar las puertas a todas las muestras del grupo.
- Repita los pasos 9.5 a 9.18 para el "grupo a-LN".
- Verifique que todas las puertas sean apropiadas para cada muestra del grupo "bazo b". Para analizar el ciclo celular entre las células BM (control positivo), haga clic en el centro de la puerta "relajada" para mostrar las células en un diagrama de puntos con ADN-A en el eje X y Ki67 en el eje y.
- Verifique que todas las puertas sean apropiadas para cada muestra de los 3 grupos (es decir, para células de bazo, LN y BM).
NOTA: La puerta de población unicelular (paso 9.7) y la puerta cuádruple para el ciclo celular (paso 9.17) pueden tener diferentes coordenadas de compuerta en diferentes muestras, principalmente debido a las posibles ligeras diferencias de intensidad del colorante hoechst entre muestras (sección 6.2). Por esta razón, podría ser necesario modificar la puerta de población de una sola célula y las puertas cuádruples para el ciclo celular en cada muestra. Esto se hará de la siguiente manera: haga doble clic en el nombre del grupo y elimine la sincronización de las propiedades del grupo. Esta operación permite la modificación de las compuertas en una muestra sin modificar las mismas compuertas en todas las demás muestras del grupo. Después de la eliminación de la sincronización, modifique las puertas cuando sea necesario. - Para visualizar los resultados obtenidos por este análisis, haga clic en Editor de diseño en la sección de la cinta del espacio de trabajo para abrirlo. Arrastre cada puerta de la estrategia de puerta en el panel de ejemplo al editor de diseño y coloque las gráficas de acuerdo con la secuencia de la estrategia de puerta. Si es necesario, cambie el tipo de gráfico haciendo doble clic en el gráfico correspondiente en el diseño y seleccionando el tipo apropiado en la ventana Definición de gráfico.
- Haga clic en el Grupo e itere por funciones en la cinta de diseño para visualizar los resultados obtenidos en cada órgano y comparar diferentes muestras.
Representative Results
Las fases del ciclo celular de las células del bazo, LNs y BM de ratones Balb/c se analizaron utilizando el colorante fluorescente de ADN, Hoechst, y un anti-Ki67 mAb, de acuerdo con el protocolo resumido en la Figura 1. Esta tinción permitió la diferenciación de células en las siguientes fases del ciclo celular: G0 (Ki67neg, con 2N de ADN definido como DNAlow), G1 (Ki67pos, DNAlow) y S-G2/M (Ki67pos, con un contenido de ADN comprendido entre 2N y 4N, o igual a 4N de ADN definido como DNAintermediato/alto).
Primero realizamos análisis del ciclo celular de células BM para reproducir resultados previamente publicados13,14 y luego analizamos las células de interés, es decir, células T CD8. La Figura 2 muestra un ejemplo típico de análisis del ciclo celular de células BM(Figura 2A). El protocolo arrojó un bajo coeficiente de variación (CV) de picos de ADN G0/G1 y G2/M,lo que indica la excelente calidad de la tinción de ADN(Figura 2B,mostrando un ejemplo con CV < 2.5; CV siempre fue < 5 en todos los experimentos).
