

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
Dieser Artikel beschreibt die Protokolle für die Probenvorbereitung, Datenreduktion und Datenanalyse in Neutronenspinechostudien (NSE) von Lipidmembranen. Eine umsichtige Deuteriummarkierung von Lipiden ermöglicht den Zugang zu unterschiedlichen Membrandynamiken auf mesoskopischen Längen- und Zeitskalen, über die lebenswichtige biologische Prozesse ablaufen.
Abstract
Lipiddoppelschichten bilden die Hauptmatrix der Zellmembranen und sind die primäre Plattform für Nährstoffaustausch, Protein-Membran-Interaktionen und virale Knospung, neben anderen lebenswichtigen zellulären Prozessen. Für eine effiziente biologische Aktivität sollten Zellmembranen starr genug sein, um die Integrität der Zelle und ihrer Kompartimente aufrechtzuerhalten, aber flüssig genug, um Membrankomponenten wie Proteine und funktionelle Domänen zu zerstreuen und interagieren zu lassen. Dieses empfindliche Gleichgewicht zwischen elastischen und flüssigen Membraneigenschaften und deren Einfluss auf die biologische Funktion erfordern ein besseres Verständnis der kollektiven Membrandynamik über mesoskopische Längen- und Zeitskalen wichtiger biologischer Prozesse, z. B. Membranverformungen und Proteinbindungsereignisse. Zu den Techniken, die diesen Dynamikbereich effektiv untersuchen können, gehört die Neutronenspinechospektroskopie (NSE). In Kombination mit der Deuterium-Markierung kann NSE verwendet werden, um direkt auf Biege- und Dickenschwankungen sowie auf die mesoskopische Dynamik ausgewählter Membranmerkmale zuzugreifen. Dieses Papier bietet eine kurze Beschreibung der NSE-Technik und beschreibt die Verfahren zur Durchführung von NSE-Experimenten an liposomalen Membranen, einschließlich Details zur Probenvorbereitung und Deuteration von Schemata sowie Anweisungen zur Datenerhebung und -reduktion. Das Papier stellt auch Datenanalysemethoden vor, die verwendet werden, um wichtige Membranparameter wie den Biegesteifigkeitsmodul, den Flächenkompressibilitätsmodul und die Viskosität in der Ebene zu extrahieren. Um die biologische Bedeutung von NSE-Studien zu veranschaulichen, werden ausgewählte Beispiele für Membranphänomene diskutiert, die von NSE untersucht wurden, nämlich die Wirkung von Additiven auf die Membranbiegesteifigkeit, den Einfluss der Domänenbildung auf Membranfluktuationen und die dynamische Signatur von Membran-Protein-Interaktionen.
Introduction
Das Verständnis von Zellmembranen und ihrer Funktion hat sich in den letzten Jahrzehnten bemerkenswert weiterentwickelt. Die frühere Sichtweise von Zellmembranen als passive Lipiddoppelschichten, die Zellgrenzen definieren und Membranproteine1 beherbergen, hat sich allmählich in ein dynamisches Modell verwandelt, in dem Lipiddoppelschichten eine wichtige Rolle bei der Regulierung lebenswichtiger biologischer Prozesse spielen, einschließlich zellulärer Signalgebung, molekularer Austausch und Proteinfunktion - um nur einige zu nennen2,3,4,5,6. Diese Erkenntnis, dass Zellmembranen hochdynamisch sind und ständig umgestaltet und molekular umverteilt werden, hat wissenschaftliche Erkundungen jenseits der Gleichgewichtsstrukturen der Membranen7,8,9vorgeben. Dementsprechend wurden mehrere Ansätze entwickelt, um die verschiedenen dynamischen Modi in biologischen und bioinspirierten Lipidmembranen zu untersuchen. Bisher konzentrierte sich die Mehrheit dieser Studien hauptsächlich auf diffusive molekulare Bewegungen10 , 11,12,13 und makroskopische Formfluktuationen14,15,16, die eine signifikante Lücke im Verständnis der mittleren Membrandynamik hinterlassen, d.h. kollektive Fluktuationen von Lipidanordnungen, die aus wenigen 10-100s Lipidmolekülen bestehen. Diese Dynamik tritt über Längenskalen von wenigen zehn bis wenigen 100 Å und über Zeitskalen von Sub-ns bis zu einigen hundert ns auf (siehe Abbildung 1),hier als mesoskopische Skalen bezeichnet. Auf diesen Skalen findet die biologische Schlüsselaktivität auf derMembranebene 17statt. Dazu gehören virale Knospen18, Kanal-Gating19und Membran-Protein-Interaktionen20. Es ist auch wichtig darauf hinzuweisen, dass die Energielandschaft der Membranproteine21,22 zeigt, dass Konformationsänderungen in Proteinen - notwendig für ihre regulatorische Rolle - über die ns-Zeitskalen23 kollektiver Membranfluktuationen stattfinden, was die Bedeutung der mesoskopischen Dynamik in der biologischen Funktion von Zellmembranen und ihren bioinspirierten Analoga weiter unterstreicht20. Dieser Beitrag konzentriert sich auf die beiden primären mesoskopischen dynamischen Modi in Lipidmembranen, nämlich Biegeschwankungen und Dickenschwankungen.
Die größte Herausforderung bei der direkten Untersuchung dieser Fluktuationsmodi ist die Schwierigkeit, gleichzeitig auf ihre räumlichen und zeitlichen Skalen mit Standardspektroskopiemethoden zuzugreifen. Die andere Herausforderung besteht darin, dass direkte Kontakttechniken die gleichen Schwankungen beeinflussen könnten, die sie messen sollen16. Dies wird durch die kompositorische und strukturelle Komplexität der biologischen Membranen24,25nochverschärft,was zu inhomogenen Membranmerkmalen führt, einschließlich der Lipiddomänenbildung26,27,28,29,30 und Membranasymmetrie31,32,33- die selektive Sonden erfordern, um die Dynamik verschiedener Membranmerkmale zu verstehen. Glücklicherweise können diese Herausforderungen mit nicht-invasiven Neutronenspektroskopiemethoden wie Neutronenspinecho (NSE) überwunden werden, die von Natur aus auf die erforderlichen Längen- und Zeitskalen zugreifen und weitere Untersuchungen selektiver Membranmerkmale ermöglichen, ohne ihre physikalisch-chemische Umgebung zu verändern34. Tatsächlich hat sich die NSE-Spektroskopie in den letzten Jahren zu einer einzigartigen und leistungsstarken Sonde der kollektiven Membrandynamik entwickelt35. Ergebnisse aus NSE-Studien an Lipidmembranen haben neue Erkenntnisse über mechanische36,37 und viskoelastische38,39 Eigenschaften von Lipidmembranen geliefert und ein neues Licht auf ihre mögliche Rolle in der biologischen Funktion geworfen40,41.
Die NSE-Spektroskopietechnik basiert auf einem interferometrischen Instrumentendesign, das erstmals von Mezei42vorgeschlagen wurde und eine Reihe von Spin-Flippern und Magnetspulen verwendet, um die Präzession des Neutronenspins zu steuern, während Neutronen das Instrument durchqueren. Das Design beruht auf der magnetischen Spiegelung der Magnetfeldelemente in Bezug auf die Probenposition (Abbildung 1A). Dies bedeutet, dass das Neutron in Abwesenheit eines Energieaustauschs zwischen dem Neutron und der Probe in der ersten und zweiten Hälfte des Instruments die gleiche Anzahl von Spin-Präzessionen in entgegengesetzte Richtungen durchführt (beachten Sie den π-Flipper zwischen den beiden Präzessionsspulen). Infolgedessen bleibt der endgültige Spinzustand des Neutrons relativ zum Ausgangszustand unverändert - ein Phänomen, das als Spin-Echo bezeichnet wird (siehe transparentes Neutron in Abbildung 1A). Wenn das Neutron jedoch energetisch mit der Probe wechselwechselt, verändert der Energieaustausch die Anzahl der Spin-Präzessionen in der zweiten Hälfte des Instruments, was zu einem anderen endgültigen Spin-Zustand führt (siehe Abbildung 1A). Dies wird experimentell als Polarisationsverlust nachgewiesen, wie später in diesem Artikel gezeigt wird. Für weitere Einzelheiten zur NSE-Technik wird der Leser auf die speziellen technischen Papiere42,43,44,45verwiesen.
Hier wird eine vereinfachte Beschreibung präsentiert, um eine grobe Schätzung der Längen- und Zeitskalen zu liefern, die mit NSE zugänglich sind. Die Längenskalen werden durch den Bereich der erreichbaren Wellenvektortransfers bestimmt, Q = 4π sin θ/λ, wobei 2θ der Streuwinkel und λ die Neutronenwellenlänge ist. Man kann sehen, dass Q durch den Wellenlängenbereich und das Ausmaß der Rotation des zweiten Arms des Spektrometers eingestellt wird (siehe Abbildung 1A). Ein typischer Q-Bereichauf NSE-Spektrometern ist ~0,02-2 Å-146,47und bis zu 0,01-4 Å-1 mit den jüngsten Upgrades48,49, entsprechend räumlichen Skalen von ~ 1-600 Å. Auf der anderen Seite wird die zugängliche Zeitskala aus dem gesamten Präzessionswinkel (oder der Phase) berechnet, der vom Neutron innerhalb der magnetischen Präzessionsspulen erfasst wird, und es wird festgestellt, dasses 50:  . In diesem Ausdruck ist t die Fourierzeit, definiert als
. In diesem Ausdruck ist t die Fourierzeit, definiert als  , wobei das
, wobei das  Neutronen-Gyromagnet-Verhältnis,
Neutronen-Gyromagnet-Verhältnis,  die Spulenlänge und
die Spulenlänge und  die Stärke des Magnetfeldes der Spule ist. Es ist erwähnenswert, dass die Fourierzeit eine Größe ist, die streng von der Instrumentengeometrie, der Magnetfeldstärke und der Neutronenwellenlänge abhängt. Zum Beispiel wird die Fourierzeit unter Verwendung von Neutronen mit einer Wellenlänge
die Stärke des Magnetfeldes der Spule ist. Es ist erwähnenswert, dass die Fourierzeit eine Größe ist, die streng von der Instrumentengeometrie, der Magnetfeldstärke und der Neutronenwellenlänge abhängt. Zum Beispiel wird die Fourierzeit unter Verwendung von Neutronen mit einer Wellenlänge  = 8 Å und Instrumenteneinstellungen von = 1,2 m und = 0,4 T als t ~ 50 ns berechnet. Experimentell wird die Fourierzeit durch Änderung des Stroms in den Präzessionsspulen (d. H. Magnetfeldstärke) oder unter Verwendung verschiedener Neutronenwellenlängen abgestimmt, was zu typischen NSE-Zeitskalen von ~ 1 ps bis 100 ns führt. Jüngste Upgrades bei NSE-Spektrometern haben jedoch den Zugriff auf längere Fourier-Zeiten ermöglicht, bis zu ~ 400 ns auf dem J-NSE-Phoenix-Spektrometer am Heinz Maier-Leibnitz Zentrum51 und dem SNS-NSE-Spektrometer am Oak Ridge National Lab48und bis zu ~ 1.000 ns am IN15 NSE-Spektrometer am Institut Laue-Langevin (ILL)49.
= 8 Å und Instrumenteneinstellungen von = 1,2 m und = 0,4 T als t ~ 50 ns berechnet. Experimentell wird die Fourierzeit durch Änderung des Stroms in den Präzessionsspulen (d. H. Magnetfeldstärke) oder unter Verwendung verschiedener Neutronenwellenlängen abgestimmt, was zu typischen NSE-Zeitskalen von ~ 1 ps bis 100 ns führt. Jüngste Upgrades bei NSE-Spektrometern haben jedoch den Zugriff auf längere Fourier-Zeiten ermöglicht, bis zu ~ 400 ns auf dem J-NSE-Phoenix-Spektrometer am Heinz Maier-Leibnitz Zentrum51 und dem SNS-NSE-Spektrometer am Oak Ridge National Lab48und bis zu ~ 1.000 ns am IN15 NSE-Spektrometer am Institut Laue-Langevin (ILL)49.
Neben dem direkten Zugriff auf die Längen- und Zeitskala der Membrandynamik verfügt NSE über die inhärenten Fähigkeiten der Neutronenisotopenempfindlichkeit52. Insbesondere die Fähigkeit von Neutronen, unterschiedlich mit den Isotopen von Wasserstoff, dem am häufigsten vorkommenden Element in biologischen Systemen, zu interagieren, führt zu einer anderen Neutronenstreulängendichte,34 oder NSLD (das Äquivalent des optischen Brechungsindex50),wenn Protium durch Deuterium ersetzt wird. Dies ermöglicht einen Ansatz, der als Kontrastvariation bekannt ist und häufig verwendet wird, um bestimmte Membranmerkmale hervorzuheben oder andere zu verbergen - das letztere Szenario wird als Kontrastabgleich bezeichnet. Eine häufige Anwendung von Kontrastvariation/Matching ist die Substitution von Wasser (NSLD = -0,56 × 10-6 Å-2) durch schweres Wasser oderD2O (NSLD = 6,4 × 10-6 Å-2), um das Neutronensignal von protiierten Lipidmembranen (NSLD ~ 0 × 10-6 Å-2) zu verstärken. Dieser Ansatz ist bei Untersuchungen der Membranstruktur sehr effektiv, da das Eindringen vonD2Oin den Kopfgruppenbereich der Membran eine genaue Bestimmung der Membrandicken (siehe Abbildung 2A, linkes Feld) und der Lage verschiedener Lipiduntergruppen ermöglicht, wenn anspruchsvollere Modelle angewendet werden53,54. Dieser Artikel hebt einige Beispiele für die Verwendung von Kontrastvariationen für Studien der kollektiven Dynamik in biomimetischen Membranen und ausgewählten Membranmerkmalen hervor.
Hier wird die Wirksamkeit von NSE bei der Bereitstellung einzigartiger Einblicke in dynamische und funktionelle Membraneigenschaften anhand konkreter Beispiele von NSE-Studien zu Modell- und biologisch relevanten Lipidmembransystemen mit Schwerpunkt auf mesoskaliger Dynamik in freistehenden Membranen in Form von liposomalen Suspensionen veranschaulicht. Für NSE-Messungen der In-Plane-Membrandynamik wird der Leser auf spezielle Publikationen zur Grazing-Incidence-Neutronen-Spin-Echo-Spektroskopie (GINSES)55,56 und andere Studien von ausgerichteten multilamellaren Membranstapeln57,58,59,60verwiesen.
