

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Este artigo descreve os protocolos de preparação de amostras, redução de dados e análise de dados em estudos de eco de giro de nêutrons (NSE) de membranas lipídicas. A rotulagem criteriosa de deutério de lipídios permite o acesso a diferentes dinâmicas de membrana em comprimento mesoscópico e escalas de tempo, sobre as quais ocorrem processos biológicos vitais.
Abstract
Bicamadas lipídicas formam a principal matriz das membranas celulares e são a principal plataforma de troca de nutrientes, interações proteína-membrana e brotamento viral, entre outros processos celulares vitais. Para uma atividade biológica eficiente, as membranas celulares devem ser rígidas o suficiente para manter a integridade da célula e seus compartimentos, mas fluidas o suficiente para permitir que componentes de membrana, como proteínas e domínios funcionais, difundam e interajam. Este delicado equilíbrio de propriedades de membrana elástica e fluida, e seu impacto na função biológica, necessitam de uma melhor compreensão da dinâmica da membrana coletiva sobre o comprimento mesoscópico e escalas de tempo dos principais processos biológicos, por exemplo, deformações de membrana e eventos de ligação de proteínas. Entre as técnicas que podem efetivamente sondar esse alcance dinâmico está a espectroscopia de nêutrons spin echo (NSE). Combinado com a rotulagem de deutério, o NSE pode ser usado para acessar diretamente flutuações de dobra e espessura, bem como dinâmica mesoscópica de características de membrana selecionadas. Este artigo fornece uma breve descrição da técnica NSE e descreve os procedimentos para a realização de experimentos NSE em membranas liposômicas, incluindo detalhes de esquemas de preparação e deuteração da amostra, juntamente com instruções para coleta e redução de dados. O artigo também introduz métodos de análise de dados usados para extrair parâmetros de membrana chave, como o módulo de rigidez de dobra, o módulo de compressão da área e a viscosidade no plano. Para ilustrar a importância biológica dos estudos da NSE, são discutidos exemplos selecionados de fenômenos de membrana sondados pela NSE, ou seja, o efeito dos aditivos sobre a rigidez de dobra de membrana, o impacto da formação de domínios sobre as flutuações da membrana e a assinatura dinâmica das interações membrana-proteína.
Introduction
A compreensão das membranas celulares e sua função evoluiu notavelmente nas últimas décadas. A visão anterior das membranas celulares como bicamadas lipídicas passivas que definem limites celulares e proteínas de membranadoméstica 1 gradualmente se transformou em um modelo dinâmico no qual bicamadas lipídicas desempenham um papel importante na regulação de processos biológicos vitais, incluindo sinalização celular, troca molecular e função proteica — para citar alguns2,3,4,5,6. Essa constatação de que as membranas celulares são altamente dinâmicas, constantemente submetidas à remodelação e redistribuição molecular, tem incitado explorações científicas além das estruturas de equilíbrio das membranas7,8,9. Assim, várias abordagens foram desenvolvidas para estudar os diversos modos dinâmicos em membranas lipídicas biológicas e bioinspiradas. Até o momento, a maioria desses estudos se concentrou principalmente nos movimentos moleculares difusivos10,11,12,13 e flutuações de forma macroscópica14,15,16, deixando uma lacuna significativa na compreensão da dinâmica da membrana intermediária, ou seja, flutuações coletivas de conjuntos lipídicos consistindo de poucos 10-100s de moléculas lipídicas. Essas dinâmicas ocorrem ao longo de escalas de comprimento de poucas dezenas a poucos 100 Å e ao longo do tempo escalas de sub-ns a algumas centenas de ns (ver Figura 1), referidas aqui como escalas mesoscópicas. É nestas escalas que a atividade biológica chave ocorre no nível de membrana17. Isso inclui o brotamento viral18, gating canal19e interações membrana-proteína20. Também é importante ressaltar que a paisagem energética das proteínas da membrana21,22 mostra que as alterações conformais nas proteínas — necessárias para seu papel regulatório — acontecem ao longo das escalas de tempons 23 de flutuações coletivas de membrana, enfatizando ainda mais a importância da dinâmica mesoscópica na função biológica das membranas celulares e seus analógicos bioinspirados20. Este artigo se concentra nos dois modos dinâmicos mesoscópicos primários em membranas lipídicas, ou seja, flutuações de dobra e flutuações de espessura.
O principal desafio em sondar diretamente esses modos de flutuação é a dificuldade em acessar simultaneamente suas escalas espaciais e temporais usando métodos de espectroscopia padrão. O outro desafio é que as técnicas de contato direto podem impactar as mesmas flutuações que se destinam a medir16. Isso é ainda mais exacerbado pela complexidade composicional e estrutural das membranas biológicas24,25, o que resulta em características de membrana não homogênea, incluindo a formação de domínio lipídudo26,27,28,29,30 e assimetria de membrana 31,32,33- exigindo sondas seletivas para entender a dinâmica de diferentes características da membrana. Felizmente, esses desafios podem ser superados com métodos não invasivos de espectroscopia de nêutrons, como o echo de giro de nêutrons (NSE), que acessam inerentemente o comprimento e as escalas de tempo necessárias, e permitem ainda estudos de características de membrana seletiva sem alterar seu ambiente físico-químico34. De fato, nos últimos anos a espectroscopia NSE evoluiu para uma sonda única e poderosa da dinâmica da membrana coletiva35. Os resultados de estudos da NSE sobre membranas lipídicas produziram novos insights sobre as propriedades mecânica36,37 e viscoelástica38,39 de membranas lipídicas e lançaram uma nova luz sobre seu potencial papel na função biológica40,41.
A técnica de espectroscopia NSE é baseada em um design de instrumento interferométrico, proposto pela primeira vez por Mezei42, usando uma série de giradoras e bobinas magnéticas para controlar a precessão do giro de nêutrons à medida que os nêutrons atravessam o instrumento. O desenho repousa no espelhamento magnético dos elementos do campo magnético em relação à posição da amostra(Figura 1A). Isso implica que, na ausência de troca de energia entre o nêutron e a amostra, o nêutron realiza o mesmo número de precessões de spin, em direções opostas, na primeira e segunda metade do instrumento (observe o π-flipper entre as duas bobinas de pré-cessão). Como resultado, o estado final de rotação do nêutron permanece inalterado em relação ao estado inicial - um fenômeno referido como spin-echo (ver nêutron transparente na Figura 1A). No entanto, quando o nêutron interage energeticamente com a amostra, a troca de energia modifica o número de precessões de spin na segunda metade do instrumento, levando a um estado de rotação final diferente (ver Figura 1A). Isso é detectado experimentalmente como uma perda de polarização, como será mostrado mais tarde neste artigo. Para mais detalhes sobre a técnica NSE, o leitor é encaminhado para documentos técnicos dedicados42,43,44,45.
Aqui, uma descrição simplificada é apresentada para fornecer uma estimativa aproximada do comprimento e escalas de tempo acessíveis com a NSE. As escalas de comprimento são determinadas pela faixa de transferências de vetores de onda alcançáveis, Q = 4π sin φ/λ, onde 2φ é o ângulo de dispersão e λ é o comprimento de onda de nêutrons. Pode-se ver que Q é definido pela faixa de comprimento de onda e a extensão da rotação do segundo braço do espectrômetro (ver Figura 1A). Uma faixa típica de Qnos espectrômetros NSE é ~0,02-2 Å-146,47, e até 0,01-4 Å-1 com atualizações recentes48,49, correspondentes a escalas espaciais de ~1-600 Å. Por outro lado, a escala de tempo acessível é calculada a partir do ângulo total de precessão (ou fase) adquirido pelo nêutron dentro das bobinas de precessão magnética, e é encontrado como50:  . Nesta expressão, t é o tempo de Fourier definido como
. Nesta expressão, t é o tempo de Fourier definido como  , onde está a razão
, onde está a razão  gyrommagnética de nêutrons,
gyrommagnética de nêutrons,  é o comprimento da bobina, e
é o comprimento da bobina, e  é a força do campo magnético da bobina. Vale ressaltar que o tempo de Fourier é uma quantidade estritamente dependente da geometria do instrumento, força do campo magnético e comprimento de onda de nêutrons. Por exemplo, usando nêutrons de comprimento de onda
é a força do campo magnético da bobina. Vale ressaltar que o tempo de Fourier é uma quantidade estritamente dependente da geometria do instrumento, força do campo magnético e comprimento de onda de nêutrons. Por exemplo, usando nêutrons de comprimento de onda  = 8 Å e configurações de instrumentos de = 1,2 m e = 0,4 T, o tempo de Fourier é calculado para ser t ~ 50 ns. Experimentalmente, o tempo de Fourier é sintonizado alterando a corrente nas bobinas de precessão (ou seja, força do campo magnético) ou usando diferentes comprimentos de onda de nêutrons, resultando em escalas típicas de tempo NSE de ~ 1 ps a 100 ns. No entanto, os upgrades recentes nos espectrômetros NSE permitiram o acesso a tempos mais longos do Fourier, até ~400 ns no espectrômetro J-NSE-Phoenix no espectrômetro Heinz Maier-Leibnitz Zentrum51 e no espectrômetro SNS-NSE no Oak Ridge National Lab48, e até ~1.000 ns no espectrômetro IN15 NSE no Institut Laue-Langevin (III)49.
= 8 Å e configurações de instrumentos de = 1,2 m e = 0,4 T, o tempo de Fourier é calculado para ser t ~ 50 ns. Experimentalmente, o tempo de Fourier é sintonizado alterando a corrente nas bobinas de precessão (ou seja, força do campo magnético) ou usando diferentes comprimentos de onda de nêutrons, resultando em escalas típicas de tempo NSE de ~ 1 ps a 100 ns. No entanto, os upgrades recentes nos espectrômetros NSE permitiram o acesso a tempos mais longos do Fourier, até ~400 ns no espectrômetro J-NSE-Phoenix no espectrômetro Heinz Maier-Leibnitz Zentrum51 e no espectrômetro SNS-NSE no Oak Ridge National Lab48, e até ~1.000 ns no espectrômetro IN15 NSE no Institut Laue-Langevin (III)49.
Além do acesso direto ao comprimento e escala de tempo da dinâmica da membrana, a NSE possui as capacidades inerentes à sensibilidade ao isótopo de nêutrons52. Especificamente, a capacidade dos nêutrons de interagir de forma diferente com os isótopos do hidrogênio, o elemento mais abundante em sistemas biológicos, resulta em uma densidade de comprimento de dispersão de nêutrons diferente,34 ou NSLD (o equivalente ao índice óptico de refração50), quando o prótium é substituído por deutério. Isso permite uma abordagem conhecida como variação de contraste, que é comumente usada para destacar características específicas da membrana ou ocultar outras — este último cenário é referido como correspondência de contraste. Uma aplicação frequente de variação/correspondência de contraste é a substituição de água (NSLD = -0,56 × 10-6 Å-2) por água pesada ou D2O (NSLD = 6,6 4 × 10-6 Å-2) para amplificar o sinal de nêutrons de membranas lipídicas protiadas (NSLD ~ 0 × 10-6 Å-2). Essa abordagem é altamente eficaz em estudos de estrutura de membrana porque a penetração de D2O na região do headgroup da membrana permite a determinação precisa das espessuras da membrana (ver Figura 2A, painel esquerdo) e da localização de diferentes subgrupos lipídes quando modelos mais sofisticados são aplicados53,54. Este artigo destaca alguns exemplos sobre o uso de variação de contraste para estudos de dinâmica coletiva em membranas biomiméticas e características de membrana selecionadas.
Aqui, a eficácia da NSE em fornecer insights únicos sobre propriedades de membrana dinâmica e funcional é ilustrada através de exemplos tangíveis de estudos NSE sobre sistemas de membrana lipídica modelo e biologicamente relevantes com ênfase na dinâmica da mesoescala em membranas autônomas, na forma de suspensões liposômicas. Para as medições NSE da dinâmica da membrana in-plane, o leitor é encaminhado para publicações dedicadas sobre espectroscopia de spin-echo de nêutrons de incidência de pastagem (GINSES)55,56 e outros estudos de pilhas de membrana multilamelaralinhadas 57,58,59,60.