Luego aplicamos el mismo protocolo a las células T CD8 específicas del antígeno de ratones vacunados. Los ratones BALB/c fueron vacunados contra la mordaza del antígeno del VIH-1 mediante el uso de Chad3-gag para el cebado y MVA-gag para el impulso, ambos diseñados para portar la mordaza del VIH-1. En el día (d) 3 post-boost, analizamos la frecuencia de las células T CD8 específicas de mordaza del bazo y las LN drenantes. Aprovechamos la estrategia de gating recientemente definida para las células T en la fase temprana de la respuesta inmune, que a diferencia de la estrategia convencional, es apropiada para detectar células T CD8 con respuesta a antígenos altamente activados12. Ejecutamos la estrategia novedosa en cinco pasos posteriores. En el paso 1, excluimos los dobletes o agregados por la puerta DNA-A / -W, y en el paso 2, identificamos las células vivas por exclusión de marcadores de células muertas. En el paso 3, identificamos la población de interés utilizando una puerta FSC-A/SSC-A "relajada" no convencional(Figura 3A)en lugar de la puerta linfocitaria estrecha canónica12. Después de la gating en células CD3+CD8+ (paso 4 de la Figura 3A),identificamos células T CD8 específicas de gag mediante el uso de dos multímetros MHC diferentes, es decir, Pent-gag y Tetr-gag (paso 5 de la Figura 3A). Se utilizaron dos multómeros en lugar de uno para mejorar la sensibilidad de la detección de células T CD8 específicas de gag en ratones vacunados, sin aumentar el fondo de tinción en ratones no tratados(Figura 3B y C,paso 5). Por lo tanto, distinguimos con éxito los ratones no tratados (0,00% y 0,00% de células T CD8 específicas de antígeno en LNs y bazo, respectivamente) de ratones vacunados (0,46% y 0,29% de células T CD8 específicas de antígeno en LNs y bazo, respectivamente, Figura 3B y C).
En particular, el protocolo nos permitió tener un fondo extremadamente bajo en la puerta de células T CD8 específica del antígeno de los LN y el bazo de ratones no tratados (generalmente 0.00% y en máximo 0.02%). La comparación de los gráficos FSC-A / SSC-A específicos de gag y no específicos de gag mostró que las células específicas de gag tenían SSC-A y FSC-A altos(Figura 3D),lo que confirma la necesidad de usar una puerta FSC-A / SSC-A "relajada" para capturar estas células. Luego evaluamos los porcentajes de células T CD8 específicas de gag en diferentes fases del ciclo celular(Figura 4A). Encontramos que las células T CD8 específicas de gag en el bazo y aún más en las LN drenantes contenían una alta proporción de células en las fases S-G2/ M en el día 3 después del impulso (18.60% y 33.52%, respectivamente).
Además, encontramos que las células T CD8 específicas de gag en las fases S-G2/ M tenían fsc-A y SSC-A altos, cuando se superponían a las células T CD8 totales del mismo órgano(Figura 4B). La expresión de CD62L por las células T CD8 específicas de gag fue baja, como se esperaba para las células T activadas, a excepción de algunas células en G0 en las LN(Figura 4C). En conjunto, estos resultados confirmaron que la puerta "relajada" (paso 3 de la Figura 3A, B y C)era necesaria para incluir todas las células T CD8 específicas del antígeno proliferante12. El protocolo fue extremadamente valioso para una evaluación "instantánea" de las fases del ciclo celular de las células T CD8 específicas del antígeno en el momento del análisis y de la expresión de CD62L por las células en diferentes fases del ciclo celular.

Figura 1: Esquema del protocolo para el análisis del ciclo celular de células T CD8 específicas de antígeno. Haga clic aquí para ver una versión más grande de esta figura.

Figura 2: Análisis del ciclo celular de células BM. Las células BM de ratones Balb/c no tratados se tiñeron y analizaron mediante citometría de flujo. (A) Ejemplo de estrategia de cierre. Nos fijamos en células individuales en la gráfica DNA-A/-W (izquierda) y posteriormente en células vivas por exclusión de colorante de células muertas (centro). Luego, se utilizó una puerta FSC-A / SSC-A "relajada" para todas las células BM (derecha). (B) Ejemplo de análisis del ciclo celular de células BM (izquierda). Utilizamos una combinación de Ki67 y tinción de ADN para identificar células en las siguientes fases del ciclo celular: G0 (cuadrante inferior izquierdo, células Ki67neg-DNAlow), G1 (cuadrante superior izquierdo, Ki67pos-DNAlow), S-G2/ M (cuadrante superior derecho, Ki67pos-DNAintermediato / alto). Se muestra el control de fluorescencia menos uno (FMO) de Ki67 mAb (centro) y el histograma de ADN (derecha). En la gráfica del histograma de ADN, las puertas izquierda y derecha corresponden al pico de ADN G0/G1 y G2/M, respectivamente, y los números representan los coeficientes de variación (CV) de cada pico. En todas las demás gráficas, los números representan porcentajes de celdas en las puertas indicadas. La figura muestra 1 experimento representativo de 5. En cada experimento, analizamos células BM agrupadas de 3 ratones. Haga clic aquí para ver una versión más grande de esta figura.