Der Einfachheit halber hebt dieser Artikel drei verschiedene Schemata der Membrandeuteration hervor, die auf einem gut untersuchten domänenbildenden oder phasenabtrennenden Lipiddoppelschichtsystem aus 1,2-Dimyristoyl-sn-glycero-3-phosphocholin (DMPC) und 1,2-Distearoyl-sn-glycero-3-phosphocholin (DSPC) -Mischungen61,62dargestellt werden. Die beiden Lipide zeichnen sich durch eine Diskrepanz in ihrer Kohlenwasserstoffkettenlänge (14 Kohlenstoffe / Schwanz in DMPC vs. 18 Kohlenstoffe / Schwanz in DSPC) und ihrer Gel-Fluid-Übergangstemperatur (Tm, DMPC = 23 ° C vs Tm, DSPC = 55 ° C) aus. Dies führt zu einer lateralen Phasentrennung in DMPC:DSPC-Membranen bei Temperaturen zwischen der oberen und unteren Übergangstemperatur des Gemisches63. Die hier betrachteten Deuterationsschemata werden ausgewählt, um die verschiedenen dynamischen Modi zu demonstrieren, die bei NSE-Messungen auf liposomalen Membranen verfügbar sind, nämlich Biegefluktuationen, Dickenschwankungen und selektive Biege-/Dickenschwankungen von lateralen Domänen. Alle Lipidzusammensetzungen werden für DMPC:DSPC-Doppelschichten berichtet, die mit einem Molanteil von 70:30 unter Verwendung handelsüblich erhältlicher protiated und perdeuterated Varianten von DMPC und DSPC hergestellt wurden. Alle Probenvorbereitungsschritte basieren auf 4 ml liposomaler Suspension inD2O mit einer Lipidkonzentration von 50 mg/ml für eine Gesamtlipidmasse von Mtot = 200 mg pro Probe.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Für das Experiment erforderliches Deuterationsschema
- Für Biegeschwankungsmessungen werden vollständig protiierte Liposomen inD2O(D 99,9%) oderD2O-Puffer (z. B. Phosphatpuffer mitD2Oanstelle vonH2Ohergestellt) hergestellt. Verwenden Sie vollständig protiierte DMPC (C36H72NO8P) und DSPC (C44H88NO8P) mit
 133,4 mg, wobei XDMPC und XDSPC die Molfraktionen von DMPC und DSPC sind, hier auf 0,7 bzw. 0,3 festgelegt, und MwDMPC und MwDSPC sind die molaren Gewichte von 677,9 g / mol bzw. 790,1 g / mol. In ähnlicher Weise mDSPC = 66,6 mg. Dieses Deuterationsschema erhöht den Streukontrast zwischen der Membran (NSLD ~ 0 × 10-6 Å-2) und dem deuterierten Puffer (NSLD ~ 6,4 × 10-6 Å-2) und verstärkt das Signal von Membranaufführungen (siehe Abbildung 2A linkes Feld).
133,4 mg, wobei XDMPC und XDSPC die Molfraktionen von DMPC und DSPC sind, hier auf 0,7 bzw. 0,3 festgelegt, und MwDMPC und MwDSPC sind die molaren Gewichte von 677,9 g / mol bzw. 790,1 g / mol. In ähnlicher Weise mDSPC = 66,6 mg. Dieses Deuterationsschema erhöht den Streukontrast zwischen der Membran (NSLD ~ 0 × 10-6 Å-2) und dem deuterierten Puffer (NSLD ~ 6,4 × 10-6 Å-2) und verstärkt das Signal von Membranaufführungen (siehe Abbildung 2A linkes Feld). - Zur Messung der Biegedynamik ausgewählter lateraler Membranmerkmale, z.B. Matrixdynamik in phasentrennenden DMPC:DSPC-Membranen, verwenden Sie protiiertes DMPC (C36H72NO8P) und deuteriertes, DSPC-d83 (C44H5NO8PD83,Mw 873,7 g/mol), so dass mDMPC = 128,8 mg und mDSPC-d83 = 71,2 mg. Dieses Deuterationsschema minimiert die Streuung aus den unerwünschten DSPC-reichen Domänen und ermöglicht so selektive Messungen von Biegefluktuationen aus der DMPC-reichen Matrix (siehe Abbildung 2B Mitte).
HINWEIS: Um die optimale Lipiddeuteration zu finden, die für ein bestimmtes Kontrastanpassungsschema erforderlich ist, verwenden Sie verfügbare webbasierte SLD-Rechner (Scattering Length Density), wie sie vom NIST Center for Neutron Research64entwickelt wurden. Diese webbasierten Schnittstellen sind mit benutzerfreundlichen Werkzeugen zur einfachen Berechnung der SLD von Lipiden mit unterschiedlichen Deuterationsgraden sowie von Lipidmischungen ausgestattet. - Für NSE-Messungen der mittleren Membrandickenschwankungen (ohne seitlichen Kontrast) werden schwanzdeuterierte Varianten der Konstituenten Lipide verwendet, d.h. DMPC-d54 (C36H18NO8PD54, 732,3 g/mol) und DSPC-d70 (C44H18NO8PD70, 860,1 g/mol)35,38, so dass mDMPC-d54 = 133,0 mg und mDSPC-d70 = 67,0 mg. Dieses Kontrastschema (Abbildung 2A, rechte Tafel) verstärkt das Streusignal von den Lipidkopfgruppen (NSLD ~ 4,5 × 10-6 Å -2) durch Kontrastanpassung der Schwanzgruppe (NSLD ~ 6,4 × 10-6 Å-2 ) zum deuterierten Puffer, was die Erkennung von Schwankungen in der Dicke der Membran ermöglicht.
- Für Dickenfluktuationsstudien ausgewählter Membrankompartimente, z. B. DMPC-reiche Matrix, verwenden Sie die gleiche Strategie, die in Schritt 1.2 beschrieben wurde, indem protiierte DMPC-Lipide durch ihre schwanzdeuterierten Analoga, d. H. DMPC-d54, ersetzt werden, so dass die DSPC-reichen Domänen kontrastreich auf den deuterierten Puffer abgestimmt sind und das primäre Streusignal aus der Headgroup-Region der tail-deuterierten DMPC-reichen Matrix stammt.
 133,4 mg, wobei XDMPC und XDSPC die Molfraktionen von DMPC und DSPC sind, hier auf 0,7 bzw. 0,3 festgelegt, und MwDMPC und MwDSPC sind die molaren Gewichte von 677,9 g / mol bzw. 790,1 g / mol. In ähnlicher Weise mDSPC = 66,6 mg. Dieses Deuterationsschema erhöht den Streukontrast zwischen der Membran (NSLD ~ 0 × 10-6 Å-2) und dem deuterierten Puffer (NSLD ~ 6,4 × 10-6 Å-2) und verstärkt das Signal von Membranaufführungen (siehe Abbildung 2A linkes Feld).
133,4 mg, wobei XDMPC und XDSPC die Molfraktionen von DMPC und DSPC sind, hier auf 0,7 bzw. 0,3 festgelegt, und MwDMPC und MwDSPC sind die molaren Gewichte von 677,9 g / mol bzw. 790,1 g / mol. In ähnlicher Weise mDSPC = 66,6 mg. Dieses Deuterationsschema erhöht den Streukontrast zwischen der Membran (NSLD ~ 0 × 10-6 Å-2) und dem deuterierten Puffer (NSLD ~ 6,4 × 10-6 Å-2) und verstärkt das Signal von Membranaufführungen (siehe Abbildung 2A linkes Feld).2. Herstellung der Lipidsuspension für die Extrusion
- Berechnen Sie die Masse jedes Bestandteils in der Probe, abhängig von der Probenzusammensetzung. Als Faustregel gilt, dass für Proben mit mehreren molekularen Komponenten die Masse einer Komponente durch ihre molare Masse Mwigegeben ist, gewichtet durch ihre Molfraktion, Xi, und über alle Komponenten normalisiert wird, so dass:
 wobei Mtot die Gesamtmasse ist, hier auf 200 mg festgelegt. Siehe das obige Beispiel für DMPC-DSPC-Lipiddoppelschichten mit unterschiedlichen Deuterationsschemata.
wobei Mtot die Gesamtmasse ist, hier auf 200 mg festgelegt. Siehe das obige Beispiel für DMPC-DSPC-Lipiddoppelschichten mit unterschiedlichen Deuterationsschemata. - Wiegen Sie mit einer digitalen Halbmikrowaage die berechneten Massen an Lipiden (und anderen Probenbestandteilen, z. B. Proteinen, Nanopartikeln usw.) und geben Sie sie in eine Durchstechflasche oder einen Rundkolben - denken Sie daran, die Durchstechflasche oder den Kolben vorher zu wiegen. Fügen Sie 1 ml Lösungsmittel hinzu, um die gewogenen Komponenten durch manuelles Mischen in einer Haube aufzulösen. Für reine Lipidproben verwenden Sie Chloroform oder Ethanol. Wählen Sie für Proben mit zusätzlichen, nicht lipidfreien Komponenten (z. B. Nanopartikel) ein gemeinsames Lösungsmittel, das alle Komponenten dispergiert.
- Für kleine Lipidmengen (<10 mg) eine Stammlösung vorbereiten und das erforderliche Volumen in die Mischung pipetten.
HINWEIS: Fügen Sie keine übermäßigen Mengen an Lösungsmittel hinzu, da dies den unten beschriebenen Lösungsmitteltrocknungsschritt erheblich verlangsamt.
- Für kleine Lipidmengen (<10 mg) eine Stammlösung vorbereiten und das erforderliche Volumen in die Mischung pipetten.
- Trocknen Sie die Lipidlösung in einer Haube, indem Sie vorsichtig ein Inertgas (z. B. Stickstoff, Argon) in die Durchstechflasche strömen lassen, während Sie die Durchstechflasche langsam in einem Winkel drehen. Halten Sie die Fläschchen in geneigter Position, um einen dünnen Film aus getrocknetem Lipid an den Wänden der Durchstechflasche zu erzeugen, der eine gleichmäßige Trocknung ermöglicht. Legen Sie die Durchstechflasche intermittierend in ein Wasserbad bei 35 °C, um die verdunstungsvermittelte Abkühlung zu umgehen, wodurch die Lösungsmittelverdampfung verlangsamt wird.
- Legen Sie die Fläschchen über Nacht in einen Vakuumofen bei ~35 °C, um das Restlösungsmittel vollständig zu entfernen. Für ungesättigte Lipide das Vakuum mit einem Inertgas reinigen, um die Oxidation zu minimieren.
- Um eine vollständige Lösungsmittelentfernung zu gewährleisten, wiegen Sie die Durchstechflasche nach der Lipidtrocknung und bestätigen Sie, dass über die gemessenen Materialmengen hinaus keine überschüssige Masse vorhanden ist. Tun Sie dies, indem Sie die Masse der Durchstechflasche nach dem Trocknen von der gemessenen Masse subtrahieren. Wenn überschüssige Masse vorhanden ist, trocknen Sie die Probe unter Vakuum für weitere 6 h. Wiederholen Sie diesen Vorgang nach Bedarf.
- Hydratisieren Sie den Lipidfilm mit 4 mLD2OoderD2O-Puffer, um eine Lipidkonzentration von 50 mg/ml zu erhalten. Bei Lipiden mit hohen Übergangstemperaturen, wie DMPC-DSPC-Mischungen, erhitzen Sie den Puffer auf über die Übergangstemperatur (60 °C), um eine gleichmäßige Durchmischung zu gewährleisten.
HINWEIS: Da NSE-Experimente relativ große Probenvolumina (~ 4 ml) erfordern, sollten Sie die Probe mit der Hälfte des erforderlichen Puffers, d. H. 2 ml, hydratisieren, um die Anzahl der Extrusionen pro Probe zu minimieren (siehe Abschnitt 3). Fügen Sie in diesem Fall die verbleibende Hälfte der Puffernachextrusion hinzu. Beachten Sie, dass die Kapazität der in der Extrusion verwendeten Spritzen auf 1 ml begrenzt ist. Daher würde die Hydratierung mit 4 ml Puffer vier Extrusionssätze erfordern. - Mischen Sie die hydratisierte Lipidlösung so lange, bis der Lipidfilm vollständig aufgelöst ist und an den Wänden der Durchstechflasche nicht mehr sichtbar ist. In diesem Stadium bilden die hydratisierten Lipide multilamellare Vesikel und mikrometergroße multilamellare Stapel und die Suspension erscheint milchig weiß.
- Um das Brechen der Lipidstapel zu erleichtern und die Multilamellität zu reduzieren, führen Sie fünf Gefrier-/Auftauzyklen durch, indem Sie die Durchstechflasche mit hydratisierter Lipidlösung in einen Gefrierschrank in Laborqualität (vorzugsweise -80 °C Gefrierschrank) legen, bis sie vollständig gefroren ist, und dann die Durchstechflasche in ein 35 ° C-Wasserbad überführen, bis die Lipidlösung vollständig aufgetaut ist. Wirbeln Sie die aufgetaute Lösung vor, bis sie homogen ist. Wiederholen Sie den Vorgang noch vier weitere Male.
HINWEIS: Alternativ kann ein Trockeneisbad durch die Kombination von Aceton und Trockeneis für ein schnelles Einfrieren vorbereitet werden.
 wobei Mtot die Gesamtmasse ist, hier auf 200 mg festgelegt. Siehe das obige Beispiel für DMPC-DSPC-Lipiddoppelschichten mit unterschiedlichen Deuterationsschemata.
wobei Mtot die Gesamtmasse ist, hier auf 200 mg festgelegt. Siehe das obige Beispiel für DMPC-DSPC-Lipiddoppelschichten mit unterschiedlichen Deuterationsschemata.3. Extrusion der hydratisierten Lipidlösung
- Montieren Sie den Extruderaufbau mit einer Polycarbonatmembran zwischen zwei Membranträgern und fügen Sie zwei Papierfilter auf jeder Seite hinzu, um zusätzliche Unterstützung zu bieten. Verwenden Sie eine Polycarbonatmembran mit einer Porengröße, die der liposomalen Zielgröße entspricht (übliche Porengrößen für NSE-Experimente sind 50 nm und 100 nm – typischerweise ermöglichen Liposomen mit einem Durchmesser von 100 nm weniger eingeschränkte Membranschwankungen, aber kleinere 50-nm-Liposomen könnten für Krümmungsstudien verwendet werden). Stellen Sie sicher, dass die Polycarbonatmembran vollständig gedehnt ist, bevor Sie die Montage abschließen und das externe Extrudergehäuse festziehen.
- Hydratisieren Sie die Polycarbonatmembran, indem Sie ~ 0,3 mLD2O oderD2O-Puffer einige Male durch die Membrananordnung mit luftdichten Glasspritzen geben. Verwenden Sie den gleichen Puffer, der auch bei der Probenvorbereitung verwendet wird. Lassen Sie es mindestens 10 Minuten ein, dann saugen Sie den Puffer vollständig aus, bevor Sie die Probe einführen.