Para simplificar, este artigo destaca três diferentes esquemas de deuteração de membrana ilustrados em uma formação de domínio bem estudada, ou separação de fase, sistema de bicamadas lipídicas de misturas 1,2-dimyristoyl-sn-glycero-3-fosphocholina (DMPC) e 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) misturas61,62. Os dois lipídios são caracterizados por uma incompatibilidade em seu comprimento da cadeia de hidrocarbonetos (14 carbonos/cauda em DMPC vs 18 carbonos/cauda em DSPC) e sua temperatura de transição gel-fluido (Tm, DMPC = 23 °C vs Tm, DSPC = 55 °C). Isso resulta na separação lateral de fases nas membranas DMPC:DSPC a temperaturas entre as temperaturas de transição superior e inferior da mistura63. Os esquemas de deuteração aqui considerados são escolhidos para demonstrar os diferentes modos dinâmicos acessíveis nas medidas NSE em membranas lipoesômicas, ou seja, flutuações de dobra, flutuações de espessura e flutuações seletivas de dobra/espessura dos domínios laterais. Todas as composições lipídicas são relatadas para bicamadas DMPC:DSPC preparadas a uma fração toupeira de 70:30, utilizando variantes protiadas e semeadas comercialmente disponíveis de DMPC e DSPC. Todas as etapas de preparação da amostra são baseadas em 4 mL de suspensão liposômica, em D2O, com concentração lipídica de 50 mg/mL, para uma massa lipídica total de Mtot = 200 mgs por amostra.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Esquema de desauteização necessário para o experimento
- Para medir a flutuação de dobra, faça lipossomos totalmente protiados em D2O (D 99,9%) ou D2O-buffer (por exemplo, tampão fosfato preparado com D2O em vez de H2O). Use DMPC totalmente protiado (C36H72NO8P) e DSPC (C44H88NO8P) com
 133,4 mgs, onde XDMPC e XDSPC são as frações moles de DMPC e DSPC, aqui definidas para 0,7 e 0,3, respectivamente, e MwDMPC e MwDSPC são os pesos molares dados por 677,9 g/mol e 790,1 g/mol, respectivamente. Da mesma forma, mDSPC = 66,6 mgs. Este esquema de deuteração aumenta o contraste de dispersão entre a membrana (NSLD ~ 0 × 10-6 Å-2) e o tampão deuterado (NSLD ~ 6,4 × 10-6 Å-2) e amplifica o sinal de ondulações de membrana (ver Figura 2A painel esquerdo).
133,4 mgs, onde XDMPC e XDSPC são as frações moles de DMPC e DSPC, aqui definidas para 0,7 e 0,3, respectivamente, e MwDMPC e MwDSPC são os pesos molares dados por 677,9 g/mol e 790,1 g/mol, respectivamente. Da mesma forma, mDSPC = 66,6 mgs. Este esquema de deuteração aumenta o contraste de dispersão entre a membrana (NSLD ~ 0 × 10-6 Å-2) e o tampão deuterado (NSLD ~ 6,4 × 10-6 Å-2) e amplifica o sinal de ondulações de membrana (ver Figura 2A painel esquerdo). - Para medir a dinâmica de dobra das características de membrana lateral selecionadas, por exemplo, dinâmica de matriz nas membranas DMPC:DSPC de separação de fases, utilizar DMPC protiado (C36H72NO8P) e deuterado, DSPC-d83 (C44H5NO8PD83, Mw 873,7 g/mol), tal que mDMPC = 128,8 mgs e mDSPC-d83 = 71,2 mgs. Este esquema de deuteração minimiza a dispersão dos domínios ricos em DSPC indesejados, permitindo medições seletivas de flutuações de dobra da matriz rica em DMPC (ver Figura 2B no meio).
NOTA: Para encontrar a deuteração lipídica ideal necessária para um esquema específico de correspondência de contraste, utilize calculadoras disponíveis de densidade de comprimento de dispersão baseada na Web (SLD), como a desenvolvida pelo NIST Center for Neutron Research64. Essas interfaces baseadas na Web são equipadas com ferramentas fáceis de usar para fácil cálculo do SLD de lipídios com vários graus de deuteração, bem como a de misturas lipídicas. - Para medições NSE de flutuações médias de espessura da membrana (sem contraste lateral), use variantes deuteradas de cauda dos lipídios constituintes, ou seja, DMPC-d54 (C36H18NO8PD54, 732,3 g/mol) e DSPC-d70 (C44H18NO8PD7 00, 860,1 g/mol)35,38, tal que mDMPC-d54 = 133,0 mgs e mDSPC-d70 = 67,0 mgs. Este esquema de contraste (Figura 2A, painel direito) amplifica o sinal de dispersão dos grupos de cabeça lipídes (NSLD ~ 4,5 × 10-6 Å-2) por contraste do grupo de cauda (NSLD ~ 6,4 × 10-6 Å-2) para o tampão deuterado que permite a detecção de flutuações na espessura da membrana.
- Para estudos de flutuação de espessura de compartimentos de membrana selecionados, por exemplo, matriz rica em DMPC, use a mesma estratégia descrita na etapa 1.2 substituindo lipídios DMPC protiados por seus análogos desuterados de cauda, ou seja, DMPC-d54, de modo que os domínios ricos em DSPC são contrastados com o buffer deuterado e o sinal de dispersão primária é da região do grupo principal da matriz DMPC-defetada de cauda.
 133,4 mgs, onde XDMPC e XDSPC são as frações moles de DMPC e DSPC, aqui definidas para 0,7 e 0,3, respectivamente, e MwDMPC e MwDSPC são os pesos molares dados por 677,9 g/mol e 790,1 g/mol, respectivamente. Da mesma forma, mDSPC = 66,6 mgs. Este esquema de deuteração aumenta o contraste de dispersão entre a membrana (NSLD ~ 0 × 10-6 Å-2) e o tampão deuterado (NSLD ~ 6,4 × 10-6 Å-2) e amplifica o sinal de ondulações de membrana (ver Figura 2A painel esquerdo).
133,4 mgs, onde XDMPC e XDSPC são as frações moles de DMPC e DSPC, aqui definidas para 0,7 e 0,3, respectivamente, e MwDMPC e MwDSPC são os pesos molares dados por 677,9 g/mol e 790,1 g/mol, respectivamente. Da mesma forma, mDSPC = 66,6 mgs. Este esquema de deuteração aumenta o contraste de dispersão entre a membrana (NSLD ~ 0 × 10-6 Å-2) e o tampão deuterado (NSLD ~ 6,4 × 10-6 Å-2) e amplifica o sinal de ondulações de membrana (ver Figura 2A painel esquerdo).2. Preparação de suspensão lipídica para extrusão
- Calcule a massa de cada constituinte na amostra, dependendo da composição da amostra. Como regra geral, para amostras com múltiplos componentes moleculares, a massa de um componente é dada por sua massa molar, Mwi, ponderada por sua fração de toupeira, Xi, e normalizada sobre todos os componentes de tal forma que: onde
 Mtot é a massa total, fixada aqui para 200 mgs. Veja o exemplo acima para bicamadas lipídicas DMPC-DSPC com diferentes esquemas de deuteração.
Mtot é a massa total, fixada aqui para 200 mgs. Veja o exemplo acima para bicamadas lipídicas DMPC-DSPC com diferentes esquemas de deuteração. - Usando um semi-microequilípmo digital, pese as massas calculadas de lipídios (e outros componentes amostrais, por exemplo, proteínas, nanopartículas, etc.) e adicione-os a um frasco de frasco ou fundo redondo – lembre-se de pesar o frasco ou frasco de antemão. Adicione 1 mL de solvente para dissolver os componentes pesados misturando manualmente dentro de um capô. Para amostras lipídicas puras, use clorofórmio ou etanol. Para amostras com componentes adicionais não lipídes (por exemplo, nanopartículas), escolha um solvente comum que disperse todos os componentes.
- Para pequenas quantidades lipídicas (<10 mgs), prepare uma solução de estoque e pipeta o volume necessário na mistura.
NOTA: Não adicione quantidades excessivas de solvente, pois irá diminuir significativamente a etapa de secagem do solvente descrita abaixo.
- Para pequenas quantidades lipídicas (<10 mgs), prepare uma solução de estoque e pipeta o volume necessário na mistura.
- Seque a solução lipídica, dentro de um capô, transmitindo suavemente um gás inerte (por exemplo, nitrogênio, argônio) no frasco enquanto gira lentamente o frasco em um ângulo. Mantenha os frascos em posição inclinada para criar uma fina película de lipídio seco nas paredes do frasco, o que permitirá até mesmo a secagem. Coloque intermitentemente o frasco em um banho de água a 35 °C para contornar o resfriamento mediado pela evaporação, o que retardará a evaporação do solvente.
- Coloque os frascos durante a noite em um forno a vácuo a ~35 °C para remover totalmente o solvente residual. Para lipídios insaturados, purga o vácuo com um gás inerte para minimizar a oxidação.
- Para garantir a remoção total do solvente, pese o frasco após a secagem lipídica e confirme que não há massa excedente além das quantidades medidas de materiais. Faça isso subtraindo a massa do frasco da massa medida após a secagem. Se houver excesso de massa, seque a amostra sob vácuo por mais 6 h. Repita este processo conforme necessário.
- Hidrate o filme lipíduo com 4 mL de D2O ou D2O-tampão para obter uma concentração lipídica de 50 mg/mL. Para lipídios com altas temperaturas de transição, como misturas DMPC-DSPC, aqueça o tampão acima da temperatura de transição (60 °C) para garantir uma mistura uniforme.
NOTA: Uma vez que os experimentos NSE requerem volumes amostrais relativamente grandes (~4 mL), considere hidratar a amostra usando metade do buffer necessário, ou seja, 2 mL, para minimizar o número de extrusões por amostra (ver seção 3). Neste caso, adicione a metade restante da extrusão do pós-tampão. Observe que a capacidade das seringas utilizadas na extrusão é limitada a 1 mL. Assim, hidratar com 4 mL de tampão exigiria quatro conjuntos de extrusão. - Misture o vórtice da solução lipídica hidratada até que o filme lipíduo esteja totalmente dissolvido e não seja mais visível nas paredes do frasco. Nesta fase, os lipídios hidratados formam vesículas multilamellar e pilhas multilamellar do tamanho de mícrons e a suspensão parece branca leitosa.
- Para facilitar a quebra das pilhas lipídicas e reduzir a multilamellaridade, realize cinco ciclos de congelamento/degelo colocando o frasco de solução lipídica hidratada em um congelador de grau de laboratório (de preferência -80 °C congelado) até congelar totalmente e, em seguida, transferir o frasco para um banho de água de 35 °C até que a solução lipídica seja totalmente descongelada. Vórtice a solução descongelada até homogênea. Repita mais quatro vezes.
NOTA: Alternativamente, um banho de gelo seco pode ser preparado para congelamento rápido, combinando acetona e gelo seco.
 Mtot é a massa total, fixada aqui para 200 mgs. Veja o exemplo acima para bicamadas lipídicas DMPC-DSPC com diferentes esquemas de deuteração.
Mtot é a massa total, fixada aqui para 200 mgs. Veja o exemplo acima para bicamadas lipídicas DMPC-DSPC com diferentes esquemas de deuteração.3. Extrusão da solução lipídica hidratada
- Monte a configuração do extrusor usando uma membrana de policarbonato entre dois suportes de membrana e adicionando dois filtros de papel em cada lado para fornecer suporte adicional. Use uma membrana de policarbonato com um tamanho de poroso que corresponda ao tamanho liposomal alvo (tamanhos de poros comuns para experimentos NSE são 50 nm e 100 nm – tipicamente, lipossomos de 100 nm de diâmetro permitem flutuações de membrana menos restritas, mas lipossomos menores de 50 nm poderiam ser usados para estudos de curvatura). Certifique-se de que a membrana de policarbonato esteja totalmente esticada antes de completar o conjunto e apertar a carcaça da extrusora externa.
- Hidrate a membrana de policarbonato passando ~0,3 mL de D2O ou D 2 O-buffer algumasvezesatravés do conjunto de membrana usando seringas de vidro herméticos. Utilize o mesmo buffer usado na preparação da amostra. Deixe-o por pelo menos 10 minutos, depois suga completamente o tampão antes de introduzir a amostra.
- Encha uma seringa de 1 mL com a solução lipídica preparada e insira em uma extremidade do aparelho extrusor. Em seguida, insira uma seringa vazia na extremidade oposta. Uma vez que as seringas estejam conectadas ao conjunto extrusor, coloque-a no bloco de extrusora.
- Se forem necessárias temperaturas elevadas para extrusão, como no caso de lipídios saturados com altas temperaturas de transição (por exemplo, DSPC, Tm = 55 °C), pré-aqueça o bloco de aquecimento extrusor acima da temperatura de transição lipídica (por exemplo, 60 °C), colocando o bloco de aquecimento em uma placa quente ou usando um banho de circulação como mostrado na Figura 3A.
NOTA: Este passo é crucial para garantir a mistura homogênea de lipídios e evitar exercer extrema pressão durante a extrusão, que poderia romper a membrana de policarbonato. Para amostras lipídicas com baixas temperaturas de transição (<25 °C), realize a extrusão à temperatura ambiente. - Para extrusir a solução lipídica, conecte o conjunto extrusor a uma bomba de seringa programável com uma estrutura de alumínio/aço, como mostrado na Figura 3A. Para extrusões controladas pela temperatura, adicione uma base de extrusor personalizada com um canal fluido e conecte-se a um banho de água circulante.
- Programe a bomba de seringa para realizar 15-20 ciclos de extrusão seguindo o manual do fabricante. Quando extrudada, a cor da solução lipídica muda de branco leitoso para azul opala transparente(Figura 3B,C),indicando um tamanho lipossômico final menor que o comprimento de onda da luz visível, como esperado. Para o tipo de bomba de seringa mostrada na Figura 3A,siga os passos abaixo.
- Comece ajustando as configurações da bomba. Mantenha pressionado o botão Taxa e digite a taxa de extrusão (50,99 mL/h), pressione o botão Diâmetro e digite o diâmetro da seringa (4,606 mm). Use as setas para cima sob cada dígito na tela para alterar esse valor de dígito.