Figura 3: Análisis de linfocitos T CD8 específicos de antígenos de LNs y bazo. Los ratones Balb/c se prepararon por vía intramuscular (i.m.) con Chad3-gag y aumentaron i.m. con MVA-gag. En el día 3 después del impulso, las células de LN y bazo drenantes de ratones de control vacunados y no tratados se tiñeron y analizaron mediante citometría de flujo. A) Esquema de la estrategia de cierre en cinco pasos para identificar células individuales (Paso 1); células vivas (Paso 2); linfocitos (Paso 3); Células T CD8 (Paso 4); y células específicas de gag (Paso 5). (B-C) Ejemplo de gráficos: análisis de células de (B) LNs y (C) bazo de ratones no tratados (arriba) y vacunados (abajo). Identificamos células individuales en la gráfica DNA-A/-W en el Paso 1. Luego, en el Paso 2, seleccionamos células vivas por exclusión de colorante de células muertas. En el Paso 3, utilizamos una puerta "relajada" no canónica para los linfocitos. En el Paso 4, identificamos las células T CD8 por su doble expresión de CD3 y CD8. Luego identificamos células específicas de gag y no específicas de gag en el Paso 5, en función de su capacidad para unirse a H-2kd-gag-Pentamer (Pent-gag) y H-2kd-gag-Tetramer (Tetr-gag) marcados con fluorocromo, o no, respectivamente. (D) Perfiles FSC-A/SSC-A de células específicas de mordaza (azul) y no específicas de mordaza (gris) después de la compuerta como se describió anteriormente. Los números representan porcentajes de celdas en las puertas indicadas. La figura muestra 1 experimento representativo de 5. En cada experimento, analizamos el bazo agrupado y las células LN agrupadas de 3 ratones vacunados y 3 ratones no tratados. Haga clic aquí para ver una versión más grande de esta figura.

Figura 4: Análisis del ciclo celular de células T CD8 específicas de antígenos. Los ratones fueron vacunados como en la Figura 3 y el análisis del ciclo celular de las células específicas de la mordaza se realizó en el día 3 post-boost, después de la apertura en 5 pasos como en la Figura 3. (A)Ejemplo de análisis del ciclo celular de células T CD8 específicas de gag de LNs (arriba) y bazo (abajo) de ratones vacunados. Las fases del ciclo celular se identificaron como en la Figura 2B. Los paneles representan celdas en G0,en G1,y en S-G2/M (izquierda) y Fluorescencia Menos Uno (FMO) control de Ki67 mAb (derecha). Los números representan porcentajes de celdas en las puertas indicadas. (B) Diagramas de puntos FSC-A/SSC-A que muestran células T CD8 específicas de gag en fases S-G2/M (en rojo) superpuestas sobre el total de células T CD3+CD8+ (en gris) de LNs (arriba) y bazo (abajo) de ratones vacunados. (C) Histogramas de desplazamiento que muestran la expresión de CD62L por células T CD8 específicas de gag en G0 (verde), en G1 (azul) y en S-G2/ M (rojo) de LNs (arriba) y bazo (abajo) de ratones vacunados. Los ejes y indican el número normalizado de eventos. La figura muestra 1 ejemplo representativo de 5 experimentos independientes con un total de 15 ratones. Haga clic aquí para ver una versión más grande de esta figura.
Material complementario: Ajustes del citómetro de flujo. Haga clic aquí para descargar este archivo.