- Füllen Sie eine 1 mL gasdichte Spritze mit der vorbereiteten Lipidlösung und führen Sie sie in ein Ende der Extruderapparatur ein. Setzen Sie dann eine leere Spritze in das gegenüberliegende Ende ein. Sobald die Spritzen mit der Extruderbaugruppe verbunden sind, legen Sie sie in den Extruderblock.
- Wenn für die Extrusion erhöhte Temperaturen erforderlich sind, wie bei gesättigten Lipiden mit hohen Übergangstemperaturen (z. B. DSPC, Tm = 55 °C), erwärmen Sie den Extruder-Heizblock über der Lipidübergangstemperatur (z. B. 60 °C) vor, indem Sie den Heizblock auf eine Heizplatte legen oder ein Zirkulationsbad verwenden, wie in Abbildung 3Agezeigt.
HINWEIS: Dieser Schritt ist entscheidend, um eine homogene Mischung der Lipide zu gewährleisten und zu vermeiden, dass während der Extrusion ein extremer Druck ausgeübt wird, der die Polycarbonatmembran brechen könnte. Bei Lipidproben mit niedrigen Übergangstemperaturen (<25 °C) wird die Extrusion bei Raumtemperatur durchgeführt. - Um die Lipidlösung zu extrudieren, befestigen Sie den Extrudersatz an einer programmierbaren Spritzenpumpe mit einem Aluminium-/Stahlrahmen, wie in Abbildung 3Adargestellt. Für temperaturgesteuerte Extrusionen fügen Sie eine speziell angefertigte Extruderbasis mit einem Flüssigkeitskanal hinzu und befestigen Sie sie an einem zirkulierenden Wasserbad.
- Programmieren Sie die Spritzenpumpe so, dass sie 15-20 Extrusionszyklen gemäß der Anleitung des Herstellers durchführt. Beim Extrudieren ändert sich die Farbe der Lipidlösung von milchigweiß zu transparentem Opalblau(Abbildung 3B,C),was auf eine endgültige liposomale Größe hinweist, die wie erwartet kleiner als die Wellenlänge des sichtbaren Lichts ist. Führen Sie für den in Abbildung 3Agezeigten Spritzenpumpentyp die folgenden Schritte aus.
- Beginnen Sie mit der Anpassung der Pumpeneinstellungen. Halten Sie die Taste Rate gedrückt, geben Sie die Extrusionsrate (50,99 ml/h) ein, drücken Sie dann die Taste Durchmesser und geben Sie den Spritzendurchmesser (4,606 mm) ein. Verwenden Sie die Pfeile nach oben unter jeder Ziffer auf dem Bildschirm, um diesen Ziffernwert zu ändern.
- Setzen Sie den Extrudersatz mit der Probenspritze rechts ein (siehe Abbildung 3A). Drücken Sie die Auszugstaste, bis die Auszugsleuchte aufleuchtet. Drücken Sie Start und warten Sie, bis die Probe in die linke (leere) Spritze abgegeben wurde.
- Drücken Sie die Stop-Taste, kurz bevor die Probespritze (rechts) vollständig leer ist. Erfassen Sie das dosierte Volumen und programmieren Sie damit den Extrusionszyklus. Halten Sie die Schaltfläche Rate gedrückt, bis Phase 1 (PH:01) auf dem Bildschirm angezeigt wird. Drücken Sie die Lautstärketaste, um die zuvor aufgezeichnete Menge einzugeben. Achten Sie in dieser Phase darauf, dass das Entnahmelicht ausgeschaltet ist – dadurch wird die Probe in die richtige Richtung abgegeben.
- Drücken Sie erneut die Taste Rate und verwenden Sie den Pfeil ganz rechts nach oben, um auf Phase 2 (PH:02) zuzugreifen. Drücken Sie Lautstärke, um den gleichen Wert des zuvor aufgezeichneten dosierten Volumens einzugeben. Drücken Sie in dieser Phase die Withdraw-Taste, bis die Withdraw-Leuchte leuchtet – dadurch wird die Probe nach links abgegeben.
- Um diesen Zyklus zu wiederholen, drücken Sie erneut die Taste Rate und verwenden Sie den Pfeil ganz rechts nach oben, um auf Phase 3 (PH:03) zuzugreifen. Drücken Sie die Lautstärketaste, bis LP:SE auf dem Bildschirm angezeigt wird, und stellen Sie sie auf 20 ein. Dies ist die Anzahl der Schleifen oder Wiederholungen, die die Pumpe ausführen wird. Drücken Sie abschließend die Rate-Taste, greifen Sie auf Phase 4 (PH:04) zu und drücken Sie die Lautstärketaste, um zur Stop-Funktion zu gelangen. Die Pumpe ist nun für die automatisierte Extrusion eingerichtet.
- Drücken Sie Start, um den Extrusionszyklus zu starten.
- Entleeren Sie die Spritze mit der extrudierten Lipidsuspension in einer sauberen Durchstechflasche und bereiten Sie die Lagerung oder Messungen vor. Bei Lipidproben mit hoher Schmelztemperatur lagern Sie die Probe über dem Flüssigkeitsphasenübergang bis zur Messung. Andernfalls bewahren Sie die Proben bei Raumtemperatur auf.
- Extrudierte Proben nicht einfrieren, da das Einfrieren dazu führt, dass die Vesikel platzen (die Suspension wird wieder milchig weiß).
4. NSE-Messungen für die Probe(n) und Reduzierung der gesammelten Daten
- Charakterisieren Sie vor dem NSE-Experiment die extrudierte liposomale Probe aus Schritt 3.7 mit verfügbaren Methoden, um eine angemessene Probenqualität sicherzustellen. Eine Liste potenzieller Verkohlungsmethoden, mit denen die Qualität liposomalen Suspensionen für NSE-Experimente beurteilt werden kann, z. B. Größenverteilung, Multilamellität, laterale Membranstruktur, ist im Diskussionsabschnitt enthalten.
- Bestimmen Sie den Q-Bereich und die entsprechenden Geräteeinstellungen, die für das Experiment erforderlich sind. Für Biegesteifigkeitsmessungen von Lipiddoppelschichten verwenden Sie einen Q-Bereich von ~(0,04 - 0,2) Å-1. Für Untersuchungen von Membrandickenschwankungen verwenden Sie einen Q-Bereich von ~(0,04 - 0,2) Å-1 entsprechend der Membrandicke35,66,67.
HINWEIS: Besprechen Sie den Versuchsaufbau mit dem Instrumentenwissenschaftler vor Beginn des Experiments. Wie bereits erwähnt, ist eine SANS-Charakterisierung der Probe notwendig, insbesondere wenn keine Vorherigeinformation des Streusignals verfügbar ist, wie bei selektiv deuterierten Membranen. Alternativ können Sie statische Messungen (auch als Beugung bezeichnet) über einen begrenzten Q-Bereich auf dem NSE-Instrument durchführen, mit dem Vorbehalt, dass solche Messungen im Vergleich zu SANS viel länger dauern. - Laden Sie die extrudierte liposomale Suspension(en) mit einer Spritze oder einer Transferpipette in die vorgesehenen Probenzellen, die an NSE-Beamlines verfügbar sind. Beachten Sie, dass Standard-NSE-Probenzellen in Dicken von 1, 2, 3 und 4 mm erhältlich sind. Wählen Sie die Zelldicke so, dass das Streusignal optimiert wird, während das inkohärente Hintergrundsignal auf einer angemessenen Intensität gehalten wird.
HINWEIS: Als Faustregel gilt, Probenzellen mit einer Pfadlänge von 1 oder 2 mm für protiierte Liposomen in deuteriertem Puffer zu verwenden – dickere Zellen können zu mehreren Streueffekten führen, die schwer zu korrigieren sind. Für Liposomen mit höheren Deuterationen (z. B. schwanzkontrastangepasste Liposomen oder asymmetrische Liposomen mit einzelnen protiierten Blättchen) sollten Sie die Verwendung einer dickeren Probenzelle (z. B. 3 oder 4 mm Pathlänge) in Betracht ziehen, um die Zählstatistik zu verbessern, wenn die Probe in größeren Mengen verfügbar ist - manchmal kann dies unerschwinglich sein. - Bereiten Sie eine identische Probenzelle für den Puffer vor. Verwenden Sie den gleichen Puffer wie in der liposomalen Suspension. Messungen am Puffer sind für intensitätsnormalisierte und Hintergrundkorrekturen (BKG) notwendig.
- Legen Sie die Probenzelle(n) in den Probenhalter des NSE-Spektrometers, programmieren Sie die Messläufe und sammeln Sie Echodaten. Konsultieren Sie den Instrumentenwissenschaftler über die Programmierung der Messungen, wenn Sie zum ersten Mal NSE-Benutzer sind.
- Führen Sie zwei zusätzliche Messsätze durch, die für die Datenreduktion erforderlich sind: Auflösung (R) und Übertragung (T) Messungen.
- Durchführung der Auflösungsmessung (R) an einer elastischen Streureferenz (z. B. Kohlenstoff) – die unter den gleichen Einstellungen ausgeführt werden soll; d.h. gleiche Wellenvektor- und Fourier-Zeiten wie die Proben- und Puffermessungen.
- Führen Sie Transmissionsmessungen (T) an probe und puffer durch, um die Intensität des übertragenen Neutronenstrahls zu berechnen (siehe Schritt 4.9. unten). Die Transmission wird berechnet als das Verhältnis der Neutronenzahlen aus der Probe oder des Puffers dividiert durch die Neutronenzahlen für einen offenen Strahl (d.h. mit einer leeren Probenposition).
- Verwenden Sie die dedizierte Datenreduktionssoftware für das NSE-Spektrometer, auf dem die Messungen durchgeführt werden, um die gesammelten Daten zu reduzieren.
HINWEIS: Verschiedene Spektrometer können unterschiedliche Software oder Benutzeroberflächen verwenden. Im Folgenden finden Sie ein Beispiel für die NSE-Datenreduktion mithilfe der Data Analysis and Visualization Environment (DAVE)68 Software, die speziell für das NSE-Spektrometer am NIST Center for Neutron Research geschrieben wurde.- Öffnen Sie die DAVE-Software und wählen Sie NSE-Daten reduzieren aus dem Menü Datenreduktion. Es werden mehrere Popup-Fenster angezeigt.
- Laden Sie die Datendateien über verschiedene Q-Werte hoch, indem Sie die Option .echo-Dateien öffnen aus dem Dateimenü verwenden. Diese Dateien entsprechen den Rohdatendateien mit den Spin-Echo-Signalen und haben die Erweiterung .echo im Dateinamen. Sobald der Datei-Upload abgeschlossen ist, werden die Dateien unter den verfügbaren Datensätzen angezeigt.
- Klicken Sie mit der rechten Maustaste auf die ausgewählte Datei und beschriften Sie sie entsprechend der Messung, der sie entspricht. d. h. Sample, Cell (für leere Zelle oder Puffer) oder Resolution.
- Gruppieren Sie die Detektorpixel in 2 x 2, um das Signal-Rausch-Verhältnis mithilfe der Registerkarte Datensatz zu verbessern. Wenden Sie die gleiche Bindung auf alle Dateien an. d. h. Auflösung, Zelle und Beispiel.
- Überprüfen Sie die Daten über alle Pixelgruppen und maskieren Sie diejenigen mit schlechten Signalen (siehe Abbildung 4B),indem Sie die m-Taste auf der Tastatur drücken. Drücken Sie die Eingabetaste, um auf ein Popup-Fenster zuzugreifen, in dem Sie dieselbe Maske auf alle Fourier-Zeiten oder nachfolgende Fourier-Zeiten anwenden können. Dies kann auch auf einzelne Pixel zu einem beliebigen Zeitpunkt während der Datenreduktion angewendet werden. Maskierte Pixel werden grün.
- Stellen Sie sicher, dass die gesammelten Daten in Form eines Echosignals, d. h. einer Kosinusfunktion in Bezug auf den Phasenstrom, über jedem Detektorpixel vorliegen (siehe Abbildung 4A).
HINWEIS: Der Phasenstrom ist proportional zum Präzessionswinkel des Neutronenspins; Daher ist es üblich, den Phasenstrom als Phasenwinkel darzustellen, wie in Abbildung 4Adargestellt. Für Messungen an gepulsten Quellen werden zusätzliche Flugzeitberechnungen auf die Daten angewendet, um die Echosignale als Funktion der einfallenden Neutronenwellenlänge innerhalb eines Neutronenpulses zu erhalten. - Beginnen Sie mit dem Anpassen der Auflösungsdatei. Wählen Sie eine Auflösungsdatei aus der Liste der hochgeladenen Dateien aus und klicken Sie mit der rechten Maustaste auf die Datei. Wählen Sie im Popupmenü die Option "Einfügevorgänge: Echos anpassen (Auflösung)".
- Stellen Sie sicher, dass die Passungen der Echosignale eine Reihe von Anpassungsparametern ergeben, einschließlich des parameters A, der in Schritt 4.8 erforderlich ist. Die Passungen werden automatisch mit dem folgenden Ausdruck ausgeführt.

Hier ist ζ die Periode des Echosignals (d.h. Kosinusfunktion in Abbildung 4A),σ die Breite der Gaußschen Hülle ist, die durch die mittlere Wellenlänge und Wellenlängenspreizung des einfallenden Neutronenstrahls bestimmt wird, Φc ist der Phasenstrom und Φ0 ist der Echopunkt, der von der Feldbahn abhängt, die Neutronen50erfahren. Physikalische Informationen über die Probe sind in der Amplitude Ader Kosinusfunktion in Gleichung (1) kodiert.
HINWEIS: Die Breite der Gaußschen Hüllkurve basiert auf Werten, die vom Instrumentenwissenschaftler vorgegeben wurden und sollte nicht geändert werden. Die anderen Parameter sind Variablen, die an das spezifische Echosignal über jedes Pixel angepasst sind. - Überprüfen Sie die Anpassungsergebnisse, indem Sie auf jedes Pixel klicken, um die resultierenden Anpassungsparameter, die Qualität der Anpassung und die mittlere quadratische Abweichung der Anpassung anzuzeigen. Um den Fehler zu überprüfen, der jedem Formstückparameter über den gesamten Detektor zugeordnet ist, wählen Sie Bildoptionen und dann den gewünschten Formstückparameter aus. Dadurch wird eine Karte mit dem Wert des passenden Parameters über jedem Pixel generiert. Klicken Sie mit der rechten Maustaste auf das Detektorbild. Es erscheint ein Popup-Fenster mit einer Fehlerleistenzuordnung des ausgewählten Formstückparameters.