- Coloque o conjunto de extrusora com a seringa amostra à direita (ver Figura 3A). Pressione o botão Retirar até que a luz de retirada se acenda. Pressione Inicie e aguarde que a amostra dispense na seringa esquerda (vazia).
- Aperte o botão Parar pouco antes da seringa da amostra (direita) estar totalmente vazia. Registo o volume dispensado e use-o para programar o ciclo de extrusão. Mantenha pressionado o botão Classificar até que a fase 1 (PH:01) apareça na tela. Pressione o botão Volume para inserir o volume dispensado registrado anteriormente. Nesta fase, certifique-se de que a luz de retirada está desligada – isso dispensa a amostra na direção certa.
- Pressione novamente o botão Classificar e use a seta mais à direita para acessar a fase 2 (PH:02). Pressione volume para inserir o mesmo valor do volume dispensado registrado anteriormente. Nesta fase, pressione o botão Retirar até que a luz Retirar esteja acesa – isso dispensa a amostra para a esquerda.
- Para repetir este ciclo, pressione novamente o botão Classificar e use a seta mais à direita para acessar a fase 3 (PH:03). Pressione o botão Volume até que o LP:SE apareça na tela e ajuste-o para 20. Este é o número de loops ou repetições que a bomba irá executar. Por fim, pressione o botão Classificar, acesse a fase 4 (PH:04) e aperte o botão Volume para chegar à função Stop. A bomba está pronta para extrusão automatizada.
- Pressione Comece a iniciar o ciclo de extrusão.
- Esvazie a seringa contendo a suspensão lipídica extrudada em um frasco limpo e prepare-se para armazenamento ou medidas. Para amostras lipídicas com alta temperatura de fusão, armazene a amostra acima da transição de fase do fluido até medir. Caso contrário, mantenha as amostras em temperatura ambiente.
- Não congele amostras extrudadas, pois o congelamento fará com que as vesículas explodam (a suspensão ficará branca leitosa novamente).
4. Medidas NSE para a amostra(s) e redução dos dados coletados
- Antes do experimento NSE, caracterize a amostra liposômica extrudada da etapa 3.7 utilizando métodos disponíveis para garantir a qualidade adequada da amostra. Uma lista de potenciais métodos de charcaterização que podem ser usados para avaliar a qualidade das suspensões liposômicas para experimentos NSE, por exemplo, distribuição de tamanho, multilamellaridade, estrutura de membrana lateral, está incluída na seção de discussão.
- Determine a faixa Q e as configurações de instrumentos correspondentes necessárias para o experimento. Para dobrar as medidas de rigidez das bicamadas lipídicas, use uma faixa Q de ~(0,04 - 0,2) Å-1. Para estudos de flutuações de espessura da membrana, utilize uma faixa Q de ~(0,04 - 0,2) Å-1 correspondente à espessura da membrana35,66,67.
NOTA: Discuta a configuração experimental com o cientista de instrumentos antes do início do experimento. Como mencionado anteriormente, a caracterização da amostra é necessária, especialmente se informações prévias do sinal de dispersão não estão disponíveis, como em membranas seletivamente deutadas. Alternativamente, execute medições estáticas (também conhecidas como difração) sobre uma faixa Q limitada no instrumento NSE, com a ressalva de que tais medidas levam muito mais tempo em comparação com a SANS. - Utilizando uma seringa ou uma pipeta de transferência, carregue a suspensão lipossômica extrudada nas células de amostra designadas disponíveis nas linhas de vigas NSE. Note que as células de amostra NSE padrão vêm em espessuras de 1, 2, 3 e 4 mm. Escolha a espessura da célula de forma a otimizar o sinal de dispersão, mantendo o sinal de fundo incoerente a uma intensidade razoável.
NOTA: Como regra geral, use células amostrais com comprimento de 1 ou 2 mm para lipossomos protiados em tampão deuterado – células mais grossas podem resultar em múltiplos efeitos de dispersão difíceis de corrigir. Para lipossomos com níveis mais elevados de deuterações (por exemplo, lipossomos com contraste de cauda ou lipossomos assimétricos com folhetos protiados único), considere usar uma célula amostral mais grossa (por exemplo, comprimento de pathlength de 3 ou 4 mm) para melhorar as estatísticas de contagem se a amostra estiver disponível em maiores quantidades – às vezes isso pode ser proibitivo de custo. - Prepare uma célula de amostra idêntica para o buffer. Use o mesmo tampão da suspensão lipossômica. As medidas no buffer são necessárias para correções de normalização de intensidade e fundo (BKG).
- Coloque as células amostrais no suporte amostral do espectrômetro NSE, programe as corridas de medição e colete dados de eco. Consulte o cientista de instrumentos sobre a programação das medições se um usuário nse pela primeira vez.
- Realize dois conjuntos adicionais de medições necessárias para a redução de dados: Medidas de Resolução (R) e transmissão(T).
- Medição de resolução de execução (R) em uma referência de dispersão elástica (por exemplo, carbono) — a ser executada sob as mesmas configurações; ou seja, o mesmo vetor de onda e fourier vezes que as medidas de amostra e tampão.
- Realizar medições de transmissão(T) na amostra e tampão para calcular a intensidade do feixe de nêutrons transmitido (ver passo 4.9. abaixo). A transmissão é calculada como a razão de contagem de nêutrons da amostra ou tampão dividido pela contagem de nêutrons para um feixe aberto (ou seja, com uma posição amostral vazia).
- Use o software dedicado de redução de dados para o espectrômetro NSE no qual as medições são realizadas para reduzir os dados coletados.
NOTA: Diferentes espectrômetros podem utilizar diferentes interfaces de software ou usuário. Abaixo está um exemplo de redução de dados da NSE usando o Ambiente de Análise e Visualização de Dados (DAVE)68 software escrito especificamente para o espectrômetro NSE no NIST Center for Neutron Research.- Abra o software DAVE e selecione Reduzir dados NSE do menu de redução de dados. Várias janelas pop-up aparecerão.
- Carregue os arquivos de dados sobre diferentes valores Q usando os Arquivos Open .echo do menu do arquivo. Esses arquivos correspondem aos arquivos de dados brutos com os sinais de eco de spin e têm a extensão .echo no nome do arquivo. Uma vez que o upload do arquivo esteja concluído, os arquivos serão exibidos nos conjuntos de dados disponíveis.
- Clique com o botão direito do mouse no arquivo selecionado e rotule-o de acordo com a medição a que corresponde; ou seja, Amostra, Célula (para célula vazia ou buffer) ou Resolução.
- Agrupar os pixles do detector em 2 x 2 para melhorar a relação sinal-ruído usando a guia Data Set. Aplicar o mesmo binning a todos os arquivos; ou seja, Resolução, Célula e Amostra.
- Inspecione os dados sobre todos os grupos de pixels e mascara aqueles com sinais ruins (ver Figura 4B) pressionando a tecla m no teclado. Pressione Enter para acessar uma janela pop-up para aplicar a mesma máscara em todas as vezes Fourier ou fourier subsequentes. Isso também pode ser aplicado a pixels individuais em qualquer momento durante a redução de dados. Pixels mascarados ficarão verdes.
- Certifique-se de que os dados coletados estão na forma de um sinal de eco, ou seja, função cosseno em termos da corrente de fase, sobre cada pixel detector (ver Figura 4A).
NOTA: A corrente de fase é proporcional ao ângulo de precessão do giro de nêutrons; portanto, é comum representar a corrente de fase como um ângulo de fase como mostrado na Figura 4A. Para medições em fontes pulsadas, cálculos adicionais de voo são aplicados aos dados para obter os sinais de eco em função do comprimento de onda de nêutrons incidente dentro de um pulso de nêutrons. - Comece ajustando o arquivo de resolução. Selecione um arquivo de resolução na lista de arquivos carregado e clique com o botão direito do mouse no arquivo. Selecione Operações de ajuste: Encaixe ecos (resolução) no menu pop-up.
- Certifique-se de que os ajustes dos sinais de eco produzam uma série de parâmetros de montagem, incluindo o parâmetro, A, necessário na etapa 4.8. Os ajustes são realizados automaticamente usando a seguinte expressão.

Aqui, ζ é o período do sinal de eco (ou seja, função cosseno na Figura 4A), σ é a largura do envelope gaussiano determinado pelo comprimento médio de onda e propagação do comprimento de onda do feixe de nêutrons incidente, Φc é a corrente de fase, e Φ0 é o ponto de eco que depende do caminho de campo experimentado pelos nêutrons50. As informações físicas sobre a amostra estão codificadas na amplitude, A, da função cosseno na equação (1).
NOTA: A largura do envelope gaussiano é baseada em valores predeterminados pelo cientista do instrumento e não deve ser alterada. Os outros paramters são variáveis que são instaladas no sinal de eco específico sobre cada pixel. - Inspecione os resultados de ajuste clicando em cada pixel para mostrar os parâmetros de encaixe resultantes, a qualidade do ajuste e o desvio quadrado médio do ajuste. Para inspecionar o erro associado a cada parâmetro de montagem sobre todo o detector, selecione Opções de imagem e selecione o parâmetro de interesse de montagem. Isso vai gerar um mapa com o valor do paramter de montagem sobre cada pixel. Clique com o botão direito do mouse na imagem do detector. Uma janela pop-up aparecerá mostrando um mapa de barra de erro do parâmetro de montagem selecionado.
- Se o ajuste sobre um pixel específico for insatisfatório (por exemplo, ajuste para-se com grandes barras de erro), reequiva o sinal sobre esse pixel específico. Selecione esse pixel,pressione a guia Encaixe e pressione o Fit Pixel. Insira novos parâmetros de partida para a fase (Φ0) e período (ζ) na guia De montagem para obter um ajuste mais satisfatório.
NOTA: É útil traçar a fase montada em função do Fourier Time. Para isso, acesse a janela principal do enredo e selecione Fit Phase v. Fourier Time. Este enredo deve ser suave e contínuo. Inspecione as descontinuidades neste gráfico e reequipa os pixels a que correspondem.
- Reduza o arquivo Amostra ou Célula selecionando o arquivo correspondente na lista de arquivos carregado e rotulado.
- Inspecione todos os pixels e mascara os com estatísticas ruins como descrito na etapa 4.7.5.
- Clique com o botão direito do mouse no arquivo e selecione Operações de ajuste: Fases de importação (Amostra, Célula). Isso importa as fases e a máscara aplicada do arquivo Resolução.
- Encaixe os sinais de eco usando o mesmo procedimento descrito anteriormente para o arquivo Resolução (etapas 4.7.8-4.7.10). Ao encaixar os arquivos Amostra e Célula, não altere os valores do período e o ponto de fase de eco importado da Resolução se encaixa. Esses parâmetros dependem das configurações instrumentais e não devem variar com as amostras.
- Antes de prosseguir para a redução de dados, insira o centro de feixe para todos os arquivos de dados. Selecione o arquivo de dados, vá para a guia Geral e digite os valores da central de feixe X e Y. Esses valores são registrados durante o experimento.
- Uma vez que os ajustes nos arquivos Amostra, Celular e Resolução estejam concluídos, calcule a função de dispersão intermediária normalizada a ser usada posteriormente na análise e interpretação dos dados. Para fazer isso, clique com o botão direito do mouse no arquivo Sample para ser reduzido da lista de arquivos instalados e selecione Calcular I(Q) no menu pop-up. Uma janela aparecerá com opções de entrada para os arquivos Resolução e Célula (ou seja, buffer) e o número de arcos Q (ver passo 4.9). Depois de inserir todas as informaitons necessárias, pressione o botão OK. Os resultados aparecerão em uma nova janela.
NOTA: A redução dos dados é realizada de acordo com a seguinte equação para obter a função de dispersão intermediária normalizada69.
onde t é o tempo fourier, N paracima e N parabaixo são as contagens de nêutrons nas configurações não-spin-flip e spin-flip (medidos com as π/2-flippers fora e o π-flipper fora e sobre, respectivamente), e os sobrescritos, BKG e R, correspondem às medições de fundo e resolução, respectivamente, conforme definido nas etapas 4.4 e 4.6. Note-se que a polarização do feixe, assim, muda no estado de rotação devido à troca de energia entre o nêutron e a amostra é detectada como uma queda na polarização (da unidade).
feixe, assim, muda no estado de rotação devido à troca de energia entre o nêutron e a amostra é detectada como uma queda na polarização (da unidade).
- Por fim, agrupar os pixels do detector em arcos Q,como mostrado na Figura 4B para obter aq -dependência da função de dispersão intermediária normalizada, S(Q,t) / S( Q,0). Isso é tecnicamente referido como binning de dados e deve ser feito criteriosamente, ou seja, levando em conta as estatísticas de contagem da amostra e o desvio padrão esperado dos dados sobre os pixels agrupados.
- Para dispersar fortemente amostras, divida o detector em mais arcos Q, mantendo barras de erro razoáveis na função de dispersão intermediária resultante, S(Q,t) / S(Q,0). Isso produz mais pontos de dados Q e é importante para o procedimento de análise de dados descrito abaixo. Esteja ciente de que para amostras fracamente dispersas, o binning excessivo resulta em sinais de decadência ruins, ou seja, grandes barras de erro em S(Q,t) / S(Q,0), o que poderia resultar em grandes incertezas.