Discussion
Aunque la expansión clonal de las células T ha sido intensamente estudiada, algunos aspectos siguen siendo desconocidos, principalmente porque las herramientas disponibles para investigarla son pocas y tienen sus propios inconvenientes. Desde esta perspectiva, establecimos un método citométrico de flujo altamente sensible para analizar el ciclo celular de las células T CD8 específicas del antígeno en los primeros momentos después de la vacunación en un modelo de ratón. El protocolo se basa en una combinación de Ki67 y tinción de ADN, que anteriormente se utilizaba para analizar el ciclo celular de las células hematopoyéticas BM en ratones13,14. Para adaptar el protocolo a las células T CD8 específicas del antígeno, tuvimos que considerar algunas cuestiones críticas, incluida la elección del colorante de ADN, las condiciones adecuadas para obtener una tinción de ADN comparable en diferentes muestras y la estrategia de cierre para el análisis de datos.
Muchos colorantes están disponibles para la tinción del ADN, incluyendo yoduro de propidio y 7-aminoactinomicina D; elegimos Hoechst porque era compatible con la tinción de membrana y el protocolo de fijación / permeabilización leve requerido para la tinción de Ki67. Al mismo tiempo, la tinción con Hoechst nos permitió obtener un histograma de ADN de excelente calidad, es decir, los picos de ADN G0/G1 y G2/Mtenían un coeficiente de variación (CV) mucho menor que los picos de ADN generalmente obtenidos con otros colorantes de ADN, por ejemplo, DRAQ519. De hecho, Hoechst puede teñir el ADN incluso en células vivas20.
Se utilizaron algunas estrategias para evitar la fluctuación en la intensidad de Hoechst en diferentes muestras del mismo experimento. La tinción de Hoechst se realizó justo antes de la adquisición de la muestra en el citómetro de flujo para minimizar la disminución de la intensidad del tinte durante el tiempo. Para aquellos interesados en reproducir el protocolo en grandes experimentos con numerosas muestras, recomendamos realizar la tinción de Hoechst en unas pocas muestras a la vez. Otro inconveniente es que la intensidad de Hoechst puede estar fuertemente influenciada por el número de células durante la incubación con el tinte. Por esta razón, recomendamos encarecidamente utilizar siempre el mismo número de células y el mismo volumen por muestra para la tinción de ADN. Si se requiere un alto número de células para la adquisición en el citómetro de flujo, recomendamos preparar dos o más muestras idénticas y luego fusionarlas justo antes del paso de tinción de Hoechst.
Un punto clave del protocolo es la estrategia de cierre para el análisis de datos. Recientemente publicamos una novedosa estrategia para el análisis de células T en los primeros momentos de la respuesta inmune, que nos permitió aumentar la sensibilidad de detección de células T específicas de antígenos12. Aplicamos esta estrategia a los datos que se muestran aquí de la siguiente manera. En primer lugar, excluimos los agregados celulares en la gráfica DNA-A/W. En segundo lugar, después de cerrar las células muertas, utilizamos una puerta de linfocitos bastante grande en la parcela FSC / SSC ("puerta relajada"). Mediante esta estrategia, pudimos incluir células T CD8 específicas de antígeno altamente activadas en S-G2/ M que generalmente se pierden en las estrategias actuales de gating, ya que estas células tienen un alto FSC-A y SSC-A. En resumen, el análisis de datos representa una parte crítica del método, que es esencial para obtener una detección sensible de células T específicas de antígenos activados / proliferantes.
El método evita la posibilidad de que falten datos críticos de células T en las primeras fases de la respuesta inmune y abre nuevas perspectivas para el monitoreo inmunológico de células T. Una mejora futura podría ser incluir la tinción para la fosfo-histona 3 que permitiría la diferenciación entre G2 y M21. Una limitación actual es que las células tienen que ser fijadas y permeabilizadas para teñir para el marcador nuclear, Ki67. Por lo tanto, las células no se pueden utilizar para otros tipos de análisis, como la clasificación y el posterior análisis funcional. Además, los colorantes de ADN, incluido Hoechst, generalmente interfieren con el análisis genómico de ADN y no son adecuados para este tipo de evaluación. La identificación de marcadores de membrana que se correlacionan con diferentes fases del ciclo celular y que pueden teñirse en células vivas podría superar esta limitación. En conclusión, el método tiene un gran potencial para la evaluación de células T activadas/proliferantes en varios contextos como la vacunación, la infección, las enfermedades inmunomediadas y la inmunoterapia.