- Wenn die Anpassung über ein bestimmtes Pixel unbefriedigend ist (z. B. Parameeter mit großen Fehlerbalken anpassen), passen Sie das Signal über dieses bestimmte Pixel an. Wählen Sie dieses Pixelaus, drücken Sie die Registerkarte Formstück, und drücken Sie dann Pixel anpassen. Geben Sie neue Startparameter für die Phase (Φ0) und den Zeitraum (ζ) in die Registerkarte Formstück ein, um eine zufriedenstellendere Anpassung zu erhalten.
HINWEIS: Es ist nützlich, die angepasste Phase als Funktion der Fourierzeit zu zeichnen. Gehen Sie dazu zum Hauptplotfenster und wählen Sie Phase v. Fourierzeit anpassen. Dieses Diagramm sollte glatt und kontinuierlich sein. Untersuchen Sie die Diskontinuitäten in diesem Diagramm, und überpassen Sie die Pixel, denen sie entsprechen.
- Reduzieren Sie die Sample- oder Cell-Datei, indem Sie die entsprechende Datei aus der Liste der hochgeladenen und beschrifteten Dateien auswählen.
- Überprüfen Sie alle Pixel und maskieren Sie die Pixel mit fehlerhaften Statistiken, wie in Schritt 4.7.5 beschrieben.
- Klicken Sie mit der rechten Maustaste auf die Datei und wählen Sie Anpassungsvorgänge: Importphasen (Beispiel, Zelle). Dadurch werden die Phasen und die angewendete Maske aus der Auflösungsdatei importiert.
- Passen Sie die Echosignale mit dem gleichen Verfahren an, das zuvor für die Auflösungsdatei beschrieben wurde (Schritte 4.7.8-4.7.10). Ändern Sie beim Anpassen der Sample- und Cell-Dateien nicht die Werte des Zeitraums und des Echophasenpunkts, die aus den Auflösungsanpassungen importiert wurden. Diese Parameter hängen von den instrumentalen Einstellungen ab und sollten nicht mit den Samples variieren.
- Bevor Sie mit der Datenreduktion fortfahren, geben Sie das Strahlzentrum für alle Datendateien ein. Wählen Sie die Datendatei aus, gehen Sie zur Registerkarte Allgemein und geben Sie X- und Y-Balkenmittelwerte ein. Diese Werte werden während des Experiments aufgezeichnet.
- Sobald die Anpassung an die Sample-, Cell- und Resolution-Dateien abgeschlossen ist, berechnen Sie die normalisierte Zwischenstreufunktion, die später bei der Datenanalyse und -interpretation verwendet werden soll. Klicken Sie dazu mit der rechten Maustaste auf die Beispieldatei, die aus der Liste der angepassten Dateien reduziert werden soll, und wählen Sie im Popupmenü die Option I(Q) berechnen aus. Es erscheint ein Fenster mit Eingabemöglichkeiten für die Dateien Auflösung und Zelle (d. h. Puffer) sowie die Anzahl der Q-Arcs (siehe Schritt 4.9). Nachdem Sie alle erforderlichen Informationen eingegeben haben, drücken Sie die Taste OK. Die Ergebnisse werden in einem neuen Fenster angezeigt.
HINWEIS: Die Datenreduktion wird gemäß der folgenden Gleichung durchgeführt, um die normalisierte Zwischenstreufunktion69zu erhalten.
wobei t die Fourierzeit ist, Noben und Nunten die Neutronenzahlen in den Non-Spin-Flip- und Spin-Flip-Konfigurationen sind (gemessen mit ausgeschalteten π/2-Flippern bzw. π-Flipper), und die hochgestellten BKG und Rentsprechen den Hintergrund- bzw. Auflösungsmessungen, wie in den Schritten 4.4 und 4.6 definiert. Beachten Sie, dass die Strahlpolarisation, also Änderungen im Spinzustand aufgrund des Energieaustauschs zwischen dem Neutron und der Probe, als Abfall der Polarisation (von der Einheit) detektiert wird.
also Änderungen im Spinzustand aufgrund des Energieaustauschs zwischen dem Neutron und der Probe, als Abfall der Polarisation (von der Einheit) detektiert wird.
- Gruppieren Sie schließlich die Detektorpixel in Q-Bögen,wie in Abbildung 4B gezeigt, um die Q-Abhängigkeitder normalisierten Zwischenstreufunktion S(Q,t) / S(Q, 0 ) zu erhalten. Dies wird technisch als Datenbinning bezeichnet und sollte mit Bedacht erfolgen, d.h. unter Berücksichtigung der Zählstatistiken aus der Stichprobe und der erwarteten Standardabweichung der Daten über die gruppierten Pixel.
- Für stark streuende Proben teilen Sie den Detektor in mehr Q-Bögen auf, wobei vernünftige Fehlerbalken auf der resultierenden Zwischenstreufunktion S(Q,t) / S(Q, 0 ) beibehalten werden. Dies ergibt mehr Q-Datenpunkte und ist wichtig für das unten beschriebene Datenanalyseverfahren. Beachten Sie, dass bei schwach streuenden Proben ein übermäßiges Binning zu schlechten Zerfallssignalen führt, d.h. zu großen Fehlerbalken auf S(Q,t) / S(Q, 0), was zu großen Unsicherheiten führen kann.


 also Änderungen im Spinzustand aufgrund des Energieaustauschs zwischen dem Neutron und der Probe, als Abfall der Polarisation (von der Einheit) detektiert wird.
also Änderungen im Spinzustand aufgrund des Energieaustauschs zwischen dem Neutron und der Probe, als Abfall der Polarisation (von der Einheit) detektiert wird.5. Datenanalyse und -interpretation
- Passen Sie die normalisierten Zwischenstreufunktionen S(Q,t) / S(Q, 0), die aus der obigen Datenreduktion erhalten wurden, auf eine gestreckte Exponentialfunktion mit einem Dehnungsexponenten von 2/370an.

HINWEIS: Ein Beispiel für diese Passungen finden Sie in Abbildung 5B. Anfälle von S(Q,t) / S(Q, 0) zu Gleichung (3) ergeben die Q-abhängigen Relaxationsraten Γ (Q). - Zeichnen Sie Γ(Q) als Funktion von Q auf und passen Sie sich an ein geeignetes Modell an, um relevante Membranparameter zu extrahieren.

Subscription Required. Please recommend JoVE to your librarian.
Representative Results
NSE-Studien, die auf Biegeschwankungen zugreifen, werden typischerweise über einen Q-Bereich von ~ (0,04 - 0,2) Å-1durchgeführt. Dieser Q-Bereich entspricht mittleren Längenskalen zwischen der Membrandicke und dem liposomalen Radius, wobei die Biegedynamik dominiert. Die Messung über einen erweiterten Q-Bereich kann den Zugang zu zusätzlichen dynamischen Modi ermöglichen, einschließlich liposomaler Diffusion und Intramembrandynamik. Weitere Einzelheiten zum Crossover in der Membrandynamik, auf das NSE zugreift, finden Sie in diesen einschlägigenPublikationen 25,71. Es ist wichtig zu betonen, dass NSE-Signale proportional sind zu:  , wobei Icoh bzw. Iinc die kohärente und inkohärente Streuintensität aus der Probe sind. Daher ist es ratsam, liposomale NSE-Proben in deuterierten Puffern (d.h. Puffern, die mitD2Oanstelle vonH2O hergestellt werden) vorzubereiten, um das inkohärente Streusignal zu minimieren, das hauptsächlich durch den Wasserstoffgehalt der Probe beigetragen wird. In einigen Fällen können jedoch intermediäre Deuterationsschemata (d. h. die Verwendung von Mischungen vonD2OundH2O)erforderlich sein, um optimale Kontrastbedingungen zu erhalten. Typischerweise werden NSE-Messungen von Membranbiegeschwankungen an vollständig protiierten Liposomen in deuteriertem Puffer durchgeführt, die in Abbildung 5als vollständig kontrastierte Liposomen bezeichnet werden. Dieses Deuterationsschema führt zu einer großen NSLD-Differenz zwischen dem Membrankern (~0 × 10-6 Å-2)und seiner deuterierten flüssigen Umgebung (~6,4 × 10-6 Å-2), was das Streusignal der liposomalen Membranen signifikant verbessert und die Messstatistik der Biegedynamik verbessert. Dieses Kontrastschema (Abbildung 2A linkes Panel) wird häufig in Studien zur Biegesteifigkeit von Lipidmembranen mit einzelnen38,72 und mehreren39,66 Lipidkomponenten und in Studien zur Membranerweichung / Versteifung durch biologische Einschlüsse (z. B. Cholesterin, Arzneimittelmoleküle, Peptide / Proteine)36,37,73,74,75und synthetische Additive (z. B. Nanopartikel)76,77verwendet.
, wobei Icoh bzw. Iinc die kohärente und inkohärente Streuintensität aus der Probe sind. Daher ist es ratsam, liposomale NSE-Proben in deuterierten Puffern (d.h. Puffern, die mitD2Oanstelle vonH2O hergestellt werden) vorzubereiten, um das inkohärente Streusignal zu minimieren, das hauptsächlich durch den Wasserstoffgehalt der Probe beigetragen wird. In einigen Fällen können jedoch intermediäre Deuterationsschemata (d. h. die Verwendung von Mischungen vonD2OundH2O)erforderlich sein, um optimale Kontrastbedingungen zu erhalten. Typischerweise werden NSE-Messungen von Membranbiegeschwankungen an vollständig protiierten Liposomen in deuteriertem Puffer durchgeführt, die in Abbildung 5als vollständig kontrastierte Liposomen bezeichnet werden. Dieses Deuterationsschema führt zu einer großen NSLD-Differenz zwischen dem Membrankern (~0 × 10-6 Å-2)und seiner deuterierten flüssigen Umgebung (~6,4 × 10-6 Å-2), was das Streusignal der liposomalen Membranen signifikant verbessert und die Messstatistik der Biegedynamik verbessert. Dieses Kontrastschema (Abbildung 2A linkes Panel) wird häufig in Studien zur Biegesteifigkeit von Lipidmembranen mit einzelnen38,72 und mehreren39,66 Lipidkomponenten und in Studien zur Membranerweichung / Versteifung durch biologische Einschlüsse (z. B. Cholesterin, Arzneimittelmoleküle, Peptide / Proteine)36,37,73,74,75und synthetische Additive (z. B. Nanopartikel)76,77verwendet.
Messungen von Biegeschwankungen führen zu Relaxationsraten, die einer Q3-Abhängigkeit folgen, wie von Zilman und Granek für thermisch wellenförmige elastische dünne Bleche vorhergesagt70. Eine verfeinerte Form dieser Q-Abhängigkeitergibt sich aus theoretischen Korrekturen von Watson und Brown78, die die von Seifert und Langer79vorgeschlagenen Auswirkungen der Intermonolayerreibung berücksichtigen. Durch zusätzliche Definition der neutralen Ebene an der Grenzfläche zwischen den hydrophilen Kopfgruppen und den hydrophoben Schwänzen der Membran können die Biegerelaxationsraten dann an den folgenden Ausdruck38angepasst werden.

wobei ηBuff die Pufferviskosität ist, kBT die thermische Energie, κ und die Biegesteifigkeit der gemessenen Membran (oder des kontrastierten Teils der Membran in selektiv deuterierten Systemen). Diese Art der Messung ermöglicht die direkte Berechnung der membranelastischen Eigenschaften in Form des Biegesteifigkeitsmoduls. Beachten Sie, dass κ aus der Steigung der linearen Anpassung von Γ vs. Q3extrahiert wird, wie in Abbildung 5Cdargestellt.
Andererseits zeigen NSE-Messungen von Membrandickenschwankungen Abweichungen von derQ3-Abhängigkeitin Γ(Q) um Q-Werte, die der Membrandicke entsprechen (siehe Abbildung 2 in Referenz66). Um das Dickenschwankungssignal zu isolieren, kann man Γ(Q) durch Q3dividieren, wie in Abbildung 5Dgezeigt. Die resultierenden Daten zeigen, dass die überschüssige Dynamik aufgrund von Dickenschwankungen einer Lorentzschen Funktion in Qfolgt, wie kürzlich in grobkörnigen Molekulardynamik-Simulationen (MD)bestätigt wurde 67. Um der beobachteten Überschussdynamik gerecht zu werden, entwickelten Nagao et al.38 einen Ausdruck, der auf dem theoretischen Rahmen der Membranfluktuationen von Bingham et al.80 wie folgt basiert.

In diesem Ausdruck ist Q0 der maximale Q-Wert,der der Membrandicke entspricht (die unabhängig von SANS-Messungen erhalten werden kann), μ die Viskosität der Ebene ist, AL die Fläche pro Lipid (gemessen mit SANS / SAXS) und KA der Flächenkompressibilitätsmodul. Unter der Annahme, dass KA aus κ mit dem Polymerbürstenmodell berechnet werden kann, reduziert sich dieser Ausdruck auf einen Anpassungsparameter, nämlich die Membranviskosität μ, was einen neuen Ansatz zur Messung der Membranviskosität darstellt, ohne dass eine Fluoreszenzmarkierung oder Partikelanbindeung / -verfolgung erforderlichist 13. Die Prämisse ist, dass nach Verformungsmodellen elastischer dünner Bleche81, κ und KA voneinander abhängig sind, so dass:  , wobei tm die mechanische (oder verformbare) Membrandicke und β eine Konstante ist, die die Interleaflet-Kopplung beschreibt. Die Annahme ist, dass β = 12 für vollständig gekoppelte Blättchen, β = 48 für vollständig entkoppelte Blättchen und β = 24 für zwischengekoppelte Blättchen. Letzteres wird als Polymerbürstenmodell81 bezeichnet und hat gezeigt, dass es in Einkomponenten- und binären Flüssigkeitslipidmembranen39angewendet wird. Dies muss jedoch mit Vorsicht angegangen werden. Zum Beispiel aktuelle Simulationen von Doktorova etal. 82 zeigte, dass für das Polymerbürstenmodell, um in ungesättigten lipidhaltigen Lipidmembranen zu halten, eine modifizierte Expression der mechanischen Membrandicke verwendet werden muss. Wenn eine unabhängige Messung von KA möglich ist, z.B. mittels Mikropipettenaspiration83,dann wäre die Kombination von KA-Ergebnissen mit NSE-Biegesteifigkeitsmessungen eine einzigartige Möglichkeit, die Interleaflet-Kopplung in Modell- und biologischen Membranen zu untersuchen – eine seit langem bestehende Frage in der Membranbiophysik und Strukturbiologie. Sobald die Werte von KA validiert sind, können sie in Gleichung 5 verwendet werden, um die mesoskopische Membranviskosität zu erhalten.