 feixe, assim, muda no estado de rotação devido à troca de energia entre o nêutron e a amostra é detectada como uma queda na polarização (da unidade).
feixe, assim, muda no estado de rotação devido à troca de energia entre o nêutron e a amostra é detectada como uma queda na polarização (da unidade).5. Análise e interpretação de dados
- Ajuste as funções de dispersão intermediária normalizadas, S(Q,t) / S(Q,0), obtidas a partir da redução de dados acima para uma função exponencial esticada com um expoente de alongamento de 2/370.

NOTA: Um exemplo desses ajustes é fornecido na Figura 5B. Os ajustes de S(Q,t) / S(Q,0) à equação (3) produzem as taxas de relaxamento dependentes de Q Γ(Q). - Plot Γ(Q)em função de Q e apto a um modelo adequado para extrair parâmetros de membrana relevantes.

Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Estudos NSE acessando flutuações de dobra são normalmente realizados ao longo de uma faixa Q de ~ (0,04 - 0,2) Å-1. Esta faixa Q corresponde a escalas de comprimento intermediário entre a espessura da membrana e o raio lipossômico, onde a dinâmica de dobra domina. A medição sobre um intervalo Q estendido pode dar acesso a modos dinâmicos adicionais, incluindo difusão liposômica e dinâmica intramembrana. Para obter mais detalhes sobre o cruzamento na dinâmica da membrana acessada pela NSE, consulte estas publicações relevantes25,71. É importante ressaltar que os sinais NSE são proporcionais a:  , onde eucoh e euinc estamos, respectivamente, a intensidade de dispersão coerente e incoerente da amostra. Portanto, é aconselhável preparar amostras liposômicas NSE em tampões deuterados (ou seja, tampões preparados com D2O em vez de H2O) para minimizar o sinal de dispersão incoerente, principalmente contribuído pelo conteúdo de hidrogênio da amostra. No entanto, em alguns casos, esquemas intermediários de deuteração (ou seja, usando misturas de D2O e H2O) podem ser necessários para obter condições de contraste ideais. Normalmente, as medidas NSE das flutuações de dobra de membrana são realizadas em lipossomos totalmente protiados em tampão deuterado, referido como lipossomos totalmente contrastados na Figura 5. Este esquema de deuteração resulta em uma grande diferença de NSLD entre o núcleo de membrana (~0 × 10-6 Å-2) e seu ambiente fluido deuterado (~6,4 × 10-6 Å-2), o que melhora significativamente o sinal de dispersão das membranas liposômicas e melhora as estatísticas de medição da dinâmica de dobra. Este esquema de contraste ( Painel esquerdofigura 2A) é frequentemente utilizado em estudos de rigidez de dobra de membranas lipídicas com único38,72 emúltiplos 39,66 componentes lipídicos e em estudos de amolecimento/endurecimento da membrana por inclusões biológicas (por exemplo, colesterol, moléculas de drogas, peptídeos/proteínas)36,37,73,74,75e aditivos sintéticos (por exemplo, nanopartículas)76,77.
, onde eucoh e euinc estamos, respectivamente, a intensidade de dispersão coerente e incoerente da amostra. Portanto, é aconselhável preparar amostras liposômicas NSE em tampões deuterados (ou seja, tampões preparados com D2O em vez de H2O) para minimizar o sinal de dispersão incoerente, principalmente contribuído pelo conteúdo de hidrogênio da amostra. No entanto, em alguns casos, esquemas intermediários de deuteração (ou seja, usando misturas de D2O e H2O) podem ser necessários para obter condições de contraste ideais. Normalmente, as medidas NSE das flutuações de dobra de membrana são realizadas em lipossomos totalmente protiados em tampão deuterado, referido como lipossomos totalmente contrastados na Figura 5. Este esquema de deuteração resulta em uma grande diferença de NSLD entre o núcleo de membrana (~0 × 10-6 Å-2) e seu ambiente fluido deuterado (~6,4 × 10-6 Å-2), o que melhora significativamente o sinal de dispersão das membranas liposômicas e melhora as estatísticas de medição da dinâmica de dobra. Este esquema de contraste ( Painel esquerdofigura 2A) é frequentemente utilizado em estudos de rigidez de dobra de membranas lipídicas com único38,72 emúltiplos 39,66 componentes lipídicos e em estudos de amolecimento/endurecimento da membrana por inclusões biológicas (por exemplo, colesterol, moléculas de drogas, peptídeos/proteínas)36,37,73,74,75e aditivos sintéticos (por exemplo, nanopartículas)76,77.
As medições das flutuações de dobra resultam em taxas de relaxamento que seguem uma dependência do 3º trimestre, como previsto por Zilman e Granek para folhas finas elásticas termicamente ondulantes70. Uma forma refinada desta -dependência Qé obtida a partir de correções teóricas por Watson e Brown78, que levam em conta os efeitos do atrito intermonolar proposto por Seifert e Langer79. Ao definir adicionalmente o plano neutro para estar na interface entre os grupos de cabeça hidrofílicos e as caudas hidrofóbicas da membrana, as taxas de relaxamento de dobra podem então ser encaixadas na seguinte expressão38.

onde ηbuff é a viscosidade tampão, kBT é a energia térmica, κ e é a rigidez de dobra da membrana medida (ou da porção contrastada da membrana em sistemas seletivamente deutados). Este tipo de medida permite o cálculo direto das propriedades elásticas da membrana na forma do módulo de rigidez de dobra. Note que κ é extraído da inclinação do ajuste linear de Γ vs. Q3, como mostrado na Figura 5C.
Por outro lado, as medidas nse das flutuações de espessura da membrana mostram desvios da dependência do Q3em Γ(Q) em torno de valores Q que correspondem à espessura da membrana (ver Figura 2 no ref.66). Para isolar o sinal de flutuação de espessura, pode-se dividir Γ(Q)pelo Q3,como mostrado na Figura 5D. Os dados resultantes mostram que o excesso de dinâmica devido às flutuações de espessura seguem uma função lorentziana em Q, como recentemente corroborado em simulações de dinâmica molecular de grãos grossos (MD)67. Para se adequar à dinâmica em excesso observada, Nagao et al.38 desenvolveram uma expressão baseada no quadro teórico das flutuações de membrana por Bingham et al.80 da seguinte forma.

Nesta expressão, Q0 é o pico Q-valor correspondente à espessura da membrana (que pode ser obtida independentemente das medições do SANS), μ é a viscosidade da membrana in-plane, AL é a área por lipídio (medido com SANS/SAXS), e KA é o módulo de compressão da área. Supondo que o KA possa ser calculado a partir de κ usando o modelo de pincel de polímero, esta expressão reduz a um parâmetro de ajuste, ou seja, a viscosidade damembrana μ , apresentando uma nova abordagem para medir a viscosidade da membrana sem a necessidade de rotulagem de fluorescência ou amarração/rastreamento de partículas13. A premissa é que de acordo com os modelos de deformação das folhas finas elásticas81, κ e KA são interdependentes de tal forma: ,  onde tm é a espessura mecânica (ou deformável) da membrana e β é uma constante que descreve o acoplamento entrelaçado. A suposição é que β = 12 para folhetos totalmente acoplados, β = 48 para folhetos completamente desacoplados, e β = 24 para folhetos acoplados intermediários. Este último é referido como o modelo de escova de polímero81 e tem sido mostrado para aplicar em membranas lipídicas de fluido único e binário39. No entanto, isso precisa ser abordado com cautela. Por exemplo, simulações recentes de Doktorova et al. 82 mostrou que para o modelo de escova de polímero segurar em membranas lipídicas insaturadas contendo colesterol, deve-se utilizar uma expressão modificada da espessura da membrana mecânica. Idealmente, se uma medição independente de KA for possível, por exemplo, usando aspiração de micropipette83, então combinar os resultados de KA com as medidas de rigidez de dobra NSE apresentaria uma oportunidade única para investigar o acoplamento entreleaflet em modelos e membranas biológicas – uma questão de longa data na biofísica da membrana e biologia estrutural. Uma vez que os valores de KA são validados, eles podem ser usados na equação 5 para obter a viscosidade da membrana mesoscópica.
onde tm é a espessura mecânica (ou deformável) da membrana e β é uma constante que descreve o acoplamento entrelaçado. A suposição é que β = 12 para folhetos totalmente acoplados, β = 48 para folhetos completamente desacoplados, e β = 24 para folhetos acoplados intermediários. Este último é referido como o modelo de escova de polímero81 e tem sido mostrado para aplicar em membranas lipídicas de fluido único e binário39. No entanto, isso precisa ser abordado com cautela. Por exemplo, simulações recentes de Doktorova et al. 82 mostrou que para o modelo de escova de polímero segurar em membranas lipídicas insaturadas contendo colesterol, deve-se utilizar uma expressão modificada da espessura da membrana mecânica. Idealmente, se uma medição independente de KA for possível, por exemplo, usando aspiração de micropipette83, então combinar os resultados de KA com as medidas de rigidez de dobra NSE apresentaria uma oportunidade única para investigar o acoplamento entreleaflet em modelos e membranas biológicas – uma questão de longa data na biofísica da membrana e biologia estrutural. Uma vez que os valores de KA são validados, eles podem ser usados na equação 5 para obter a viscosidade da membrana mesoscópica.

Figura 1: Design de instrumentos NSE e sobreposição sinérgica com extensão/tempo de dinâmica da membrana mesoscópica. (A) Esquema dos diferentes elementos magnéticos de um instrumento NSE, usado para manipular o giro de nêutrons atravessando o instrumento da esquerda para a direita. O nêutron destacado indica mudança na orientação de giro (ou perda de polarização) devido à troca de energia entre o nêutron e a amostra, enquanto o nêutron transparente representa spin-echo, ou seja, nenhuma mudança no giro de nêutrons devido à troca de energia zero. A seta cinza indica a possibilidade de girar o segundo braço do espectrômetro para acessar ângulos de dispersão maiores. (B) Representação pictórica da dinâmica hierárquica em membranas lipídicas, mostrando vários modos dinâmicos que abrangem múltiplos comprimentos e escalas de tempo. A área sombreada representa as escalas de comprimento e tempo acessadas pela NSE, que se sobrepõem às mesoestoes das flutuações coletivas da membrana, ou seja, flutuações de dobra e espessura. Clique aqui para ver uma versão maior desta figura.

Figura 2: Exemplos de possíveis esquemas de deuteração em experimentos NSE em membranas lipídicas. (A) Esquerda: Membranas totalmente contrastadas, por exemplo, membranas protiadas em tampão deuterado, mostrando o perfil NSLD ao longo do normal para a superfície da membrana. A diferença no NSLD entre a região da cauda (~0 × 10-2 Å-2) e a região do headgroup (~4,5 × 10-6 Å-2) da membrana é devido à hidratação do grupo de cabeça com tampão deuterado. Direito: Membranas combinadas com contraste de cauda de modo que a região da cauda de hidrocarbonetos da membrana tenha o mesmo NSLD que o tampão, como mostrado no perfil NSLD correspondente ao longo da membrana normal. (B) Membranas formadoras de domínio com dois esquemas de contraste de nêutrons onde os domínios (centro) ou a matriz (esquerda) são contrastados com o buffer, permitindo estudos seletivos de matriz ou dinâmica de domínio, respectivamente. Este número foi modificado a partir de Nickels et al., JACS 201541. (C) Membranas assimétricas preparadas pela troca de ciclodextrinas entre vesículas lipídicas protiadas e deuteradas, resultando na deuteração de um folheto de membrana, mantendo o outro folheto protiado. Isso permite estudos da dinâmica de dobra do folheto protiado e fornece insights sobre o acoplamento mecânico entre folhetos opostos em membranas assimétricas. Este número foi modificado de Rickeard et al., Nanoscale 202040. Clique aqui para ver uma versão maior desta figura.

Figura 3: Ilustração da configuração para extrusão automatizada de lipossomos. (A) Extrusor automatizado personalizado usando uma bomba de seringa, um conjunto de mini extrusor e uma estrutura de alumínio/aço para permitir extrusões cíclicas. (B) e (C) mostram a diferença na aparência visual das suspensões lipídicas antes (branco leitoso) e depois (azul opala transparente) extrusão. Isso se deve à formação inicial de pilhas lipídicas do tamanho de micron ou vesículas gigantes que estão na ordem, ou maior do que, o comprimento de onda da luz visível. Após a extrusão, a suspensão será composta por vesículas nanoscópicas (~100 nm), que são menores que o comprimento de onda da luz visível, gerando uma suspensão transparente. Clique aqui para ver uma versão maior desta figura.

Figura 4: Dados representativos de experimentos NSE em suspensões liposômicas. (A) Exemplo de um sinal de eco sobre um único pixel detector (pixel marcado no painel B), mostrando os ajustes do sinal de eco usando equação (1), com uma ilustração dos diferentes parâmetros exigidos no ajuste de eco. Observe que o sinal de eco é plotado em função do ângulo de fase em vez da corrente de fase como discutido na etapa 4.7 do protocolo. (B) Imagem do detector NSE mostrando a variação na contagem de nêutrons por pixel. A imagem também mostra pixels de detector eliminados (verde) devido a sinais de eco fracos. O binning dos pixels do detector em arcos Q (também conhecidos como anéis Debye-Scherrer) produz a dependência Q da função de dispersão intermediária, necessária para analisar e interpretar dados NSE. Este valor foi modificado a partir de Ashkar, J. Appl. Phys. 202050. Clique aqui para ver uma versão maior desta figura.