Disclosures
A. Folgori y S. Capone son empleados de Reithera Srl. A. Nicosia es nombrado inventor en la solicitud de patente WO 2005071093 (A3) "Portadores de la vacuna contra el adenovirus de chimpancé". Los otros autores no tienen nada que revelar.
Acknowledgments
Este trabajo fue apoyado por Reithera, por el proyecto MIUR 2017K55HLC_006, y por 5 × subvención 1000 de la Associazione Italiana Ricerca sul Cancro (AIRC). El siguiente tetrámero se obtuvo a través de la Instalación de Tetrámeros de los NIH: H-2K conjugado con APC (d) mordaza del VIH 197-205 AMQMLKETI.
Materials
| Name | Company | Catalog Number | Comments |
| 1-200 μL universal fit bulk packed pipet tips | Corning | CLS4866-1000EA | |
| 2.4G2 anti-FcγR mAb | BD | 553141 | 10 μg/ml final concentration |
| 5 ml syringe plunger | BD Emerald | 307733 | |
| 15 ml conical tubes | MercK Millipore | SBHA025SB | |
| 60 mm TC-treated Cell Culture Dish | Falcon | 353002 | |
| 70 μm cell strainer | Falcon | 352097 | |
| 96-well Clear Round Bottom TC-treated Culture Microplate | Falcon | 353077 | |
| Anti-Rat/Hamster Ig,k/Negative Control Compensation Particles | BD- Bioscience | 552845 | |
| Beta-mercaptoethanol | Sigma | M3148 | |
| Bovine Serum Albumin | Sigma | A07030 | |
| BUV805 Rat Anti-Mouse CD8a | BD- Bioscience | 564920 | 4 μg/ml final concentration |
| Dulbecco's Phosphate Buffer Saline w/o Calcium w/o Magnesium | Euroclone | ECB4004L | |
| Eppendorf Safe-Lock Tubes, 1.5 mL | Eppendorf | 30120159 | |
| Ethanol | Sigma | 34852-1L-M | |
| Ethylenediaminetetraacetic Acid Disodium Salt solution (EDTA) | Sigma | E7889 | |
| Fetal Bovine Serum | Corning | 35-079-CV | |
| Filcon, Sterile, Syringe-Type 70 μm | Falcon | 352350 | |
| Fixable Viability Dye eFluor 780 | eBioscience | 65-0865-14 | 1:1000 final concentration |
| Foxp3 / Transcription Factor Staining Buffer Set | eBioscience | 00-5523-00 | This Set contains fixation/permeabilization concentrate and diluent, and permeabilization buffer 10x |
| H-2k(d) AMQMLKETI allophycocyanin (APC)-labelled tetramer | provided by NIH Tetramer Core Facility | 6 μg/ml final concentration | |
| H-2k(d) AMQMLKETI phycoerythrine (PE) labelled pentamer | Proimmune | F176-2A-E - 176 | 10 μL / sample |
| Hoechst 33342, Trihydrochloride, Trihydrate - 10 mg/mL Solution in Water | ThermoFisher | H3570 | |
| Ki-67 Monoclonal Antibody (SolA15), FITC | eBioscience | 11-5698-82 | 5 μg/ml final concentration |
| L-Glutamine 100X (200 mM) | Euroclone | ECB3000D | |
| Millex-HA Filters 0,45 µm | BD | 340606 | |
| Penicillin/Streptomycin 100X | Euroclone | ECB3001D | |
| PE/Cyanine7 anti-mouse CD62L Antibody | Biolegend | 104418 | 0.2 μg/ml final concentration |
| PerCP-Cy™5.5 Hamster Anti-Mouse CD3e | BD- Bioscience | 551163 | 4.4 μg/ml final concentration |
| Red Blood Cell Lysis Buffer | Sigma | R7757 | |
| Round-Bottom Polystyrene Tubes, 5 mL | Falcon | 352058 | |
| RPMI 1640 Medium without L-Glutamine with Phenol Red | Euroclone | ECB9006L | |
| Software package for analyzing flow cytometry data | FlowJo | v.10 | |
| Software for acquisition of samples at flowcytometer | BD FACSDiva | v 6.2 | |
| Trypan Blue Solution | Euroclone | ECM0990D |
References
- Castellino, F., et al. Chemokines enhance immunity by guiding naive CD8+ T cells to sites of CD4+ T cell-dendritic cell interaction. Nature. 440 (7086), 890-895 (2006).