, wobei tm die mechanische (oder verformbare) Membrandicke und β eine Konstante ist, die die Interleaflet-Kopplung beschreibt. Die Annahme ist, dass β = 12 für vollständig gekoppelte Blättchen, β = 48 für vollständig entkoppelte Blättchen und β = 24 für zwischengekoppelte Blättchen. Letzteres wird als Polymerbürstenmodell81 bezeichnet und hat gezeigt, dass es in Einkomponenten- und binären Flüssigkeitslipidmembranen39angewendet wird. Dies muss jedoch mit Vorsicht angegangen werden. Zum Beispiel aktuelle Simulationen von Doktorova etal. 82 zeigte, dass für das Polymerbürstenmodell, um in ungesättigten lipidhaltigen Lipidmembranen zu halten, eine modifizierte Expression der mechanischen Membrandicke verwendet werden muss. Wenn eine unabhängige Messung von KA möglich ist, z.B. mittels Mikropipettenaspiration83,dann wäre die Kombination von KA-Ergebnissen mit NSE-Biegesteifigkeitsmessungen eine einzigartige Möglichkeit, die Interleaflet-Kopplung in Modell- und biologischen Membranen zu untersuchen – eine seit langem bestehende Frage in der Membranbiophysik und Strukturbiologie. Sobald die Werte von KA validiert sind, können sie in Gleichung 5 verwendet werden, um die mesoskopische Membranviskosität zu erhalten.

Abbildung 1: NSE-Instrumentendesign und synergistische Überlappung mit Längen-/Zeitskalen der mesoskopischen Membrandynamik. (A) Schematische Darstellung der verschiedenen magnetischen Elemente eines NSE-Instruments, das verwendet wird, um den Spin von Neutronen zu manipulieren, die das Instrument von links nach rechts durchqueren. Das hervorgehobene Neutron zeigt eine Änderung der Spinorientierung (oder Polarisationsverlust) aufgrund des Energieaustauschs zwischen dem Neutron und der Probe an, während das transparente Neutron Spin-Echo darstellt, dh keine Änderung des Neutronenspins aufgrund von Nullenergieaustausch. Der graue Pfeil zeigt die Möglichkeit an, den zweiten Arm des Spektrometers zu drehen, um auf größere Streuwinkel zuzugreifen. (B) Bildliche Darstellung der hierarchischen Dynamik in Lipidmembranen, die verschiedene dynamische Modi zeigt, die sich über mehrere Längen- und Zeitskalen erstrecken. Der schattierte Bereich stellt die von NSE abgerufenen Längen- und Zeitskalen dar, die sich mit den Mesoskalen kollektiver Membranfluktuationen überlappen, nämlich Biege- und Dickenschwankungen. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 2: Beispiele für mögliche Deuterationsschemata in NSE-Experimenten an Lipidmembranen. (A) Links: Vollständig kontrastierte Membranen, z. B. protiierte Membranen in deuteriertem Puffer, die das NSLD-Profil entlang der Senkrechten zur Membranoberfläche zeigen. Die Differenz in der NSLD zwischen dem Schwanzbereich (~0 × 10-2 Å-2 ) und dem Kopfgruppenbereich (~4,5 × 10-6 Å-2)der Membran ist auf die Hydratation der Kopfgruppe mit deuteriertem Puffer zurückzuführen. Rechts: Tail-Contrast-abgestimmte Membranen, so dass der Kohlenwasserstoff-Tail-Bereich der Membran die gleiche NSLD wie der Puffer hat, wie im entsprechenden NSLD-Profil entlang der Membrannormalen gezeigt. (B) Domänenbildende Membranen mit zwei Neutronenkontrastschemata, bei denen die Domänen (Mitte) oder die Matrix (links) kontrastabgeglichen auf den Puffer abgestimmt sind, was selektive Untersuchungen der Matrix- bzw. Domänendynamik ermöglicht. Diese Zahl wurde modifiziert von Nickels et al., JACS 201541. (C) Asymmetrische Membranen, die durch Cyclodextrinaustausch zwischen protiierten und deuterierten Lipidvesikeln hergestellt werden, was zur Deuteration einer Membranblättchen führt, während die andere Blättchenprossion erhalten bleibt. Dies ermöglicht Untersuchungen der Biegedynamik des protiierten Blättchens und gibt Einblicke in die mechanische Kopplung zwischen gegenüberliegenden Blättchen in asymmetrischen Membranen. Diese Zahl wurde modifiziert von Rickeard et al., Nanoscale 202040. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 3: Abbildung des Aufbaus für die automatisierte Extrusion von Liposomen. (A) Maßgeschneiderter automatisierter Extruder mit einer Spritzenpumpe, einem Mini-Extruder-Set und einem Aluminium-/Stahlrahmen, um zyklische Extrusionen zu ermöglichen. (B) und (C) zeigen den Unterschied im visuellen Erscheinungsbild von Lipidsuspensionen vor (milchig weiß) und nach (transparentes opalblau) Extrusion. Dies ist auf die anfängliche Bildung von mikrometergroßen Lipidstapeln oder riesigen Vesikeln zurückzuführen, die in der Größenordnung oder größer als die Wellenlänge des sichtbaren Lichts liegen. Nach der Extrusion besteht die Suspension aus nanoskopischen Vesikeln (~ 100 nm), die kleiner als die Wellenlänge des sichtbaren Lichts sind, was zu einer transparenten Suspension führt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 4: Repräsentative Daten aus NSE-Experimenten an liposomalen Suspensionen. (A) Beispiel eines Echosignals über einem einzelnen Detektorpixel (markiertes Pixel in Feld B), das die Passung des Echosignals anhand der Gleichung (1) zeigt, mit einer Illustration der verschiedenen Parameter, die für die Echopassung erforderlich sind. Beachten Sie, dass das Echosignal als Funktion des Phasenwinkels und nicht des Phasenstroms dargestellt wird, wie in Schritt 4.7 des Protokolls erläutert. (B) NSE-Detektorbild, das die Variation der Neutronenzahlen pro Pixel zeigt. Das Bild zeigt auch eliminierte Detektorpixel (grün) aufgrund schlechter Echosignale. Das Binning der Detektorpixel in Q-Arcs (auch Debye-Scherrer-Ringe genannt) ergibt die Q-Abhängigkeit der Zwischenstreufunktion, die für die Analyse und Interpretation von NSE-Daten benötigt wird. Diese Zahl wurde modifiziert von Ashkar, J. Appl. Phys. 202050. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 5: Repräsentative Ergebnisse von NSE-Experimenten an liposomalen Suspensionen mit unterschiedlichen Deuterationsschemata. (A) Streugeometrie eines Neutrons, das mit einem Liposom interagiert, zeigt den Streuwinkel von 2θund den Wellenvektortransfer,  . (B) Zwischenstreufunktionen, S(Q,t) / S(Q,0), zeigen Zerfälle als Funktion der Fourierzeit. Die Anpassung der gemessenen Zerfälle an eine durch Gleichung 3 gegebene gestreckte Exponentialfunktion ergibt die Relaxationsraten, Γ(Q). (C) Für vollständig protiierte Liposomen in deuteriertem Puffer folgt Γ(Q) einerQ3-Abhängigkeit, die typisch für die Biegedynamik ist. Die lineare Anpassung der erhaltenen Daten an ein Zilman-Granek-Modell ergibt den Biegesteifigkeitsmodul der Membran. (D) Bei tail deuterierten Liposomen werden neben Biegeschwankungen auch überhöhte Dynamik beobachtet und sind bei Q-Werten, die der Membrandicke entsprechen, am ausgeprägtesten. Die Anpassung der überschüssigen Dynamik an eine Lorentzsche Funktion (Gleichung 5) ermöglicht die Extraktion der Membranviskosität. Datensätze wurden auf dem NSE-Spektrometer am NIST gesammelt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
. (B) Zwischenstreufunktionen, S(Q,t) / S(Q,0), zeigen Zerfälle als Funktion der Fourierzeit. Die Anpassung der gemessenen Zerfälle an eine durch Gleichung 3 gegebene gestreckte Exponentialfunktion ergibt die Relaxationsraten, Γ(Q). (C) Für vollständig protiierte Liposomen in deuteriertem Puffer folgt Γ(Q) einerQ3-Abhängigkeit, die typisch für die Biegedynamik ist. Die lineare Anpassung der erhaltenen Daten an ein Zilman-Granek-Modell ergibt den Biegesteifigkeitsmodul der Membran. (D) Bei tail deuterierten Liposomen werden neben Biegeschwankungen auch überhöhte Dynamik beobachtet und sind bei Q-Werten, die der Membrandicke entsprechen, am ausgeprägtesten. Die Anpassung der überschüssigen Dynamik an eine Lorentzsche Funktion (Gleichung 5) ermöglicht die Extraktion der Membranviskosität. Datensätze wurden auf dem NSE-Spektrometer am NIST gesammelt. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
NSE ist eine leistungsstarke und einzigartige Technik zur Messung der mesoskopischen Dynamik von Lipidmembranen unter verschiedenen Bedingungen. Die effektive Nutzung von NSE hängt von der Probenqualität, dem Neutronenkontrast und dem Bereich der zugänglichen Dynamik ab, die für eine bestimmte Probe untersucht werden kann. Daher sind mehrere kritische Schritte erforderlich, um erfolgreiche NSE-Experimente durchzuführen und qualitativ hochwertige Daten zu sammeln. Ein wichtiger Schritt, um die effektive Nutzung der Neutronenstrahlzeit während eines NSE-Experiments sicherzustellen, besteht darin, die liposomalen Suspensionen vor dem NSE-Experiment mit laborbasierten Methoden zu charakterisieren. Zum Beispiel kann die Größenverteilung (oder Diffusionskonstante) von extrudierten Liposomen durch dynamische Lichtstreuung (DLS) bestimmt werden, die in einzelnen Laboren oder in gemeinsamen Einrichtungen leicht verfügbar ist84. Die Kryo-Elektronenmikroskopie ist eine weitere kürzlich an liposomalen Proben validierte Verfärbungsmethode, bei der hochauflösende Mikroskopiebilder an kryomektomierten Abschnitten liposomaler Suspension effektiv verwendet werden können, um liposomale Unilamellarität65, Domänenbildung85,86oder den Einbau von Additiven wie Nanopartikeln76 und Proteinen87zu untersuchen . Alternativ kann die Kleinwinkel-Röntgenstreuung (SAXS) verwendet werden, um dieMembranstruktur 88zu charakterisieren, die liposomale Multilamellität65zu beurteilen oder die Auswirkungen von Additiven auf die strukturellen Eigenschaften der Membran zu bewerten89. Neben diesen laborbasierten Techniken ist es sehr ratsam, dass NSE-Messungen an liposomalen Proben mit Strukturstudien unter Verwendung der Kleinwinkel-Neutronenstreuung (SANS)54,90gepaart werden. SANS ist eine hervorragende Ergänzung zu NSE, nicht nur für die Erfassung struktureller Membraninformationen, sondern auch für die Untersuchung der Intensität des Neutronenstreusignals aus der Probe, die Bestätigung des Kontrastschemas und die fundierte Wahl des Q-Bereichs, über den die NSE-Messungen durchgeführt werden sollen. Daher wird empfohlen, dass NSE-Benutzer SANS-Beamtime anfordern, wenn sie sich für NSE-Experimente bewerben.
NSE leidet jedoch unter Probenbeschränkungen bei Studien an biologischen Membranen. Einer der wichtigsten limitierenden Faktoren solcher Experimente ist die Standardmenge an Proben, die für NSE-Messungen (2-4 ml) erforderlich ist, und die hohen Probenkonzentrationen in Höhe von 100-200 mg Membranmaterial (Lipide und Proteine), um qualitativ hochwertige Daten zu erhalten. In vielen Fällen ist die Herstellung solcher Mengen an biologischem Material nicht machbar oder unerschwinglich. In solchen Szenarien ist es möglich, die Konzentration auf 20-25 mg/ml zu reduzieren, dies würde jedoch mindestens eine 4-fache Erhöhung der Erfassungszeit erfordern, um vergleichbare Statistiken mit Proben mit 50 mg/ml Konzentrationen zu erhalten. Diese strengen Anforderungen an Probenvolumen und -konzentration könnten mit der nächsten Generation von NSE-Spektrometern an Neutronenquellen mit höherem Fluss, wie der zweiten Zielstation am Oak Ridge National Lab und der European Spallation Source, gemildert werden. Eine weitere kritische Einschränkung bei der Durchführung von NSE-Experimenten an Lipidmembranen, die selektive Deuterationsschemata erfordern, ist die fehlende kommerzielle Verfügbarkeit einiger deuterierter Varianten von Lipidmolekülen oder deren exorbitante Preise, falls verfügbar. In einigen Fällen können diese Einschränkungen umgangen werden, indem die Synthese deuterierter Lipide (oder Cholesterin, Proteine) durch Benutzerdeuterationseinrichtungen wie das Biodeuterationslabor am Oak Ridge National Lab, die nationale Deuterationseinrichtung an der ANSTO oder die Deuterationseinrichtung an der ISIS-Neutronen- und Myonenquelle angefordert wird. Der Zugang zu diesen Einrichtungen und ihren Synthesefähigkeiten ist durch eingereichte Benutzervorschläge möglich, die auf der Grundlage des wissenschaftlichen Werts der vorgeschlagenen Materialsynthese und ihrer beabsichtigten Verwendung in isotopensensitiven Studien von Experten begutachtet werden.
Trotz dieser Einschränkungen hat die Anwendung der NSE-Spektroskopie in Studien der Membranmechanik zur Bestimmung der Biegesteifigkeitsmodule von Membranen mit unterschiedlichen Komplexitätsgraden geführt, von Einkomponenten-Lipidmembranen35,38 bis zu mehrkomponentigen biomimetischen Membranen41,66,91, die alle unser Verständnis der dynamischen Natur von Lipidmembranen erweitert haben. Zum Beispiel haben NSE-Biegesteifigkeitsmessungen von Lipidmembranen mit unterschiedlichen molekularen Einheiten, z. B. Lipide unterschiedlicher Acylkettenlängen und Kettensättigung38,72,92, wesentliche Informationen über die Rolle der Molekularchemie in der Membranmechanik geliefert. Gepaart mit Strukturinformationen wie Membrandicke oder molekularer Packung93eröffnen diese Messungen neue Perspektiven auf die Interdependenz zwischen Membranstruktur und Dynamik und wie sie die Membranfunktion beeinflussen. Die mesoskopischen Skalen von NSE positionieren es einzigartig für solche grundlegenden Untersuchungen von Struktur-Eigenschafts-Beziehungen, die am relevantesten auf der Längenskala molekularer Anordnungen sind. Dieses Thema wurde kürzlich in zwei NSE-Studien an cholesterinreichen Lipidmembranen36 und in binären Lipidmembranen mit hydrophobem Missverhältnis zwischen den beiden Lipidkomponenten39untersucht. Beide Studien fanden starke Beweise dafür, dass die Membranmechanik mit der Fläche pro Lipid skaliert, was die Schlussfolgerungen aus einer kürzlich durchgeführten All-Atom-MD-Simulation von Doktorova et al.82bestätigt. Diese Ergebnisse betonen die selbstorganisierte Natur von Lipidmembranen und liefern ein einheitliches Bild der molekularen Packung als Schlüsselparameter bei der Definition membrandynamischer und funktioneller Eigenschaften.