Figura 5: Resultados representativos de experimentos NSE em suspensões liposômicas com diferentes esquemas de deuteração. (A) Geometria dispersa de um nêutron interagindo com um liposomático, mostrando o ângulo de dispersão, 2φ, e a transferência de vetores de onda,  . (B) Funções de dispersão intermediárias, S(Q,t) / S(Q,0), exibem decaimentos em função do tempo de Fourier. O ajuste das decaimentos medidos a uma função exponencial esticada dada pela equação 3 produz as taxas de relaxamento, Γ(Q). (C) Para lipossomos totalmente protiados em tampão deuterado, Γ(Q) segue uma dependência Q3, típica da dinâmica de dobra. O ajuste linear dos dados obtidos para um modelo Zilman-Granek produz o módulo de rigidez de dobra da membrana. (D) Para lipossomos desuterados na cauda, o excesso de dinâmica é observado, além de flutuações de dobra e são mais pronunciados em valores Q que correspondem à espessura da membrana. Encaixar a dinâmica em excesso a uma função lorentziana (equação 5) permite a extração da viscosidade da membrana. Os conjuntos de dados foram coletados no espectrômetro NSE no NIST. Clique aqui para ver uma versão maior desta figura.
. (B) Funções de dispersão intermediárias, S(Q,t) / S(Q,0), exibem decaimentos em função do tempo de Fourier. O ajuste das decaimentos medidos a uma função exponencial esticada dada pela equação 3 produz as taxas de relaxamento, Γ(Q). (C) Para lipossomos totalmente protiados em tampão deuterado, Γ(Q) segue uma dependência Q3, típica da dinâmica de dobra. O ajuste linear dos dados obtidos para um modelo Zilman-Granek produz o módulo de rigidez de dobra da membrana. (D) Para lipossomos desuterados na cauda, o excesso de dinâmica é observado, além de flutuações de dobra e são mais pronunciados em valores Q que correspondem à espessura da membrana. Encaixar a dinâmica em excesso a uma função lorentziana (equação 5) permite a extração da viscosidade da membrana. Os conjuntos de dados foram coletados no espectrômetro NSE no NIST. Clique aqui para ver uma versão maior desta figura.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
NSE é uma técnica poderosa e única na medição da dinâmica mesoscópica das membranas lipídicas sob várias condições. A utilização efetiva da NSE depende da qualidade da amostra, do contraste de nêutrons e da gama de dinâmicas acessíveis que podem ser sondadas para uma determinada amostra. Assim, várias etapas críticas são necessárias para a realização de experimentos NSE bem-sucedidos e a coleta de dados de alta qualidade. Um passo fundamental para garantir o uso efetivo do tempo de feixe de nêutrons durante um experimento NSE é caracterizar as suspensões lipossômicas com métodos baseados em laboratório antes do experimento NSE. Para exmaple, a distribuição de tamanho (ou constante de difusão) de lipossomos extrudados pode ser determinada por dispersão dinâmica de luz (DLS), prontamente disponível em laboratórios individuais ou em instalações compartilhadas84. A microscopia crio-elétron é outro método de charcaterização recentemente validado em amostras liposômicas, onde imagens de microscopia de alta resolução em seções criomicrotomed de suspensão liposômica podem ser efetivamente usadas para examinar a unilamellaridade liposômica65, formação de domínio85,86, ou a incorporação de aditivos como nanopartículas76 e proteínas87. Alternativamente, a dispersão de raios-X de pequeno ângulo (SAXS) pode ser usada para caracterizar a estrutura da membrana88, avaliar a multilamellaridade liposômica65, ou avaliar os efeitos dos aditivos nas propriedades estruturais da membrana89. Além dessas técnicas baseadas em laboratório, é altamente aconselhável que as medidas NSE em amostras liposômicas sejam emparelhadas com estudos estruturais utilizando dispersão de nêutrons de pequeno ângulo (SANS)54,90. O SANS é um excelente complemento para a NSE, não apenas para a aquisição de informações de membrana estrutural, mas também para examinar a intensidade do sinal de dispersão de nêutrons da amostra, confirmando o esquema de contraste, e fazendo uma escolha informada sobre a faixa Q sobre a qual as medidas NSE devem ser realizadas. Portanto, recomenda-se que os usuários da NSE solicitem o tempo de feixe SANS ao solicitar experimentos NSE.
No entanto, a NSE sofre de limitações amostrais em estudos de membranas biológicas. Um dos principais fatores limitantes desses experimentos é a quantidade padrão de amostra necessária para as medições de NSE (2-4 mL) e as altas concentrações amostrais que totalizam 100-200 mg de material de membrana (lipídios e proteínas) para obter dados de alta qualidade. Em muitos casos, a produção de tais quantidades de material biológico não é viável ou é proibitiva de custos. Nesses cenários, é possível reduzir a concentração para 20-25 mg/mL, mas isso exigiria pelo menos um aumento de 4 vezes no tempo de aquisição, a fim de obter estatísticas comparáveis a amostras com concentrações de 50 mg/mL. Esses requisitos rigorosos sobre volume e concentração amostral poderiam ser aliviados com a próxima geração de espectrômetros NSE em fontes de nêutrons de maior fluxo, como a segunda estação-alvo no Oak Ridge National Lab e a European Spallation Source. Outra limitação crítica na realização de experimentos NSE em membranas lipídicas que requerem esquemas seletivos de deuteração é a falta de disponibilidade comercial de algumas variantes deuteradas de moléculas lipídicas ou seus preços exorbitantes, se disponíveis. Em alguns casos, essas limitações podem ser contornadas solicitando a síntese de lipídios deuterados (ou colesterol, proteínas) através de instalações de deuteração de usuários, como o laboratório de bio-deuteração no Oak Ridge National Lab, a instalação nacional de deuteração na ANSTO, ou a instalação de deuteração no ISIS Neutron e Muon Source. O acesso a essas instalações e suas capacidades de síntese está disponível por meio de propostas de usuário submetidas que são revisadas por pares com base no mérito científico da síntese material proposta e seu uso pretendido em estudos sensíveis a isótopos.
Apesar dessas limitações, a aplicação da espectroscopia NSE em estudos de mecânica de membranas levou à determinação do moduli de rigidez de dobra de membranas com vários graus de complexidade, desde membranas lipídicas de componente único35,38 até membranas biomiméticas multicomponentes41,66,91, todas as quais avançaram em nossa compreensão da natureza dinâmica das membranas lipídicas. Por exemplo, as medidas de rigidez de dobra nse de membranas lipídicas com diferentes unidades moleculares, por exemplo, lipídios de diferentes comprimentos da cadeia de aciis e saturação da cadeia38,72,92, forneceram informações essenciais sobre o papel da química molecular na mecânica da membrana. Quando emparelhadas com informações estruturais, como a espessura da membrana ou a embalagem molecular93,essas medidas começam a fornecer novas perspectivas sobre a interdependência entre estrutura e dinâmica da membrana e como influenciam a função da membrana. As escalas mesoscópicas da NSE posicionam-na exclusivamente para investigações fundamentais das relações estruturais-propriedade, mais relevantes na escala de comprimento dos conjuntos moleculares. Este tema foi recentemente explorado em dois estudos da NSE sobre membranas lipídicas ricas emcolesterol 36 e em membranas lipídicas binárias com incompatibilidade hidrofóbica entre os dois componentes lipídicos39. Ambos os estudos encontraram fortes evidências de que a mecânica da membrana escala com a área por lipídio, corroborando as conclusões de uma recente simulação de MD de todos os átomos por Doktorova et al.82. Esses achados enfatizam a natureza auto-montada das membranas lipídicas e fornecem uma imagem unificadora da embalagem molecular como parâmetro-chave na definição de propriedades dinâmicas e funcionais da membrana.
Outras aplicações da NSE envolvem estudos da resposta mecânica de membranas a pequenos aditivos, incluindo moléculas biológicas comocolesterol 36,37, trehalose92e melittina73,94, ou aditivos inorgânicos, como nanopartículas para aplicações de entrega de medicamentos76. A NSE também tem sido usada para entender como a mecânica da membrana responde às mudanças em seu ambiente, incluindo temperatura92, pH74, e a presença de macromoléculas de aglomeração96. Tais estudos estão contribuindo para uma melhor compreensão dos fatores que influenciam o amolecimento ou enrijecimento das membranas lipídicas, em condições biológicas relacionadas à saúde e doença, e em ambientes controlados para aplicações terapêuticas. Notavelmente, as medidas NSE também têm sido utilizadas para sondar o efeito de peptídeos antimicrobianos na dinâmica da membrana73,94,95. Outros exemplos de aplicações NSE em biomembranos incluem estudos da dinâmica das estruturas de membrana achatada, chamadas de timkoóides, que abrigam o maquinário fotossintético em células cianobacterianas97,98.
Pode-se também utilizar a deuteração lipídica seletiva em estudos NSE para investigar a dinâmica de características de membrana específicas que são relevantes para a função biológica. Por exemplo, Nickels et al. utilizaram deuteração lipídica seletiva em membranas lipídicas formadoras de domínio para gerar contraste lateral dentro da membrana, como ilustrado anteriormente por Heberle et al.28. Este esquema de deuteração possibilitou medições independentes da rigidez de dobra dos domínios lipídios e da matriz lipídica hospedeira41 (ver Figura 2B). Os achados confirmaram que os dois compartimentos de membrana possuem moduli de rigidez de dobra distinta, que poderia ser um mecanismo de condução para a formação de domínio em membranas celulares. Em um estudo mais recente, Rickeard et al. utilizaram a troca de ciclodextrinas entre lipossomos protiados e deuterados para obter lipossomos assimétricos com folhetos isototicamente rotulados40 (Figura 2C). Seus lipossomos finais tinham um folheto protiado e um folheto deuterado que é contrastado com o tampão, permitindo estudos da dinâmica individual do folheto e fornecendo um primeiro relato experimental direto do efeito da assimetria e do acoplamento de folhetos sobre flutuações de dobra de membrana.
A deuteração de membrana seletiva também tem sido utilizada em estudos NSE de flutuações de espessura de membrana, um modo dinâmico há muito previsto nas membranas lipídicas99 que só foi observado recentemente com o advento da espectroscopia NSE35,100. Essas medidas utilizam membranas deuteradas na cauda para amplificar o sinal das regiões do grupo da cabeça da membrana e resolver o sinal de flutuação da espessura. Este tipo de experimentos NSE é relativamente recente, mas tem sido efetivamente utilizado para entender a interdependência das propriedades elásticas e viscosas da membrana38,para explorar o dimensionamento da rigidez e viscosidade de dobra com embalagem molecular em membranas lipídicas mistas39, e para sondar os efeitos locais do colesterol na viscosidade da membrana36. Outra área de significância biológica em que esse modo dinâmico pode ter implicações de longo alcance são as interações membrana-proteína mesoscópica95. Sabe-se que a função das proteínas de membrana está fortemente ligada à correspondência hidrofóbica entre a proteína e a membrana hospedeira. Assim, variações na espessura da membrana, devido às flutuações de espessura, poderiam atuar como um mecanismo regulatório para a função das proteínas de membrana. O NSE é extremamente adequado para estudos como pode sondar diretamente os efeitos da ligação proteica e inserção em flutuações de espessura da membrana. Medições recentes da NSE do nosso grupo (inéditas) sugerem que a inserção da proteína transmembrana poderia suprimir significativamente as flutuações de espessura da membrana e poderia apresentar um mecanismo potencial para regular eventos de sinalização. Trata-se de uma área de pesquisa premente, porém subdesenvolvida, onde a NSE pode ter impacto significativo na compreensão das respostas dinâmicas das membranas à ligação proteica e à inserção na extensão e escalas de tempo das principais funções biológicas transmitidas pelas interações das proteínas com membranas celulares.
Em resumo, a NSE evoluiu ao longo dos últimos anos como uma poderosa ferramenta para interrogar a dinâmica da membrana sobre escalas espaciais e temporais de funções biológicas vitais. A técnica está rapidamente ganhando interesse generalizado e seu potencial em responder a perguntas-chave na função de membrana está se tornando bem reconhecido. As capacidades de variação de contraste dentro da NSE posicionaram-na como uma abordagem única para medir propriedades de membrana mesoscópica que de outra forma seria desafiadora de obter. Outra vantagem significativa da NSE em relação aos métodos tradicionais de espectroscopia em estudos de dinâmica de membrana é sua sobreposição com o comprimento e escalas de tempo acessíveis com simulações de MD, permitindo que estudos experimentais/computacionais sinérgicos obtenham uma compreensão de nível molecular dos diferentes componentes moleculares que compõem as membranas. Apesar de sua promessa, ainda há algumas limitações no uso de NSE em estudos de membrana biológica, incluindo a exigência de grandes volumes amostrais, a dificuldade em darteração seletiva em sistemas biológicos e o fluxo de nêutrons relativamente baixo nos espectrômetros NSE, o que resulta em tempos de medição mais longos e disponibilidade limitada de feixes. No entanto, essas deficiências poderiam ser superadas em um futuro próximo com desenvolvimentos constantes em fontes de nêutrons e instrumentação, juntamente com avanços nas instalações de deuteração.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Os autores não declaram conflitos de interesse e não têm nada a revelar.