- Zhang, N., Bevan, M. J. CD8(+) T cells: foot soldiers of the immune system. Immunity. 35 (2), 161-168 (2011).
- Bajénoff, M., et al. Highways, byways and breadcrumbs: directing lymphocyte traffic in the lymph node. Trends Immunology. 28 (8), 346-352 (2007).
- Bevan, M. J., Fink, P. J. The CD8 response on autopilot. Nature Immunology. 2 (5), 381-382 (2001).
- Van Stipdonk, M. J., Lemmens, E. E., Schoenberger, S. P. Naïve CTLs require a single brief period of antigenic stimulation for clonal expansion and differentiation. Nature Immunology. 2 (5), 423-429 (2001).
- Kaech, S. M., Wherry, E. J., Ahmed, R. Effector and memory T-cell differentiation: implications for vaccine development. Nature Review Immunology. 2 (4), 251-262 (2002).
- Beverley, P. C. Primer: making sense of T-cell memory. Nature Clinical Practice Rheumatology. 4 (1), 43-49 (2008).
- Parretta, E., et al. CD8 cell division maintaining cytotoxic memory occurs predominantly in the bone marrow. Journal of Immunology. 174 (12), 7654-7664 (2005).
- Di Rosa, F. Maintenance of memory T cells in the bone marrow: survival or homeostatic proliferation. Nature Review Immunology. 16 (4), 271 (2016).
- Di Rosa, F. Two niches in the bone marrow: a hypothesis on life-long T cell memory. Trends in Immunology. 37 (8), 503-512 (2016).
- Di Rosa, F. Commentary: Memory CD8(+) T cells colocalize with IL-7(+) stromal cells in bone marrow and rest in terms of proliferation and transcription. Frontiers in Immunology. 7, 102 (2016).
- Simonetti, S., et al. Antigen-specific CD8 T cells in cell cycle circulate in the blood after vaccination. Scandinavian Journal of Immunology. 89 (2), 12735 (2019).
- Wilson, A., et al. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes & Development. 18 (22), 2747-2763 (2004).
- Hirche, C., et al. Systemic virus infections differentially modulate cell cycle state and functionality of long-term hematopoietic stem cells in vivo. Cell Report. 19 (11), 2345-2356 (2017).
- Colloca, S., et al. Vaccine vectors derived from a large collection of simian adenoviruses induce potent cellular immunity across multiple species. Science Translational Medicine. 4 (115), (2012).
- Di Lullo, G., et al. Marker gene swapping facilitates recombinant Modified Vaccinia Virus Ankara production by host-range selection. Journal of Virological Methods. 156 (1-2), 37-43 (2009).
- Di Lullo, G., et al. The combination of marker gene swapping and fluorescence-activated cell sorting improves the efficiency of recombinant modified vaccinia virus Ankara vaccine production for human use. Journal of Virological Methods. 163 (2), 195-204 (2010).
- Mouse phenotype. , Available from: https://www.mousephenotype.org/data/secondaryproject/3i (2020).
- Yoon, H., Kim, T. S., Braciale, T. J. The cell cycle time of CD8+ T cells responding in vivo is controlled by the type of antigenic stimulus. PLoS One. 5 (11), 15423 (2010).
- Pauklin, S., Vallier, L. The cell-cycle state of stem cells determines cell fate propensity. Cell. 155 (1), 135-147 (2013).
- Vignon, C., et al. Flow cytometric quantification of all phases of the cell cycle and apoptosis in a two-color fluorescence plot. PLoS One. 8 (7), 68425 (2013).