Andere Anwendungen von NSE umfassen Untersuchungen der mechanischen Reaktion von Membranen auf kleine Additive, einschließlich biologischer Moleküle wie Cholesterin36,37, Trehalose92und Melittin73,94oder anorganische Additive wie Nanopartikel für Arzneimittelabgabeanwendungen76. NSE wurde auch verwendet, um zu verstehen, wie die Membranmechanik auf Veränderungen in ihrer Umgebung reagiert, einschließlich Temperatur92, pH74und das Vorhandensein von Überfüllungsmakromolekülen96. Solche Studien tragen zu einem besseren Verständnis der Faktoren bei, die die Erweichung oder Versteifung von Lipidmembranen unter biologischen Bedingungen im Zusammenhang mit Gesundheit und Krankheit und in kontrollierten Umgebungen für therapeutische Anwendungen beeinflussen. Bemerkenswerterweise wurden NSE-Messungen auch verwendet, um die Wirkung antimikrobieller Peptide auf die Membrandynamik zu untersuchen73,94,95. Weitere Beispiele für NSE-Anwendungen an Biomembranen sind Untersuchungen der Dynamik abgeflachten Membranstrukturen, sogenannte Thylakoide, die die photosynthetische Maschinerie in Cyanobakterienzellenbeherbergen 97,98.
Man kann auch selektive Lipiddeuteration in NSE-Studien verwenden, um die Dynamik spezifischer Membranmerkmale zu untersuchen, die für die biologische Funktion relevant sind. Zum Beispiel verwendeten Nickels et al. selektive Lipiddeuteration in domänenbildenden Lipidmembranen, um einen lateralen Kontrast innerhalb der Membran zu erzeugen, wie zuvor von Heberle et al.28veranschaulicht. Dieses Deuterationsschema ermöglichte unabhängige Messungen der Biegesteifigkeit der Lipiddomänen und der Wirtslipidmatrix41 (siehe Abbildung 2B). Die Ergebnisse bestätigten, dass die beiden Membrankompartimente unterschiedliche Biegesteifigkeitsmodule aufweisen, die ein treibender Mechanismus für die Domänenbildung in Zellmembranen sein könnten. In einer neueren Studie verwendeten Rickeard et al. den Cyclodextrinaustausch zwischen protiierten und deuterierten Liposomen, um asymmetrische Liposomen mit isotopisch markierten Blättchen zu erhalten40 (Abbildung 2C). Ihre Endliposomen hatten ein protiiertes Blättchen und ein deuteriertes Blättchen, das kontrastreich auf den Puffer abgestimmt ist, was Untersuchungen der Dynamik einzelner Blättchen ermöglichte und eine erste direkte experimentelle Darstellung der Wirkung von Asymmetrie und Blättchenkopplung auf Membranbiegeschwankungen lieferte.
Selektive Membrandeuteration wurde auch in NSE-Studien von Membrandickenschwankungen verwendet, einem lange vorhergesagten dynamischen Modus in Lipidmembranen99, der erst kürzlich mit dem Aufkommen der NSE-Spektroskopie35,100beobachtet wurde. Diese Messungen verwenden schwanzdeuterierte Membranen, um das Signal aus den Membrankopfgruppenregionen zu verstärken und das Dickenschwankungssignal aufzulösen. Diese Art von NSE-Experimenten ist relativ neu, aber sie wurde effektiv verwendet, um die Interdependenz von membranelastischen und viskosen Eigenschaften zu verstehen38, um die Skalierung der Biegesteifigkeit und Viskosität mit molekularer Packung in gemischten Lipidmembranen zu untersuchen39, und um die lokalen Auswirkungen von Cholesterin auf die Membranviskosität zu untersuchen36. Ein weiterer Bereich von biologischer Bedeutung, in dem dieser dynamische Modus weitreichende Auswirkungen haben könnte, sind mesoskopische Membran-Protein-Interaktionen95. Es ist bekannt, dass die Funktion von Membranproteinen eng mit dem hydrophoben Matching zwischen dem Protein und der Wirtsmembran verbunden ist. So könnten Variationen in der Membrandicke aufgrund von Dickenschwankungen als Regulationsmechanismus für die Funktion von Membranproteinen wirken. NSE eignet sich sehr gut für solche Studien, da es die Auswirkungen von Proteinbindung und -insertion auf Membrandickenschwankungen direkt untersuchen kann. Jüngste NSE-Messungen aus unserer Gruppe (unveröffentlicht) deuten darauf hin, dass die Insertion von Transmembranproteinen Membrandickenschwankungen signifikant unterdrücken und einen potenziellen Mechanismus zur Regulierung von Signalereignissen darstellen könnte. Dies ist ein dringendes, aber unterentwickeltes Forschungsgebiet, in dem NSE einen signifikanten Einfluss auf das Verständnis der dynamischen Reaktionen von Membranen auf die Proteinbindung und -insertion auf der Länge und Zeitskala wichtiger biologischer Funktionen haben kann, die durch die Wechselwirkungen von Proteinen mit Zellmembranen vermittelt werden.
Zusammenfassend lässt sich sagen, dass sich NSE in den letzten Jahren zu einem leistungsstarken Werkzeug entwickelt hat, um die Membrandynamik über räumliche und zeitliche Skalen lebenswichtiger biologischer Funktionen zu untersuchen. Die Technik gewinnt schnell an breitem Interesse und ihr Potenzial zur Beantwortung von Schlüsselfragen in der Membranfunktion wird immer besser erkannt. Die Kontrastvariationsfähigkeiten innerhalb von NSE haben es als einen einzigartigen Ansatz zur Messung mesoskopischer Membraneigenschaften positioniert, die sonst schwierig zu erhalten wären. Ein weiterer wesentlicher Vorteil von NSE gegenüber herkömmlichen Spektroskopiemethoden bei Studien der Membrandynamik ist seine Überlappung mit den Längen- und Zeitskalen, die mit MD-Simulationen zugänglich sind, was synergistische experimentelle / Berechnungsstudien ermöglicht, um ein Verständnis der verschiedenen molekularen Komponenten, aus denen Membranen bestehen, auf molekularer Ebene zu erlangen. Trotz seines Versprechens gibt es immer noch einige Einschränkungen bei der Verwendung von NSE in biologischen Membranstudien, einschließlich der Anforderung großer Probenvolumina, der Schwierigkeit der selektiven Deuteration in biologischen Systemen und des relativ geringen Neutronenflusses auf NSE-Spektrometern, was zu längeren Messzeiten und begrenzter Strahlzeitverfügbarkeit führt. Diese Mängel könnten jedoch in naher Zukunft durch ständige Entwicklungen bei Neutronenquellen und Instrumenten sowie Fortschritten bei Deuterationsanlagen überwunden werden.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Die Autoren erklären keine Interessenkonflikte und haben nichts offenzulegen.
Acknowledgments
R. Ashkar dankt M. Nagao, L.-R. Stingaciu und P. Zolnierczuk für viele nützliche Diskussionen und für ihre häufige Unterstützung bei NSE-Experimenten auf ihren jeweiligen Beamlines. Die Autoren erkennen den Einsatz von Neutronenspin-Echospektrometern am NIST und ORNL an. Das NSE-Spektrometer am NIST wird vom Center for High Resolution Neutron Scattering unterstützt, einer Partnerschaft zwischen dem National Institute of Standards and Technology und der National Science Foundation unter der Vereinbarung Nr. DMR-1508249. Das NSE-Spektrometer an der Spallations-Neutronenquelle des ORNL wird von der Scientific User Facilities Division, Office of Basic Energy Sciences, US Department of Energy, unterstützt. Das Oak Ridge National Laboratory wird von UT-Battelle, LLC unter der US DOE Contract No. DE-AC05-00OR22725.
Materials
| Name | Company | Catalog Number | Comments |
| Chloroform (biotech grade) | Sigma Aldrich | 496189 | Biotech. grade, ≥99.8%, contains 0.5-1.0% ethanol as stabilizer |
| Circulating water bath | Julabo | SE-12 | Heating Circulator with smart pump, programmable temperature settings, and external sensor connection for measurement and control |
| Deuterium Oxide | Cambridge Isotopes Laboratories | DLM-4 | Deuterated water; Heavy water (D2O) (D, 99.9%) |
| Digital Semi-Microbalance | Mettler Toledo | MS105 | Semi-micro balance with 120 g capacity, 0.01 mg readability, high resolution weighing cell, ergonomic doors, and pipette-check application |
| Ethanol (molecular biology grade) | Sigma Aldrich | E7023 | 200 proof ethanol for molecular biology applications |
| Glass Pipets | VWR | 36360-536 | Disposable Soda Lime glass Pasteur pipets |
| Glass Vials | Thermo Scientific | B7990-1 | Borosilicate glass vials with PTFE/Silione septum caps |
| Lab grade freezer | Fisher Scientific | IU2886D | Ultra-low temprature freezer (-86 to -50 C) for long-term storage of lipids and proteins |
| Lipids (protaited or perdeuterated) | Avanti Polar Lipids | varies by lipid | Lipids can be purchased from Avanti in powder form or in a chloroform solution with the required amounts and deuteration schemes. |
| Millipore water purifier | Millipore Sigma | ZRQSVP3US | Direct-Q® 3 UV Water Purification System which deliver both pure and ultrapure water with a built-in UV lamp to reduce the levels of organics for biological applications |
| Mini Extruder Set | Avanti Polar Lipids | 610020 | Mini-extruder set includes mini-extruder, heating block, 2 GasTight Syringes, and 2 O-rings, Polycarbonate Membranes, and Filter Supports |
| Quick Connect Fittings | Grainger | 2YDA1 and 2YDA7 | Push-button tube fittings for QuickConnect water circulation applications, e.g. high temperature vesicle extrusion |
| Syringe Pump | SyringePump.com | New Era-1000 | Fully programmable syringe pump for infusion and withdrawal; programs up to 41 pumping phases with adjustable pumping rates, dispensed volumes, and extrusion cycles |
| Ultrasonic bath | Fisher Scientific | CPX2800 | Temperature controlled ultra sonic bath with programmable functionality for degassing and ultrasonic applications |
| Vacuum Oven | Thermo Scientific | 3608 | 0.7 cu ft vaccum oven with built-in-high-limit thermostat guards against overheating |
| Vortex Mixer | Fisher Scientific | 02-215-414 | Variable speed, analog control that allows low rpm start-up for gentle shaking or high-speed mixing for vigorous vortexing of samples |
References
- Singer, S. J., Nicolson, G. L. The fluid mosaic model of the structure of cell membranes. Science. 175 (4023), 720-731 (1972).
- Andersen, O. S., Koeppe, R. E. Bilayer thickness and membrane protein function: an energetic perspective. Annual Review of Biophysics and Biomolecular Structure. 36, 107-130 (2007).
- Lundbæk, J. A., Collingwood, S. A., Ingólfsson, H. I., Kapoor, R., Andersen, O. S. Lipid bilayer regulation of membrane protein function: gramicidin channels as molecular force probes. Journal of The Royal Society Interface. 7 (44), 373-395 (2010).
- Bradley, R. P., Radhakrishnan, R. Curvature-undulation coupling as a basis for curvature sensing and generation in bilayer membranes. Proceedings of the National Academy of Sciences of the United States of America. 113 (35), 117-124 (2016).
- Perozo, E., Cortes, D. M., Sompornpisut, P., Kloda, A., Martinac, B. Open channel structure of MscL and the gating mechanism of mechanosensitive channels. Nature. 418 (6901), 942-948 (2002).
- Jensen, M. Ø, Mouritsen, O. G. Lipids do influence protein function-the hydrophobic matching hypothesis revisited. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1666 (1-2), 205-226 (2004).
- Rajendran, L., Simons, K. Lipid rafts and membrane dynamics. Journal of Cell Science. 118 (6), 1099-1102 (2005).
- Katchalsky, A., Spangler, R. Dynamics of membrane processes. Quarterly Reviews of Biophysics. 1 (2), 127-175 (1968).
- Rheinstädter, M. C. Collective molecular dynamics in proteins and membranes (Review). Biointerphases. 3 (2), 83-90 (2008).
- Fujiwara, T., Ritchie, K., Murakoshi, H., Jacobson, K., Kusumi, A. Phospholipids undergo hop diffusion in compartmentalized cell membrane. The Journal of Cell Biology. 157 (6), 1071-1082 (2002).
- Hac, A. E., Seeger, H. M., Fidorra, M., Heimburg, T. Diffusion in two-component lipid membranes--a fluorescence correlation spectroscopy and monte carlo simulation study. Biophysical Journal. 88 (1), 317-333 (2005).
- Heinrich, M., Tian, A., Esposito, C., Baumgart, T. Dynamic sorting of lipids and proteins in membrane tubes with a moving phase boundary. Proceedings of the National Academy of Sciences of the United States of America. 107 (16), 7208-7213 (2010).
- Hormel, T. T., Kurihara, S. Q., Brennan, M. K., Wozniak, M. C., Parthasarathy, R. Measuring lipid membrane viscosity using rotational and translational probe diffusion. Physical Review Letters. 112 (18), 188101 (2014).
- Dimova, R. Recent developments in the field of bending rigidity measurements on membranes. Advances in Colloid and Interface Science. 208, 225-234 (2014).
- Bassereau, P., Sorre, B., Lévy, A. Bending lipid membranes: Experiments after W. Helfrich's model. Advances in Colloid and Interface Science. 208, 47-57 (2014).
- Monzel, C., Sengupta, K. Measuring shape fluctuations in biological membranes. Journal of Physics D: Applied Physics. 49 (24), 243002 (2016).
- Deserno, M. Mesoscopic membrane physics: concepts, simulations, and selected applications. Macromolecular Rapid Communications. 30 (9-10), 752-771 (2009).
- Reynwar, B. J., et al. Aggregation and vesiculation of membrane proteins by curvature-mediated interactions. Nature. 447 (7143), 461-464 (2007).
- Haswell, E. S., Phillips, R., Rees, D. C. Mechanosensitive channels: what can they do and how do they do it. Structure. 19 (10), 1356-1369 (2011).
- Phillips, R., Ursell, T., Wiggins, P., Sens, P. Emerging roles for lipids in shaping membrane-protein function. Nature. 459 (7245), 379-385 (2009).
- Dill, K. A., Chan, H. S. From Levinthal to pathways to funnels. Nature Structural Biology. 4 (1), 10-19 (1997).
- Henzler-Wildman, K., Kern, D. Dynamic personalities of proteins. Nature. 450 (7172), 964-972 (2007).