Acknowledgments
R. Ashkar agradece M. Nagao, L.-R. Stingaciu e P. Zolnierczuk para muitas discussões úteis e por sua assistência frequente com experimentos NSE em suas respectivas linhas de trave. Os autores reconhecem o uso de espectrômetros de eco de spin de nêutrons no NIST e ORNL. O espectrômetro NSE no NIST é apoiado pelo Centro de Dispersão de Nêutrons de Alta Resolução, uma parceria entre o Instituto Nacional de Padrões e Tecnologia e a Fundação Nacional de Ciência sob o acordo nº. DMR-1508249. O espectrômetro NSE na Fonte de Nêutrons spallation da ORNL é apoiado pela Divisão de Instalações de Usuários Científicos, Escritório de Ciências Básicas de Energia, Departamento de Energia dos EUA. Oak Ridge National Laboratory é gerenciado pela UT-Battelle, LLC sob o contrato us doe no. DE-AC05-00OR22725.
Materials
| Name | Company | Catalog Number | Comments |
| Chloroform (biotech grade) | Sigma Aldrich | 496189 | Biotech. grade, ≥99.8%, contains 0.5-1.0% ethanol as stabilizer |
| Circulating water bath | Julabo | SE-12 | Heating Circulator with smart pump, programmable temperature settings, and external sensor connection for measurement and control |
| Deuterium Oxide | Cambridge Isotopes Laboratories | DLM-4 | Deuterated water; Heavy water (D2O) (D, 99.9%) |
| Digital Semi-Microbalance | Mettler Toledo | MS105 | Semi-micro balance with 120 g capacity, 0.01 mg readability, high resolution weighing cell, ergonomic doors, and pipette-check application |
| Ethanol (molecular biology grade) | Sigma Aldrich | E7023 | 200 proof ethanol for molecular biology applications |
| Glass Pipets | VWR | 36360-536 | Disposable Soda Lime glass Pasteur pipets |
| Glass Vials | Thermo Scientific | B7990-1 | Borosilicate glass vials with PTFE/Silione septum caps |
| Lab grade freezer | Fisher Scientific | IU2886D | Ultra-low temprature freezer (-86 to -50 C) for long-term storage of lipids and proteins |
| Lipids (protaited or perdeuterated) | Avanti Polar Lipids | varies by lipid | Lipids can be purchased from Avanti in powder form or in a chloroform solution with the required amounts and deuteration schemes. |
| Millipore water purifier | Millipore Sigma | ZRQSVP3US | Direct-Q® 3 UV Water Purification System which deliver both pure and ultrapure water with a built-in UV lamp to reduce the levels of organics for biological applications |
| Mini Extruder Set | Avanti Polar Lipids | 610020 | Mini-extruder set includes mini-extruder, heating block, 2 GasTight Syringes, and 2 O-rings, Polycarbonate Membranes, and Filter Supports |
| Quick Connect Fittings | Grainger | 2YDA1 and 2YDA7 | Push-button tube fittings for QuickConnect water circulation applications, e.g. high temperature vesicle extrusion |
| Syringe Pump | SyringePump.com | New Era-1000 | Fully programmable syringe pump for infusion and withdrawal; programs up to 41 pumping phases with adjustable pumping rates, dispensed volumes, and extrusion cycles |
| Ultrasonic bath | Fisher Scientific | CPX2800 | Temperature controlled ultra sonic bath with programmable functionality for degassing and ultrasonic applications |
| Vacuum Oven | Thermo Scientific | 3608 | 0.7 cu ft vaccum oven with built-in-high-limit thermostat guards against overheating |
| Vortex Mixer | Fisher Scientific | 02-215-414 | Variable speed, analog control that allows low rpm start-up for gentle shaking or high-speed mixing for vigorous vortexing of samples |
References
- Singer, S. J., Nicolson, G. L. The fluid mosaic model of the structure of cell membranes. Science. 175 (4023), 720-731 (1972).
- Andersen, O. S., Koeppe, R. E. Bilayer thickness and membrane protein function: an energetic perspective. Annual Review of Biophysics and Biomolecular Structure. 36, 107-130 (2007).
- Lundbæk, J. A., Collingwood, S. A., Ingólfsson, H. I., Kapoor, R., Andersen, O. S. Lipid bilayer regulation of membrane protein function: gramicidin channels as molecular force probes. Journal of The Royal Society Interface. 7 (44), 373-395 (2010).
- Bradley, R. P., Radhakrishnan, R. Curvature-undulation coupling as a basis for curvature sensing and generation in bilayer membranes. Proceedings of the National Academy of Sciences of the United States of America. 113 (35), 117-124 (2016).
- Perozo, E., Cortes, D. M., Sompornpisut, P., Kloda, A., Martinac, B. Open channel structure of MscL and the gating mechanism of mechanosensitive channels. Nature. 418 (6901), 942-948 (2002).
- Jensen, M. Ø, Mouritsen, O. G. Lipids do influence protein function-the hydrophobic matching hypothesis revisited. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1666 (1-2), 205-226 (2004).
- Rajendran, L., Simons, K. Lipid rafts and membrane dynamics. Journal of Cell Science. 118 (6), 1099-1102 (2005).
- Katchalsky, A., Spangler, R. Dynamics of membrane processes. Quarterly Reviews of Biophysics. 1 (2), 127-175 (1968).
- Rheinstädter, M. C. Collective molecular dynamics in proteins and membranes (Review). Biointerphases. 3 (2), 83-90 (2008).
- Fujiwara, T., Ritchie, K., Murakoshi, H., Jacobson, K., Kusumi, A. Phospholipids undergo hop diffusion in compartmentalized cell membrane. The Journal of Cell Biology. 157 (6), 1071-1082 (2002).
- Hac, A. E., Seeger, H. M., Fidorra, M., Heimburg, T. Diffusion in two-component lipid membranes--a fluorescence correlation spectroscopy and monte carlo simulation study. Biophysical Journal. 88 (1), 317-333 (2005).
- Heinrich, M., Tian, A., Esposito, C., Baumgart, T. Dynamic sorting of lipids and proteins in membrane tubes with a moving phase boundary. Proceedings of the National Academy of Sciences of the United States of America. 107 (16), 7208-7213 (2010).
- Hormel, T. T., Kurihara, S. Q., Brennan, M. K., Wozniak, M. C., Parthasarathy, R. Measuring lipid membrane viscosity using rotational and translational probe diffusion. Physical Review Letters. 112 (18), 188101 (2014).
- Dimova, R. Recent developments in the field of bending rigidity measurements on membranes. Advances in Colloid and Interface Science. 208, 225-234 (2014).
- Bassereau, P., Sorre, B., Lévy, A. Bending lipid membranes: Experiments after W. Helfrich's model. Advances in Colloid and Interface Science. 208, 47-57 (2014).
- Monzel, C., Sengupta, K. Measuring shape fluctuations in biological membranes. Journal of Physics D: Applied Physics. 49 (24), 243002 (2016).
- Deserno, M. Mesoscopic membrane physics: concepts, simulations, and selected applications. Macromolecular Rapid Communications. 30 (9-10), 752-771 (2009).
- Reynwar, B. J., et al. Aggregation and vesiculation of membrane proteins by curvature-mediated interactions. Nature. 447 (7143), 461-464 (2007).
- Haswell, E. S., Phillips, R., Rees, D. C. Mechanosensitive channels: what can they do and how do they do it. Structure. 19 (10), 1356-1369 (2011).
- Phillips, R., Ursell, T., Wiggins, P., Sens, P. Emerging roles for lipids in shaping membrane-protein function. Nature. 459 (7245), 379-385 (2009).
- Dill, K. A., Chan, H. S. From Levinthal to pathways to funnels. Nature Structural Biology. 4 (1), 10-19 (1997).
- Henzler-Wildman, K., Kern, D. Dynamic personalities of proteins. Nature. 450 (7172), 964-972 (2007).
- Grimaldo, M., Roosen-Runge, F., Zhang, F., Schreiber, F., Seydel, T. Dynamics of proteins in solution. Quarterly Reviews of Biophysics. 52, 7 (2019).
- Lyman, E., Hsieh, C. -L., Eggeling, C. From dynamics to membrane organization: experimental breakthroughs occasion a "modeling manifesto". Biophysical Journal. 115 (4), 595-604 (2018).
- Arriaga, L. R., et al. Dissipative curvature fluctuations in bilayer vesicles: Coexistence of pure-bending and hybrid curvature-compression modes. The European Physical Journal. E, Soft Matter. 31 (1), 105-113 (2010).
- Honerkamp-Smith, A. R., Veatch, S. L., Keller, S. L. An introduction to critical points for biophysicists; observations of compositional heterogeneity in lipid membranes. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1788 (1), 53-63 (2009).
- Veatch, S. L., Keller, S. L. Organization in lipid membranes containing cholesterol. Physical Review Letters. 89 (26), 268101 (2002).
- Heberle, F. A., et al. Bilayer thickness mismatch controls domain size in model membranes. Journal of the American Chemical Society. 135 (18), 6853-6859 (2013).
- Nickels, J. D., et al. The in vivo structure of biological membranes and evidence for lipid domains. PLOS Biology. 15 (5), 2002214 (2017).
- Simons, K., Ikonen, E. Functional rafts in cell membranes. Nature. 387 (6633), 569-572 (1997).
- van Meer, G., Voelker, D. R., Feigenson, G. W. Membrane lipids: where they are and how they behave. Nature Reviews. Molecular Cell Biology. 9 (2), 112-124 (2008).
- Liu, S. -L., et al. Orthogonal lipid sensors identify transbilayer asymmetry of plasma membrane cholesterol. Nature Chemical Biology. 13, 268 (2016).
- Rothman, J., Lenard, J. Membrane asymmetry. Science. 195 (4280), 743-753 (1977).
- Ashkar, R., et al. Neutron scattering in the biological sciences: progress and prospects. Acta Crystallographica Section D. 74 (12), 1129-1168 (2018).
- Woodka, A. C., Butler, P. D., Porcar, L., Farago, B., Nagao, M. Lipid bilayers and membrane dynamics: insight into thickness fluctuations. Physical Review Letters. 109 (5), 058102 (2012).
- Chakraborty, S., et al. How cholesterol stiffens unsaturated lipid membranes. Proceedings of the National Academy of Sciences of the United States of America. 117 (36), 21896-21905 (2020).
- Arriaga, L. R., et al. Stiffening effect of cholesterol on disordered lipid phases: a combined neutron spin echo + dynamic light scattering analysis of the bending elasticity of large unilamellar vesicles. Biophysical Journal. 96 (9), 3629-3637 (2009).
- Nagao, M., Kelley, E. G., Ashkar, R., Bradbury, R., Butler, P. D. Probing elastic and viscous properties of phospholipid bilayers using neutron spin echo spectroscopy. The Journal of Physical Chemistry Letters. 8 (19), 4679-4684 (2017).
- Kelley, E. G., Butler, P. D., Ashkar, R., Bradbury, R., Nagao, M. Scaling relationships for the elastic moduli and viscosity of mixed lipid membranes. Proceedings of the National Academy of Sciences of the United States of America. 117 (38), 23365-23373 (2020).
- Rickeard, B. W., et al. Transverse lipid organization dictates bending fluctuations in model plasma membranes. Nanoscale. 12 (3), 1438-1447 (2020).
- Nickels, J. D., et al. Mechanical properties of nanoscopic lipid domains. Journal of the American Chemical Society. 137 (50), 15772-15780 (2015).
- Mezei, F. Neutron spin echo: A new concept in polarized thermal neutron techniques. Zeitschrift für Physik A Hadrons and Nuclei. 255 (2), 146-160 (1972).
- Hayter, J. B., Penfold, J. Neutron spin-echo integral transform spectroscopy. Zeitschrift für Physik B Condensed Matter. 35 (2), 199-205 (1979).
- Monkenbusch, M., Richter, D. Neutrons in Soft Matter. Imae, T., Kanaya, T., Furusaka, M., Torikai, N. , Wiley. ch6 147-182 (2011).
- Pynn, R. Neutron Spin Echo. Mezei, F., Pappas, C., Gutberlet, T. , Springer. Berlin Heidelberg. 159-177 (2003).
- Holderer, O., et al. The JCNS neutron spin-echo spectrometer J-NSE at the FRM II. Measurement Science and Technology. 19 (3), 034022 (2008).
- Schleger, P., et al. The long-wavelength neutron spin-echo spectrometer IN15 at the Institut Laue-Langevin. Physica B: Condensed Matter. 241-243, 164-165 (1997).
- Holderer, O., Zolnierczuk, P., Pasini, S., Stingaciu, L., Monkenbusch, M. A better view through new glasses: Developments at the Jülich neutron spin echo spectrometers. Physica B: Condensed Matter. 562, 9-12 (2019).