Tags
Inmunología e infección Número 167 células T CD8 específicas de antígeno ciclo celular Ki67 colorante de ADN citometría de flujo bazo ganglios linfáticos ratónErratum
Formal Correction: Erratum: A DNA/Ki67-Based Flow Cytometry Assay for Cell Cycle Analysis of Antigen-Specific CD8 T Cells in Vaccinated Mice
Posted by JoVE Editors on 11/03/2021.
Citeable Link.
An erratum was issued for: A DNA/Ki67-Based Flow Cytometry Assay for Cell Cycle Analysis of Antigen-Specific CD8 T Cells in Vaccinated Mice. The Authors section and a figure were updated.
The authors section was updated from:
Sonia Simonetti*1,2, Ambra Natalini*1,2, Giovanna Peruzzi3, Alfredo Nicosia4, Antonella Folgori5, Stefania Capone5, Angela Santoni2, Francesca Di Rosa1
1Institute of Molecular Biology and Pathology, National Research Council of Italy (CNR),
2Department of Molecular Medicine, University of Rome “Sapienza”,
3Center for Life Nano Science, Istituto Italiano di Tecnologia,
4Department of Molecular Medicine and Medical Biotechnology, University of Naples Federico II,
5Reithera Srl
* These authors contributed equally
To:
Sonia Simonetti*1,2, Ambra Natalini*1,2, Giovanna Peruzzi3, Alfredo Nicosia4, Antonella Folgori5, Stefania Capone5, Angela Santoni2,6, Francesca Di Rosa1
1Institute of Molecular Biology and Pathology, National Research Council of Italy (CNR),
2Department of Molecular Medicine, University of Rome “Sapienza”,
3Center for Life Nano Science, Istituto Italiano di Tecnologia,
4Department of Molecular Medicine and Medical Biotechnology, University of Naples Federico II,
5Reithera Srl
6IRCCS, Neuromed
* These authors contributed equally
Figure 1 was updated from:
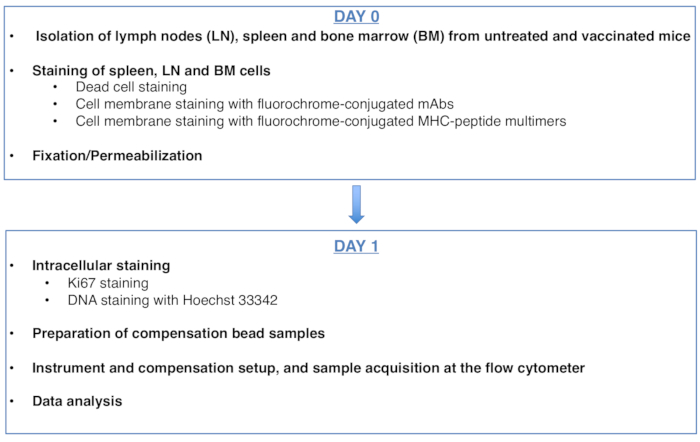
Figure 1: Scheme of the protocol for cell cycle analysis of antigen-specific CD8 T cells. Please click here to view a larger version of this figure.
To:
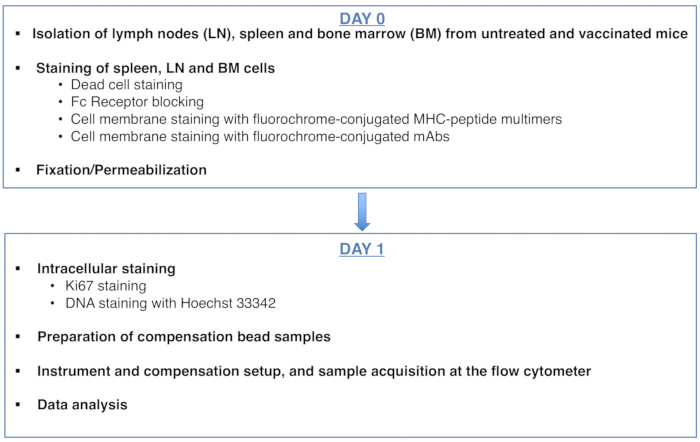
Figure 1: Scheme of the protocol for cell cycle analysis of antigen-specific CD8 T cells. Please click here to view a larger version of this figure.