- Grimaldo, M., Roosen-Runge, F., Zhang, F., Schreiber, F., Seydel, T. Dynamics of proteins in solution. Quarterly Reviews of Biophysics. 52, 7 (2019).
- Lyman, E., Hsieh, C. -L., Eggeling, C. From dynamics to membrane organization: experimental breakthroughs occasion a "modeling manifesto". Biophysical Journal. 115 (4), 595-604 (2018).
- Arriaga, L. R., et al. Dissipative curvature fluctuations in bilayer vesicles: Coexistence of pure-bending and hybrid curvature-compression modes. The European Physical Journal. E, Soft Matter. 31 (1), 105-113 (2010).
- Honerkamp-Smith, A. R., Veatch, S. L., Keller, S. L. An introduction to critical points for biophysicists; observations of compositional heterogeneity in lipid membranes. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1788 (1), 53-63 (2009).
- Veatch, S. L., Keller, S. L. Organization in lipid membranes containing cholesterol. Physical Review Letters. 89 (26), 268101 (2002).
- Heberle, F. A., et al. Bilayer thickness mismatch controls domain size in model membranes. Journal of the American Chemical Society. 135 (18), 6853-6859 (2013).
- Nickels, J. D., et al. The in vivo structure of biological membranes and evidence for lipid domains. PLOS Biology. 15 (5), 2002214 (2017).
- Simons, K., Ikonen, E. Functional rafts in cell membranes. Nature. 387 (6633), 569-572 (1997).
- van Meer, G., Voelker, D. R., Feigenson, G. W. Membrane lipids: where they are and how they behave. Nature Reviews. Molecular Cell Biology. 9 (2), 112-124 (2008).
- Liu, S. -L., et al. Orthogonal lipid sensors identify transbilayer asymmetry of plasma membrane cholesterol. Nature Chemical Biology. 13, 268 (2016).
- Rothman, J., Lenard, J. Membrane asymmetry. Science. 195 (4280), 743-753 (1977).
- Ashkar, R., et al. Neutron scattering in the biological sciences: progress and prospects. Acta Crystallographica Section D. 74 (12), 1129-1168 (2018).
- Woodka, A. C., Butler, P. D., Porcar, L., Farago, B., Nagao, M. Lipid bilayers and membrane dynamics: insight into thickness fluctuations. Physical Review Letters. 109 (5), 058102 (2012).
- Chakraborty, S., et al. How cholesterol stiffens unsaturated lipid membranes. Proceedings of the National Academy of Sciences of the United States of America. 117 (36), 21896-21905 (2020).
- Arriaga, L. R., et al. Stiffening effect of cholesterol on disordered lipid phases: a combined neutron spin echo + dynamic light scattering analysis of the bending elasticity of large unilamellar vesicles. Biophysical Journal. 96 (9), 3629-3637 (2009).
- Nagao, M., Kelley, E. G., Ashkar, R., Bradbury, R., Butler, P. D. Probing elastic and viscous properties of phospholipid bilayers using neutron spin echo spectroscopy. The Journal of Physical Chemistry Letters. 8 (19), 4679-4684 (2017).
- Kelley, E. G., Butler, P. D., Ashkar, R., Bradbury, R., Nagao, M. Scaling relationships for the elastic moduli and viscosity of mixed lipid membranes. Proceedings of the National Academy of Sciences of the United States of America. 117 (38), 23365-23373 (2020).
- Rickeard, B. W., et al. Transverse lipid organization dictates bending fluctuations in model plasma membranes. Nanoscale. 12 (3), 1438-1447 (2020).
- Nickels, J. D., et al. Mechanical properties of nanoscopic lipid domains. Journal of the American Chemical Society. 137 (50), 15772-15780 (2015).
- Mezei, F. Neutron spin echo: A new concept in polarized thermal neutron techniques. Zeitschrift für Physik A Hadrons and Nuclei. 255 (2), 146-160 (1972).
- Hayter, J. B., Penfold, J. Neutron spin-echo integral transform spectroscopy. Zeitschrift für Physik B Condensed Matter. 35 (2), 199-205 (1979).
- Monkenbusch, M., Richter, D. Neutrons in Soft Matter. Imae, T., Kanaya, T., Furusaka, M., Torikai, N. , Wiley. ch6 147-182 (2011).
- Pynn, R. Neutron Spin Echo. Mezei, F., Pappas, C., Gutberlet, T. , Springer. Berlin Heidelberg. 159-177 (2003).
- Holderer, O., et al. The JCNS neutron spin-echo spectrometer J-NSE at the FRM II. Measurement Science and Technology. 19 (3), 034022 (2008).
- Schleger, P., et al. The long-wavelength neutron spin-echo spectrometer IN15 at the Institut Laue-Langevin. Physica B: Condensed Matter. 241-243, 164-165 (1997).
- Holderer, O., Zolnierczuk, P., Pasini, S., Stingaciu, L., Monkenbusch, M. A better view through new glasses: Developments at the Jülich neutron spin echo spectrometers. Physica B: Condensed Matter. 562, 9-12 (2019).
- Farago, B., et al. The IN15 upgrade. Neutron News. 26 (3), 15-17 (2015).
- Ashkar, R. Selective dynamics in polymeric materials: Insights from quasi-elastic neutron scattering spectroscopy. Journal of Applied Physics. 127 (15), 151101 (2020).
- Pasini, S., Holderer, O., Kozielewski, T., Richter, D., Phoenix Monkenbusch, M. J-NSE- Phoenix, a neutron spin-echo spectrometer with optimized superconducting precession coils at the MLZ in Garching. Review of Scientific Instruments. 90 (4), 043107 (2019).
- Svergun, D. I., Koch, M. H. J., Timmins, P. A., May, R. P. Small Angle X-Ray and Neutron Scattering from Solutions of Biological Macromolecules. , Oxford University Press. (2013).
- Eicher, B., et al. Joint small-angle X-ray and neutron scattering data analysis of asymmetric lipid vesicles. Journal of Applied Crystallography. 50 (2), 419-429 (2017).
- Heberle, F. A., et al. Model-based approaches for the determination of lipid bilayer structure from small-angle neutron and X-ray scattering data. European Biophysics Journal. 41 (10), 875-890 (2012).
- Jaksch, S., Koutsioubas, A., Mattauch, S., Holderer, O., Frielinghaus, H. Long-range excitations in phospholipid membranes. Chemistry and Physics of Lipids. 225, 104788 (2019).
- Jaksch, S., et al. Influence of ibuprofen on phospholipid membranes. Physical Review E. 91 (2), 022716 (2015).
- Armstrong, C. L., et al. Effect of cholesterol on the lateral nanoscale dynamics of fluid membranes. European Biophysics Journal. 41 (10), 901-913 (2012).
- Rheinstädter, M. C., Häußler, W., Salditt, T. Dispersion relation of lipid membrane shape fluctuations by neutron spin-echo spectrometry. Physical Review Letters. 97 (4), 048103 (2006).
- Armstrong, C. L., Häußler, W., Seydel, T., Katsaras, J., Rheinstädter, M. C. Nanosecond lipid dynamics in membranes containing cholesterol. Soft Matter. 10 (15), 2600-2611 (2014).
- Nickels, J. D., et al. Lipid rafts: buffers of cell membrane physical properties. The Journal of Physical Chemistry B. 123 (9), 2050-2056 (2019).
- Michonova-Alexova, E. I., Sugár, I. P. Component and state separation in DMPC/DSPC lipid bilayers: a Monte Carlo simulation study. Biophysical Journal. 83 (4), 1820-1833 (2002).
- Sugár, I. P., Thompson, T. E., Biltonen, R. L. Monte Carlo simulation of two-component bilayers: DMPC/DSPC mixtures. Biophysical Journal. 76 (4), 2099-2110 (1999).
- Mabrey, S., Sturtevant, J. M. Investigation of phase transitions of lipids and lipid mixtures by sensitivity differential scanning calorimetry. Proceedings of the National Academy of Sciences. 73 (11), 3862-3866 (1976).
- Neutron activation and scattering calculator. , Available from: https://www.ncnr.nist.gov/resources/activation/ (2021).
- Scott, H. L., et al. On the mechanism of bilayer separation by extrusion, or why your LUVs are not really unilamellar. Biophysical Journal. 117 (8), 1381-1386 (2019).
- Ashkar, R., et al. Tuning membrane thickness fluctuations in model lipid bilayers. Biophysical Journal. 109 (1), 106-112 (2015).
- Carrillo, J. -M. Y., Katsaras, J., Sumpter, B. G., Ashkar, R. A computational approach for modeling neutron scattering data from lipid bilayers. Journal of Chemical Theory and Computation. 13 (2), 916-925 (2017).
- Azuah, R. T. DAVE: a comprehensive software suite for the reduction, visualization, and analysis of low energy neutron spectroscopic data. Journal of Research of the National Institute of Standards and Technology. 114 (6), 341-358 (2009).
- Van Hove, L. Correlations in space and time and born approximation scattering in systems of interacting particles. Physical Review. 95 (1), 249-262 (1954).
- Zilman, A. G., Granek, R. Undulations and dynamic structure factor of membranes. Physical Review Letters. 77 (23), 4788-4791 (1996).
- Kelley, E. G., Butler, P. D., Nagao, M. Collective dynamics in model biological membranes measured by neutron spin echo spectroscopy. , Walter de Gruyter, Inc. 131-176 (2019).
- Zheng, Y., Michihiro, N., Dobrin, P. B. Bending elasticity of saturated and monounsaturated phospholipid membranes studied by the neutron spin echo technique. Journal of Physics: Condensed Matter. 21 (15), 155104 (2009).
- Sharma, V. K., Qian, S. Effect of an antimicrobial peptide on lateral segregation of lipids: a structure and dynamics study by neutron scattering. Langmuir. 35 (11), 4152-4160 (2019).
- Boggara, M. B., Faraone, A., Krishnamoorti, R. Effect of pH and Ibuprofen on the Phospholipid Bilayer Bending Modulus. The Journal of Physical Chemistry B. 114 (24), 8061-8066 (2010).
- Lee, J. -H., et al. Thermal fluctuation and elasticity of lipid vesicles interacting with pore-forming peptides. Physical Review Letters. 105 (3), 038101 (2010).
- Chakraborty, S., Abbasi, A., Bothun, G. D., Nagao, M., Kitchens, C. L. Phospholipid bilayer softening due to hydrophobic gold nanoparticle inclusions. Langmuir. 34 (44), 13416-13425 (2018).
- Hoffmann, I., et al. Softening of phospholipid membranes by the adhesion of silica nanoparticles - as seen by neutron spin-echo (NSE). Nanoscale. 6 (12), 6945-6952 (2014).
- Watson, M. C., Brown, F. L. H. Interpreting membrane scattering experiments at the mesoscale: the contribution of dissipation within the bilayer. Biophysical Journal. 98 (6), 9-11 (2010).
- Seifert, U., Langer, S. A. Viscous modes of fluid bilayer membranes. Europhysics Letters (EPL). 23 (1), 71-76 (1993).
- Bingham, R. J., Smye, S. W., Olmsted, P. D. Dynamics of an asymmetric bilayer lipid membrane in a viscous solvent. EPL (Europhysics Letters). 111 (1), 18004 (2015).
- Rawicz, W., Olbrich, K. C., McIntosh, T., Needham, D., Evans, E. Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophysical Journal. 79 (1), 328-339 (2000).
- Doktorova, M., LeVine, M. V., Khelashvili, G., Weinstein, H. A new computational method for membrane compressibility: bilayer mechanical thickness revisited. Biophysical Journal. 116 (3), 487-502 (2019).
- Evans, E., Needham, D. Physical properties of surfactant bilayer membranes: thermal transitions, elasticity, rigidity, cohesion and colloidal interactions. The Journal of Physical Chemistry. 91 (16), 4219-4228 (1987).
- Lesieur, S., Grabielle-Madelmont, C., Paternostre, M. T., Ollivon, M. Size analysis and stability study of lipid vesicles by high-performance gel exclusion chromatography, turbidity, and dynamic light scattering. Analytical Biochemistry. 192 (2), 334-343 (1991).
- Heberle, F. A., et al. Direct label-free imaging of nanodomains in biomimetic and biological membranes by cryogenic electron microscopy. Proceedings of the National Academy of Sciences of the United States of America. 117 (33), 19943-19952 (2020).
- Cornell, C. E., Mileant, A., Thakkar, N., Lee, K. K., Keller, S. L. Direct imaging of liquid domains in membranes by cryo-electron tomography. Proceedings of the National Academy of Sciences of the United States of America. 117 (33), 19713-19719 (2020).
- Yao, X., Fan, X., Yan, N. Cryo-EM analysis of a membrane protein embedded in the liposome. Proceedings of the National Academy of Sciences of the United States of America. 117 (31), 18497-18503 (2020).
- Kučerka, N., Nieh, M. -P., Katsaras, J. Fluid phase lipid areas and bilayer thicknesses of commonly used phosphatidylcholines as a function of temperature. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1808 (11), 2761-2771 (2011).
- Nielsen, J. E., Bjørnestad, V. A., Lund, R. Resolving the structural interactions between antimicrobial peptides and lipid membranes using small-angle scattering methods: the case of indolicidin. Soft Matter. 14 (43), 8750-8763 (2018).
- Kučerka, N., et al. Lipid bilayer structure determined by the simultaneous analysis of neutron and X-ray scattering data. Biophysical Journal. 95 (5), 2356-2367 (2008).
- Kelley, E. G., Butler, P. D., Nagao, M. Scaling of lipid membrane rigidity with domain area fraction. Soft Matter. 15 (13), 2762-2767 (2019).
- Brüning, B. -A., et al. Bilayer undulation dynamics in unilamellar phospholipid vesicles: Effect of temperature, cholesterol and trehalose. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1838 (10), 2412-2419 (2014).
- Kučerka, N., et al. Areas of monounsaturated diacylphosphatidylcholines. Biophysical Journal. 97 (7), 1926-1932 (2009).
- Sharma, V. K., Mamontov, E., Anunciado, D. B., O'Neill, H., Urban, V. S. Effect of antimicrobial peptide on the dynamics of phosphocholine membrane: role of cholesterol and physical state of bilayer. Soft Matter. 11 (34), 6755-6767 (2015).
- Kelley, E. G., Butler, P. D., Nagao, M. Collective dynamics in lipid membranes containing transmembrane peptides. Soft Matter. , (2021).
- Yu, J., et al. Structure and dynamics of lipid membranes interacting with antivirulence end-phosphorylated polyethylene glycol block copolymers. Soft Matter. 16 (4), 983-989 (2020).
- Stingaciu, L. -R., et al. Revealing the dynamics of thylakoid membranes in living cyanobacterial cells. Scientific Reports. 6 (1), 19627 (2016).