- Farago, B., et al. The IN15 upgrade. Neutron News. 26 (3), 15-17 (2015).
- Ashkar, R. Selective dynamics in polymeric materials: Insights from quasi-elastic neutron scattering spectroscopy. Journal of Applied Physics. 127 (15), 151101 (2020).
- Pasini, S., Holderer, O., Kozielewski, T., Richter, D., Phoenix Monkenbusch, M. J-NSE- Phoenix, a neutron spin-echo spectrometer with optimized superconducting precession coils at the MLZ in Garching. Review of Scientific Instruments. 90 (4), 043107 (2019).
- Svergun, D. I., Koch, M. H. J., Timmins, P. A., May, R. P. Small Angle X-Ray and Neutron Scattering from Solutions of Biological Macromolecules. , Oxford University Press. (2013).
- Eicher, B., et al. Joint small-angle X-ray and neutron scattering data analysis of asymmetric lipid vesicles. Journal of Applied Crystallography. 50 (2), 419-429 (2017).
- Heberle, F. A., et al. Model-based approaches for the determination of lipid bilayer structure from small-angle neutron and X-ray scattering data. European Biophysics Journal. 41 (10), 875-890 (2012).
- Jaksch, S., Koutsioubas, A., Mattauch, S., Holderer, O., Frielinghaus, H. Long-range excitations in phospholipid membranes. Chemistry and Physics of Lipids. 225, 104788 (2019).
- Jaksch, S., et al. Influence of ibuprofen on phospholipid membranes. Physical Review E. 91 (2), 022716 (2015).
- Armstrong, C. L., et al. Effect of cholesterol on the lateral nanoscale dynamics of fluid membranes. European Biophysics Journal. 41 (10), 901-913 (2012).
- Rheinstädter, M. C., Häußler, W., Salditt, T. Dispersion relation of lipid membrane shape fluctuations by neutron spin-echo spectrometry. Physical Review Letters. 97 (4), 048103 (2006).
- Armstrong, C. L., Häußler, W., Seydel, T., Katsaras, J., Rheinstädter, M. C. Nanosecond lipid dynamics in membranes containing cholesterol. Soft Matter. 10 (15), 2600-2611 (2014).
- Nickels, J. D., et al. Lipid rafts: buffers of cell membrane physical properties. The Journal of Physical Chemistry B. 123 (9), 2050-2056 (2019).
- Michonova-Alexova, E. I., Sugár, I. P. Component and state separation in DMPC/DSPC lipid bilayers: a Monte Carlo simulation study. Biophysical Journal. 83 (4), 1820-1833 (2002).
- Sugár, I. P., Thompson, T. E., Biltonen, R. L. Monte Carlo simulation of two-component bilayers: DMPC/DSPC mixtures. Biophysical Journal. 76 (4), 2099-2110 (1999).
- Mabrey, S., Sturtevant, J. M. Investigation of phase transitions of lipids and lipid mixtures by sensitivity differential scanning calorimetry. Proceedings of the National Academy of Sciences. 73 (11), 3862-3866 (1976).
- Neutron activation and scattering calculator. , Available from: https://www.ncnr.nist.gov/resources/activation/ (2021).
- Scott, H. L., et al. On the mechanism of bilayer separation by extrusion, or why your LUVs are not really unilamellar. Biophysical Journal. 117 (8), 1381-1386 (2019).
- Ashkar, R., et al. Tuning membrane thickness fluctuations in model lipid bilayers. Biophysical Journal. 109 (1), 106-112 (2015).
- Carrillo, J. -M. Y., Katsaras, J., Sumpter, B. G., Ashkar, R. A computational approach for modeling neutron scattering data from lipid bilayers. Journal of Chemical Theory and Computation. 13 (2), 916-925 (2017).
- Azuah, R. T. DAVE: a comprehensive software suite for the reduction, visualization, and analysis of low energy neutron spectroscopic data. Journal of Research of the National Institute of Standards and Technology. 114 (6), 341-358 (2009).
- Van Hove, L. Correlations in space and time and born approximation scattering in systems of interacting particles. Physical Review. 95 (1), 249-262 (1954).
- Zilman, A. G., Granek, R. Undulations and dynamic structure factor of membranes. Physical Review Letters. 77 (23), 4788-4791 (1996).
- Kelley, E. G., Butler, P. D., Nagao, M. Collective dynamics in model biological membranes measured by neutron spin echo spectroscopy. , Walter de Gruyter, Inc. 131-176 (2019).
- Zheng, Y., Michihiro, N., Dobrin, P. B. Bending elasticity of saturated and monounsaturated phospholipid membranes studied by the neutron spin echo technique. Journal of Physics: Condensed Matter. 21 (15), 155104 (2009).
- Sharma, V. K., Qian, S. Effect of an antimicrobial peptide on lateral segregation of lipids: a structure and dynamics study by neutron scattering. Langmuir. 35 (11), 4152-4160 (2019).
- Boggara, M. B., Faraone, A., Krishnamoorti, R. Effect of pH and Ibuprofen on the Phospholipid Bilayer Bending Modulus. The Journal of Physical Chemistry B. 114 (24), 8061-8066 (2010).
- Lee, J. -H., et al. Thermal fluctuation and elasticity of lipid vesicles interacting with pore-forming peptides. Physical Review Letters. 105 (3), 038101 (2010).
- Chakraborty, S., Abbasi, A., Bothun, G. D., Nagao, M., Kitchens, C. L. Phospholipid bilayer softening due to hydrophobic gold nanoparticle inclusions. Langmuir. 34 (44), 13416-13425 (2018).
- Hoffmann, I., et al. Softening of phospholipid membranes by the adhesion of silica nanoparticles - as seen by neutron spin-echo (NSE). Nanoscale. 6 (12), 6945-6952 (2014).
- Watson, M. C., Brown, F. L. H. Interpreting membrane scattering experiments at the mesoscale: the contribution of dissipation within the bilayer. Biophysical Journal. 98 (6), 9-11 (2010).
- Seifert, U., Langer, S. A. Viscous modes of fluid bilayer membranes. Europhysics Letters (EPL). 23 (1), 71-76 (1993).
- Bingham, R. J., Smye, S. W., Olmsted, P. D. Dynamics of an asymmetric bilayer lipid membrane in a viscous solvent. EPL (Europhysics Letters). 111 (1), 18004 (2015).
- Rawicz, W., Olbrich, K. C., McIntosh, T., Needham, D., Evans, E. Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophysical Journal. 79 (1), 328-339 (2000).
- Doktorova, M., LeVine, M. V., Khelashvili, G., Weinstein, H. A new computational method for membrane compressibility: bilayer mechanical thickness revisited. Biophysical Journal. 116 (3), 487-502 (2019).
- Evans, E., Needham, D. Physical properties of surfactant bilayer membranes: thermal transitions, elasticity, rigidity, cohesion and colloidal interactions. The Journal of Physical Chemistry. 91 (16), 4219-4228 (1987).
- Lesieur, S., Grabielle-Madelmont, C., Paternostre, M. T., Ollivon, M. Size analysis and stability study of lipid vesicles by high-performance gel exclusion chromatography, turbidity, and dynamic light scattering. Analytical Biochemistry. 192 (2), 334-343 (1991).
- Heberle, F. A., et al. Direct label-free imaging of nanodomains in biomimetic and biological membranes by cryogenic electron microscopy. Proceedings of the National Academy of Sciences of the United States of America. 117 (33), 19943-19952 (2020).
- Cornell, C. E., Mileant, A., Thakkar, N., Lee, K. K., Keller, S. L. Direct imaging of liquid domains in membranes by cryo-electron tomography. Proceedings of the National Academy of Sciences of the United States of America. 117 (33), 19713-19719 (2020).
- Yao, X., Fan, X., Yan, N. Cryo-EM analysis of a membrane protein embedded in the liposome. Proceedings of the National Academy of Sciences of the United States of America. 117 (31), 18497-18503 (2020).
- Kučerka, N., Nieh, M. -P., Katsaras, J. Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1808 (11), 2761-2771 (2011).
- Nielsen, J. E., Bjørnestad, V. A., Lund, R. Resolving the structural interactions between antimicrobial peptides and lipid membranes using small-angle scattering methods: the case of indolicidin. Soft Matter. 14 (43), 8750-8763 (2018).
- Kučerka, N., et al. Lipid bilayer structure determined by the simultaneous analysis of neutron and X-ray scattering data. Biophysical Journal. 95 (5), 2356-2367 (2008).
- Kelley, E. G., Butler, P. D., Nagao, M. Scaling of lipid membrane rigidity with domain area fraction. Soft Matter. 15 (13), 2762-2767 (2019).
- Brüning, B. -A., et al. Bilayer undulation dynamics in unilamellar phospholipid vesicles: Effect of temperature, cholesterol and trehalose. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1838 (10), 2412-2419 (2014).
- Kučerka, N., et al. Areas of monounsaturated diacylphosphatidylcholines. Biophysical Journal. 97 (7), 1926-1932 (2009).
- Sharma, V. K., Mamontov, E., Anunciado, D. B., O'Neill, H., Urban, V. S. Effect of antimicrobial peptide on the dynamics of phosphocholine membrane: role of cholesterol and physical state of bilayer. Soft Matter. 11 (34), 6755-6767 (2015).
- Kelley, E. G., Butler, P. D., Nagao, M. Collective dynamics in lipid membranes containing transmembrane peptides. Soft Matter. , (2021).
- Yu, J., et al. Structure and dynamics of lipid membranes interacting with antivirulence end-phosphorylated polyethylene glycol block copolymers. Soft Matter. 16 (4), 983-989 (2020).
- Stingaciu, L. -R., et al. Revealing the dynamics of thylakoid membranes in living cyanobacterial cells. Scientific Reports. 6 (1), 19627 (2016).
- Stingaciu, L. -R., O'Neill, H. M., Liberton, M., Pakrasi, H. B., Urban, V. S. Influence of chemically disrupted photosynthesis on cyanobacterial thylakoid dynamics in synechocystis sp. PCC 6803. Scientific Reports. 9 (1), 5711 (2019).
- Miller, I. R. Energetics of fluctuation in lipid bilayer thickness. Biophysical Journal. 45 (3), 643-644 (1984).
- Nagao, M. Observation of local thickness fluctuations in surfactant membranes using neutron spin echo. Physical Review E. 80 (3), 031606 (2009).
Tags
Biologia Edição 171 dinâmica lipídica coletiva módulo de dobra compressão de área flutuações de espessura viscosidade da membrana deuteração seletiva preparação lipossaErratum
Formal Correction: Erratum: Neutron Spin Echo Spectroscopy as a Unique Probe for Lipid Membrane Dynamics and Membrane-Protein Interactions
Posted by JoVE Editors on 08/06/2021.
Citeable Link.
An erratum was issued for: Neutron Spin Echo Spectroscopy as a Unique Probe for Lipid Membrane Dynamics and Membrane-Protein Interactions. The Introduction, Protocol, and Representative Results sections have been updated.
In the Introduction, the fith pargraph was updated from:
Besides direct access to the length and time scale of membrane dynamics, NSE has the inherent capabilities of neutron isotope sensitivity52. Specifically, the ability of neutrons to interact differently with the isotopes of hydrogen, the most abundant element in biological systems, results in a different neutron scattering length density,34 or NSLD (the equivalent of the optical index of refraction50), when protium is substituted by deuterium. This enables an approach known as contrast variation, which is commonly used to highlight specific membrane features or conceal others — the latter scenario is referred to as contrast matching. A frequent application of contrast variation/matching is the substitution of water (NSLD = -0.56 × 10-6 Å-2) by heavy water or D2O (NSLD = 6.4 × 10-6 Å-2) to amplify the neutron signal from protiated lipid membranes (NSLD ~ 2 × 10-6 Å-2). This approach is highly effective in studies of membrane structure because the penetration of D2O into the headgroup region of the membrane allows accurate determination of the membrane thicknesses (see Figure 2A, left panel) and of the location of different lipid subgroups when more sophisticated models are applied53,54. This paper highlights some examples on the use of contrast variation for studies of collective dynamics in biomimetic membranes and select membrane features.
to:
Besides direct access to the length and time scale of membrane dynamics, NSE has the inherent capabilities of neutron isotope sensitivity52. Specifically, the ability of neutrons to interact differently with the isotopes of hydrogen, the most abundant element in biological systems, results in a different neutron scattering length density,34 or NSLD (the equivalent of the optical index of refraction50), when protium is substituted by deuterium. This enables an approach known as contrast variation, which is commonly used to highlight specific membrane features or conceal others — the latter scenario is referred to as contrast matching. A frequent application of contrast variation/matching is the substitution of water (NSLD = -0.56 × 10-6 Å-2) by heavy water or D2O (NSLD = 6.4 × 10-6 Å-2) to amplify the neutron signal from protiated lipid membranes (NSLD ~ 0 × 10-6 Å-2). This approach is highly effective in studies of membrane structure because the penetration of D2O into the headgroup region of the membrane allows accurate determination of the membrane thicknesses (see Figure 2A, left panel) and of the location of different lipid subgroups when more sophisticated models are applied53,54. This paper highlights some examples on the use of contrast variation for studies of collective dynamics in biomimetic membranes and select membrane features.
In the Protocol, step 1.1 was updated from:
For bending fluctuation measurements, make fully protiated liposomes in D2O (D 99.9%) or D2O-buffer (e.g., phosphate buffer prepared with D2O instead of H2O). Use fully protiated DMPC (C36H72NO8P) and DSPC (C44H88NO8P) with 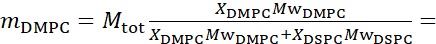 133.4 mg, where XDMPC and XDSPC are the mole fractions of DMPC and DSPC, here set to 0.7 and 0.3, respectively, and MwDMPC and MwDSPC are the molar weights given by 677.9 g/mol and 790.1 g/mol, respectively. Similarly, mDSPC = 66.6 mg. This deuteration scheme increases the scattering contrast between the membrane (NSLD ~ 2 × 10-6 Å-2) and the deuterated buffer (NSLD ~ 6.4 × 10-6 Å-2) and amplifies the signal from membrane undulations (see Figure 2A left panel).
133.4 mg, where XDMPC and XDSPC are the mole fractions of DMPC and DSPC, here set to 0.7 and 0.3, respectively, and MwDMPC and MwDSPC are the molar weights given by 677.9 g/mol and 790.1 g/mol, respectively. Similarly, mDSPC = 66.6 mg. This deuteration scheme increases the scattering contrast between the membrane (NSLD ~ 2 × 10-6 Å-2) and the deuterated buffer (NSLD ~ 6.4 × 10-6 Å-2) and amplifies the signal from membrane undulations (see Figure 2A left panel).
to:
For bending fluctuation measurements, make fully protiated liposomes in D2O (D 99.9%) or D2O-buffer (e.g., phosphate buffer prepared with D2O instead of H2O). Use fully protiated DMPC (C36H72NO8P) and DSPC (C44H88NO8P) with 133.4 mg, where XDMPC and XDSPC are the mole fractions of DMPC and DSPC, here set to 0.7 and 0.3, respectively, and MwDMPC and MwDSPC are the molar weights given by 677.9 g/mol and 790.1 g/mol, respectively. Similarly, mDSPC = 66.6 mg. This deuteration scheme increases the scattering contrast between the membrane (NSLD ~ 0 × 10-6 Å-2) and the deuterated buffer (NSLD ~ 6.4 × 10-6 Å-2) and amplifies the signal from membrane undulations (see Figure 2A left panel).
In the Representative Results, the fist pagargaph was updted from:
NSE studies accessing bending fluctuations are typically performed over a Q-range of ~ (0.04 - 0.2) Å-1. This Q-range corresponds to intermediate length scales between the membrane thickness and the liposomal radius, where bending dynamics dominate. Measurement over an extended Q-range can give access to additional dynamic modes, including liposomal diffusion and intramembrane dynamics. For more details on the cross-over in membrane dynamics accessed by NSE, check these relevant publications25,71. It is important to emphasize that NSE signals are proportional to: 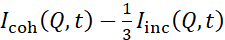 , where Icoh and Iinc are, respectively, the coherent and incoherent scattering intensity from the sample. Therefore, it is advisable to prepare NSE liposomal samples in deuterated buffers (i.e., buffers prepared with D2O instead of H2O) to minimize the incoherent scattering signal, mainly contributed by the hydrogen content of the sample. However, in some cases intermediate deuteration schemes (i.e., using mixtures of D2O and H2O) might be necessary to obtain optimal contrast conditions. Typically, NSE measurements of membrane bending fluctuations are performed on fully protiated liposomes in deuterated buffer, referred to as fully contrasted liposomes in Figure 5. This deuteration scheme results in a large NSLD difference between the membrane core (~2 × 10-6 Å-2) and its deuterated fluid environment (~6.4 × 10-6 Å-2), which significantly enhances the scattering signal from the liposomal membranes and improves the measurement statistics of bending dynamics. This contrast scheme (Figure 2A left panel) is frequently utilized in studies of bending rigidity of lipid membranes with single38,72 and multiple39,66 lipid components and in studies of membrane softening/stiffening by biological inclusions (e.g., cholesterol, drug molecules, peptides/proteins)36,37,73,74,75, and synthetic additives (e.g., nanoparticles)76,77.
, where Icoh and Iinc are, respectively, the coherent and incoherent scattering intensity from the sample. Therefore, it is advisable to prepare NSE liposomal samples in deuterated buffers (i.e., buffers prepared with D2O instead of H2O) to minimize the incoherent scattering signal, mainly contributed by the hydrogen content of the sample. However, in some cases intermediate deuteration schemes (i.e., using mixtures of D2O and H2O) might be necessary to obtain optimal contrast conditions. Typically, NSE measurements of membrane bending fluctuations are performed on fully protiated liposomes in deuterated buffer, referred to as fully contrasted liposomes in Figure 5. This deuteration scheme results in a large NSLD difference between the membrane core (~2 × 10-6 Å-2) and its deuterated fluid environment (~6.4 × 10-6 Å-2), which significantly enhances the scattering signal from the liposomal membranes and improves the measurement statistics of bending dynamics. This contrast scheme (Figure 2A left panel) is frequently utilized in studies of bending rigidity of lipid membranes with single38,72 and multiple39,66 lipid components and in studies of membrane softening/stiffening by biological inclusions (e.g., cholesterol, drug molecules, peptides/proteins)36,37,73,74,75, and synthetic additives (e.g., nanoparticles)76,77.
to:
NSE studies accessing bending fluctuations are typically performed over a Q-range of ~ (0.04 - 0.2) Å-1. This Q-range corresponds to intermediate length scales between the membrane thickness and the liposomal radius, where bending dynamics dominate. Measurement over an extended Q-range can give access to additional dynamic modes, including liposomal diffusion and intramembrane dynamics. For more details on the cross-over in membrane dynamics accessed by NSE, check these relevant publications25,71. It is important to emphasize that NSE signals are proportional to: , where Icoh and Iinc are, respectively, the coherent and incoherent scattering intensity from the sample. Therefore, it is advisable to prepare NSE liposomal samples in deuterated buffers (i.e., buffers prepared with D2O instead of H2O) to minimize the incoherent scattering signal, mainly contributed by the hydrogen content of the sample. However, in some cases intermediate deuteration schemes (i.e., using mixtures of D2O and H2O) might be necessary to obtain optimal contrast conditions. Typically, NSE measurements of membrane bending fluctuations are performed on fully protiated liposomes in deuterated buffer, referred to as fully contrasted liposomes in Figure 5. This deuteration scheme results in a large NSLD difference between the membrane core (~0 × 10-6 Å-2) and its deuterated fluid environment (~6.4 × 10-6 Å-2), which significantly enhances the scattering signal from the liposomal membranes and improves the measurement statistics of bending dynamics. This contrast scheme (Figure 2A left panel) is frequently utilized in studies of bending rigidity of lipid membranes with single38,72 and multiple39,66 lipid components and in studies of membrane softening/stiffening by biological inclusions (e.g., cholesterol, drug molecules, peptides/proteins)36,37,73,74,75, and synthetic additives (e.g., nanoparticles)76,77.
In the Representative Reults, Figure 2 was updated from:
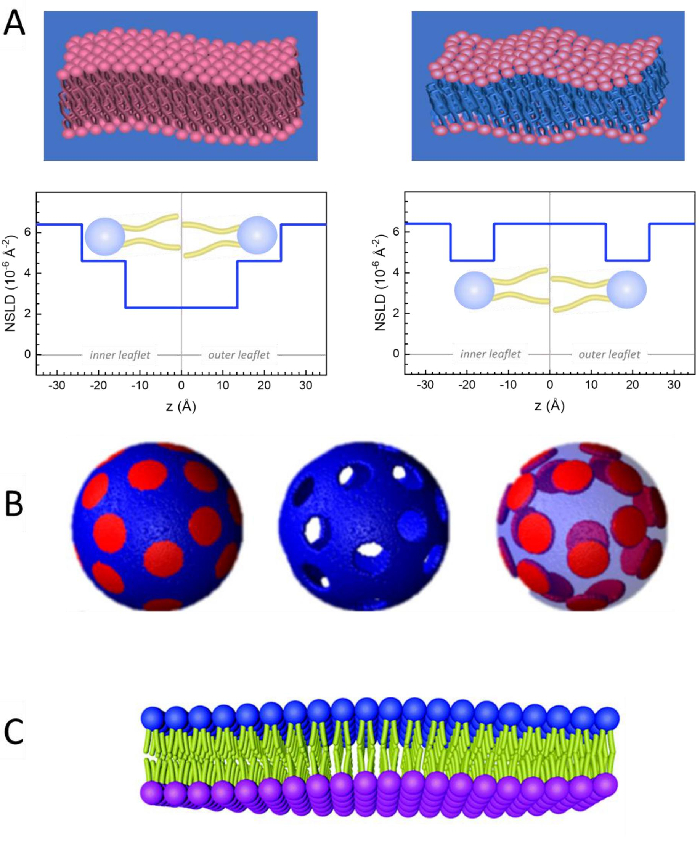
Figure 2: Examples of possible deuteration schemes in NSE experiments on lipid membranes. (A) Left: Fully contrasted membranes, e.g., protiated membranes in deuterated buffer, showing the NSLD profile along the normal to the membrane surface. The difference in the NSLD between the headgroup (~2 × 10-2 Å-2) and tail region (~4.5 × 10-6 Å-2) of the membrane is due to the headgroup hydration with deuterated buffer. Right: Tail-contrast matched membranes such that the hydrocarbon tail region of the membrane has the same NSLD as the buffer, as shown in the corresponding NSLD profile along the membrane normal. (B) Domain-forming membranes with two neutron contrast schemes where the domains (center) or the matrix (left) are contrast-matched to the buffer, enabling selective studies of matrix or domain dynamics, respectively. This figure has been modified from Nickels et al., JACS 201541. (C) Asymmetric membranes prepared by cyclodextrin exchange between protiated and deuterated lipid vesicles, resulting in the deuteration of one membrane leaflet while keeping the other leaflet protiated. This allows studies of the bending dynamics of the protiated leaflet and provides insights into the mechanical coupling between opposing leaflets in asymmetric membranes. This figure has been modified from Rickeard et al., Nanoscale 202040. Please click here to view a larger version of this figure.
to:
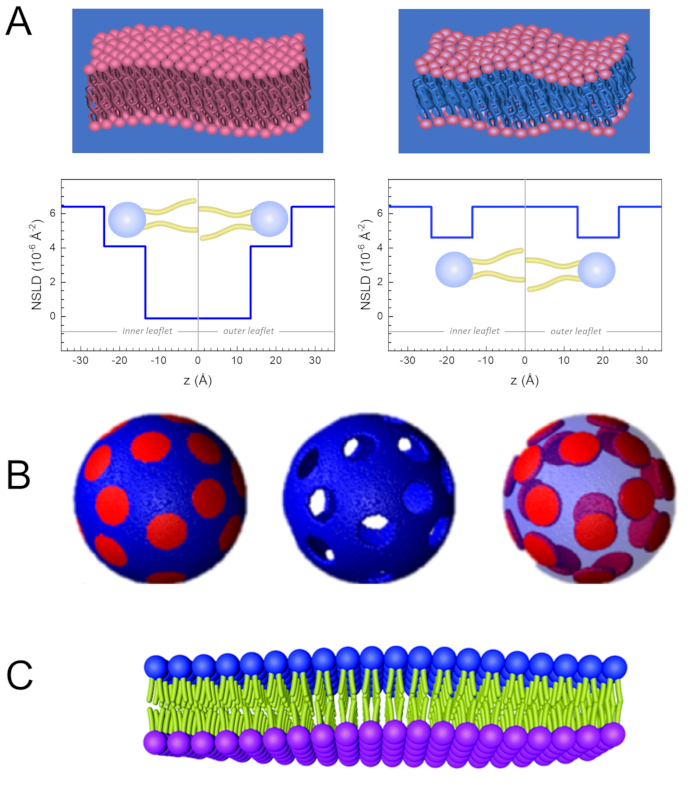
Figure 2: Examples of possible deuteration schemes in NSE experiments on lipid membranes. (A) Left: Fully contrasted membranes, e.g., protiated membranes in deuterated buffer, showing the NSLD profile along the normal to the membrane surface. The difference in the NSLD between the tail region (~0 × 10-2 Å-2) and headgroup region (~4.5 × 10-6 Å-2) of the membrane is due to the headgroup hydration with deuterated buffer. Right: Tail-contrast matched membranes such that the hydrocarbon tail region of the membrane has the same NSLD as the buffer, as shown in the corresponding NSLD profile along the membrane normal. (B) Domain-forming membranes with two neutron contrast schemes where the domains (center) or the matrix (left) are contrast-matched to the buffer, enabling selective studies of matrix or domain dynamics, respectively. This figure has been modified from Nickels et al., JACS 201541. (C) Asymmetric membranes prepared by cyclodextrin exchange between protiated and deuterated lipid vesicles, resulting in the deuteration of one membrane leaflet while keeping the other leaflet protiated. This allows studies of the bending dynamics of the protiated leaflet and provides insights into the mechanical coupling between opposing leaflets in asymmetric membranes. This figure has been modified from Rickeard et al., Nanoscale 202040. Please click here to view a larger version of this figure.