- Stingaciu, L. -R., O'Neill, H. M., Liberton, M., Pakrasi, H. B., Urban, V. S. Influence of chemically disrupted photosynthesis on cyanobacterial thylakoid dynamics in synechocystis sp. PCC 6803. Scientific Reports. 9 (1), 5711 (2019).
- Miller, I. R. Energetics of fluctuation in lipid bilayer thickness. Biophysical Journal. 45 (3), 643-644 (1984).
- Nagao, M. Observation of local thickness fluctuations in surfactant membranes using neutron spin echo. Physical Review E. 80 (3), 031606 (2009).
Tags
Biologie Heft 171 kollektive Lipiddynamik Biegemodul Flächenkompressibilität Dickenschwankungen Membranviskosität selektive Deuteration LiposomenpräparationErratum
Formal Correction: Erratum: Neutron Spin Echo Spectroscopy as a Unique Probe for Lipid Membrane Dynamics and Membrane-Protein Interactions
Posted by JoVE Editors on 08/06/2021.
Citeable Link.
An erratum was issued for: Neutron Spin Echo Spectroscopy as a Unique Probe for Lipid Membrane Dynamics and Membrane-Protein Interactions. The Introduction, Protocol, and Representative Results sections have been updated.
In the Introduction, the fith pargraph was updated from:
Besides direct access to the length and time scale of membrane dynamics, NSE has the inherent capabilities of neutron isotope sensitivity52. Specifically, the ability of neutrons to interact differently with the isotopes of hydrogen, the most abundant element in biological systems, results in a different neutron scattering length density,34 or NSLD (the equivalent of the optical index of refraction50), when protium is substituted by deuterium. This enables an approach known as contrast variation, which is commonly used to highlight specific membrane features or conceal others — the latter scenario is referred to as contrast matching. A frequent application of contrast variation/matching is the substitution of water (NSLD = -0.56 × 10-6 Å-2) by heavy water or D2O (NSLD = 6.4 × 10-6 Å-2) to amplify the neutron signal from protiated lipid membranes (NSLD ~ 2 × 10-6 Å-2). This approach is highly effective in studies of membrane structure because the penetration of D2O into the headgroup region of the membrane allows accurate determination of the membrane thicknesses (see Figure 2A, left panel) and of the location of different lipid subgroups when more sophisticated models are applied53,54. This paper highlights some examples on the use of contrast variation for studies of collective dynamics in biomimetic membranes and select membrane features.
to:
Besides direct access to the length and time scale of membrane dynamics, NSE has the inherent capabilities of neutron isotope sensitivity52. Specifically, the ability of neutrons to interact differently with the isotopes of hydrogen, the most abundant element in biological systems, results in a different neutron scattering length density,34 or NSLD (the equivalent of the optical index of refraction50), when protium is substituted by deuterium. This enables an approach known as contrast variation, which is commonly used to highlight specific membrane features or conceal others — the latter scenario is referred to as contrast matching. A frequent application of contrast variation/matching is the substitution of water (NSLD = -0.56 × 10-6 Å-2) by heavy water or D2O (NSLD = 6.4 × 10-6 Å-2) to amplify the neutron signal from protiated lipid membranes (NSLD ~ 0 × 10-6 Å-2). This approach is highly effective in studies of membrane structure because the penetration of D2O into the headgroup region of the membrane allows accurate determination of the membrane thicknesses (see Figure 2A, left panel) and of the location of different lipid subgroups when more sophisticated models are applied53,54. This paper highlights some examples on the use of contrast variation for studies of collective dynamics in biomimetic membranes and select membrane features.
In the Protocol, step 1.1 was updated from:
For bending fluctuation measurements, make fully protiated liposomes in D2O (D 99.9%) or D2O-buffer (e.g., phosphate buffer prepared with D2O instead of H2O). Use fully protiated DMPC (C36H72NO8P) and DSPC (C44H88NO8P) with 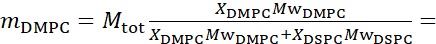 133.4 mg, where XDMPC and XDSPC are the mole fractions of DMPC and DSPC, here set to 0.7 and 0.3, respectively, and MwDMPC and MwDSPC are the molar weights given by 677.9 g/mol and 790.1 g/mol, respectively. Similarly, mDSPC = 66.6 mg. This deuteration scheme increases the scattering contrast between the membrane (NSLD ~ 2 × 10-6 Å-2) and the deuterated buffer (NSLD ~ 6.4 × 10-6 Å-2) and amplifies the signal from membrane undulations (see Figure 2A left panel).
133.4 mg, where XDMPC and XDSPC are the mole fractions of DMPC and DSPC, here set to 0.7 and 0.3, respectively, and MwDMPC and MwDSPC are the molar weights given by 677.9 g/mol and 790.1 g/mol, respectively. Similarly, mDSPC = 66.6 mg. This deuteration scheme increases the scattering contrast between the membrane (NSLD ~ 2 × 10-6 Å-2) and the deuterated buffer (NSLD ~ 6.4 × 10-6 Å-2) and amplifies the signal from membrane undulations (see Figure 2A left panel).
to:
For bending fluctuation measurements, make fully protiated liposomes in D2O (D 99.9%) or D2O-buffer (e.g., phosphate buffer prepared with D2O instead of H2O). Use fully protiated DMPC (C36H72NO8P) and DSPC (C44H88NO8P) with 133.4 mg, where XDMPC and XDSPC are the mole fractions of DMPC and DSPC, here set to 0.7 and 0.3, respectively, and MwDMPC and MwDSPC are the molar weights given by 677.9 g/mol and 790.1 g/mol, respectively. Similarly, mDSPC = 66.6 mg. This deuteration scheme increases the scattering contrast between the membrane (NSLD ~ 0 × 10-6 Å-2) and the deuterated buffer (NSLD ~ 6.4 × 10-6 Å-2) and amplifies the signal from membrane undulations (see Figure 2A left panel).
In the Representative Results, the fist pagargaph was updted from:
NSE studies accessing bending fluctuations are typically performed over a Q-range of ~ (0.04 - 0.2) Å-1. This Q-range corresponds to intermediate length scales between the membrane thickness and the liposomal radius, where bending dynamics dominate. Measurement over an extended Q-range can give access to additional dynamic modes, including liposomal diffusion and intramembrane dynamics. For more details on the cross-over in membrane dynamics accessed by NSE, check these relevant publications25,71. It is important to emphasize that NSE signals are proportional to:  , where Icoh and Iinc are, respectively, the coherent and incoherent scattering intensity from the sample. Therefore, it is advisable to prepare NSE liposomal samples in deuterated buffers (i.e., buffers prepared with D2O instead of H2O) to minimize the incoherent scattering signal, mainly contributed by the hydrogen content of the sample. However, in some cases intermediate deuteration schemes (i.e., using mixtures of D2O and H2O) might be necessary to obtain optimal contrast conditions. Typically, NSE measurements of membrane bending fluctuations are performed on fully protiated liposomes in deuterated buffer, referred to as fully contrasted liposomes in Figure 5. This deuteration scheme results in a large NSLD difference between the membrane core (~2 × 10-6 Å-2) and its deuterated fluid environment (~6.4 × 10-6 Å-2), which significantly enhances the scattering signal from the liposomal membranes and improves the measurement statistics of bending dynamics. This contrast scheme (Figure 2A left panel) is frequently utilized in studies of bending rigidity of lipid membranes with single38,72 and multiple39,66 lipid components and in studies of membrane softening/stiffening by biological inclusions (e.g., cholesterol, drug molecules, peptides/proteins)36,37,73,74,75, and synthetic additives (e.g., nanoparticles)76,77.
, where Icoh and Iinc are, respectively, the coherent and incoherent scattering intensity from the sample. Therefore, it is advisable to prepare NSE liposomal samples in deuterated buffers (i.e., buffers prepared with D2O instead of H2O) to minimize the incoherent scattering signal, mainly contributed by the hydrogen content of the sample. However, in some cases intermediate deuteration schemes (i.e., using mixtures of D2O and H2O) might be necessary to obtain optimal contrast conditions. Typically, NSE measurements of membrane bending fluctuations are performed on fully protiated liposomes in deuterated buffer, referred to as fully contrasted liposomes in Figure 5. This deuteration scheme results in a large NSLD difference between the membrane core (~2 × 10-6 Å-2) and its deuterated fluid environment (~6.4 × 10-6 Å-2), which significantly enhances the scattering signal from the liposomal membranes and improves the measurement statistics of bending dynamics. This contrast scheme (Figure 2A left panel) is frequently utilized in studies of bending rigidity of lipid membranes with single38,72 and multiple39,66 lipid components and in studies of membrane softening/stiffening by biological inclusions (e.g., cholesterol, drug molecules, peptides/proteins)36,37,73,74,75, and synthetic additives (e.g., nanoparticles)76,77.
to:
NSE studies accessing bending fluctuations are typically performed over a Q-range of ~ (0.04 - 0.2) Å-1. This Q-range corresponds to intermediate length scales between the membrane thickness and the liposomal radius, where bending dynamics dominate. Measurement over an extended Q-range can give access to additional dynamic modes, including liposomal diffusion and intramembrane dynamics. For more details on the cross-over in membrane dynamics accessed by NSE, check these relevant publications25,71. It is important to emphasize that NSE signals are proportional to: , where Icoh and Iinc are, respectively, the coherent and incoherent scattering intensity from the sample. Therefore, it is advisable to prepare NSE liposomal samples in deuterated buffers (i.e., buffers prepared with D2O instead of H2O) to minimize the incoherent scattering signal, mainly contributed by the hydrogen content of the sample. However, in some cases intermediate deuteration schemes (i.e., using mixtures of D2O and H2O) might be necessary to obtain optimal contrast conditions. Typically, NSE measurements of membrane bending fluctuations are performed on fully protiated liposomes in deuterated buffer, referred to as fully contrasted liposomes in Figure 5. This deuteration scheme results in a large NSLD difference between the membrane core (~0 × 10-6 Å-2) and its deuterated fluid environment (~6.4 × 10-6 Å-2), which significantly enhances the scattering signal from the liposomal membranes and improves the measurement statistics of bending dynamics. This contrast scheme (Figure 2A left panel) is frequently utilized in studies of bending rigidity of lipid membranes with single38,72 and multiple39,66 lipid components and in studies of membrane softening/stiffening by biological inclusions (e.g., cholesterol, drug molecules, peptides/proteins)36,37,73,74,75, and synthetic additives (e.g., nanoparticles)76,77.
In the Representative Reults, Figure 2 was updated from:
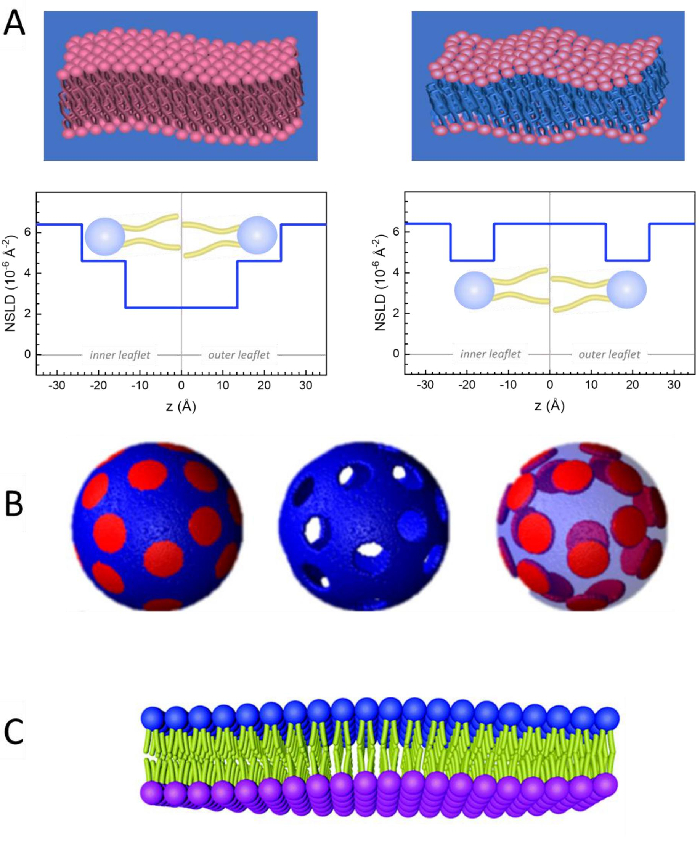
Figure 2: Examples of possible deuteration schemes in NSE experiments on lipid membranes. (A) Left: Fully contrasted membranes, e.g., protiated membranes in deuterated buffer, showing the NSLD profile along the normal to the membrane surface. The difference in the NSLD between the headgroup (~2 × 10-2 Å-2) and tail region (~4.5 × 10-6 Å-2) of the membrane is due to the headgroup hydration with deuterated buffer. Right: Tail-contrast matched membranes such that the hydrocarbon tail region of the membrane has the same NSLD as the buffer, as shown in the corresponding NSLD profile along the membrane normal. (B) Domain-forming membranes with two neutron contrast schemes where the domains (center) or the matrix (left) are contrast-matched to the buffer, enabling selective studies of matrix or domain dynamics, respectively. This figure has been modified from Nickels et al., JACS 201541. (C) Asymmetric membranes prepared by cyclodextrin exchange between protiated and deuterated lipid vesicles, resulting in the deuteration of one membrane leaflet while keeping the other leaflet protiated. This allows studies of the bending dynamics of the protiated leaflet and provides insights into the mechanical coupling between opposing leaflets in asymmetric membranes. This figure has been modified from Rickeard et al., Nanoscale 202040. Please click here to view a larger version of this figure.
to:
Figure 2: Examples of possible deuteration schemes in NSE experiments on lipid membranes. (A) Left: Fully contrasted membranes, e.g., protiated membranes in deuterated buffer, showing the NSLD profile along the normal to the membrane surface. The difference in the NSLD between the tail region (~0 × 10-2 Å-2) and headgroup region (~4.5 × 10-6 Å-2) of the membrane is due to the headgroup hydration with deuterated buffer. Right: Tail-contrast matched membranes such that the hydrocarbon tail region of the membrane has the same NSLD as the buffer, as shown in the corresponding NSLD profile along the membrane normal. (B) Domain-forming membranes with two neutron contrast schemes where the domains (center) or the matrix (left) are contrast-matched to the buffer, enabling selective studies of matrix or domain dynamics, respectively. This figure has been modified from Nickels et al., JACS 201541. (C) Asymmetric membranes prepared by cyclodextrin exchange between protiated and deuterated lipid vesicles, resulting in the deuteration of one membrane leaflet while keeping the other leaflet protiated. This allows studies of the bending dynamics of the protiated leaflet and provides insights into the mechanical coupling between opposing leaflets in asymmetric membranes. This figure has been modified from Rickeard et al., Nanoscale 202040. Please click here to view a larger version of this figure.
