

Summary
Questo protocollo descrive un metodo non invasivo per identificare in modo efficiente le cellule in fase S per studi di microscopia a valle, come la misurazione del reclutamento di proteine di riparazione del DNA mediante microirraggiamento laser.
Abstract
La riparazione del danno al DNA mantiene l'integrità genetica delle cellule in un ambiente altamente reattivo. Le cellule possono accumulare vari tipi di danni al DNA a causa di fonti sia endogene che esogene come attività metaboliche o radiazioni UV. Senza la riparazione del DNA, il codice genetico della cellula viene compromesso, minando le strutture e le funzioni delle proteine e potenzialmente causando malattie.
Comprendere le dinamiche spaziotemporali dei diversi percorsi di riparazione del DNA in varie fasi del ciclo cellulare è cruciale nel campo della riparazione del danno al DNA. Le attuali tecniche di microscopia fluorescente forniscono ottimi strumenti per misurare la cinetica di reclutamento di diverse proteine di riparazione dopo l'induzione del danno al DNA. La sintesi del DNA durante la fase S del ciclo cellulare è un punto peculiare nel destino cellulare per quanto riguarda la riparazione del DNA. Fornisce una finestra unica per schermare l'intero genoma alla ricerca di errori. Allo stesso tempo, gli errori di sintesi del DNA rappresentano anche una minaccia per l'integrità del DNA che non si incontra nelle cellule che non si dividono. Pertanto, i processi di riparazione del DNA differiscono significativamente nella fase S rispetto ad altre fasi del ciclo cellulare e tali differenze sono poco comprese.
Il seguente protocollo descrive la preparazione di linee cellulari e la misurazione della dinamica delle proteine di riparazione del DNA in fase S in siti di danno al DNA indotti localmente, utilizzando un microscopio confocale a scansione laser dotato di una linea laser a 405 nm. Tagged PCNA (con mPlum) è usato come marcatore del ciclo cellulare combinato con una proteina di riparazione di interesse marcata AcGFP (cioè EXO1b) per misurare il reclutamento del danno al DNA in fase S.
Introduction
Diversi percorsi di riparazione del DNA si sono evoluti per affrontare i diversi tipi di lesioni del DNA che possono insorgere nelle cellule, che sono tutte altamente regolate sia nello spazio che nel tempo. Uno dei periodi più vulnerabili del ciclo cellulare è la fase S, quando avviene la sintesi del DNA. Mentre la proliferazione è fondamentale per la vita, fornisce anche una grande sfida. Le cellule devono garantire una replica fedele del loro genoma per evitare che le mutazioni vengano trasmesse alle generazioni future. Di conseguenza, la proliferazione fornisce un punto di intervento terapeutico che è stato impiegato per lo sviluppo di approcci terapeutici nel campo dell'oncologia.
Tutte le principali tecniche utilizzate per studiare il reclutamento proteico nelle lesioni del DNA hanno i loro punti di forza e limiti. La microirraggiamento ha una migliore risoluzione spaziale e temporale1 rispetto alla maggior parte dei metodi alternativi come l'imaging immunofluorescente di focolai indotti da radiazioni ionizzanti (IRIF), la cromatina-immunoprecipitazione (ChIP) o il frazionamento biochimico. Tuttavia, la micro-irradiazione la robustezza delle tecniche di cui sopra che possono campionare un gran numero di cellule contemporaneamente.
Per studiare la riparazione del DNA in fase S, si deve essere in grado di distinguere le cellule della fase S in una popolazione asincrona di coltura cellulare. Esistono molti metodi ben noti per affrontare questo problema, che coinvolgono la sincronizzazione delle cellule o la visualizzazione delle diverse fasi del ciclo cellulare. Tuttavia, entrambi gli approcci introducono sfide significative e possibili artefatti. I metodi di sincronizzazione chimica ampiamente utilizzati per arricchire le cellule nella fase S iniziale (ad esempio, doppio blocco di timidina, aphidicolin e trattamento con idrossiurea) ottengono la sincronizzazione attraverso l'induzione dello stress di replicazione e alla fine del danno al DNA stesso. Ciò limita l'uso di questi metodi per studiare i processi di riparazione del DNA nella fase S2. La sincronizzazione attraverso la fame e il rilascio di siero è applicabile solo a un numero limitato di linee cellulari, in gran parte escluse le linee cellulari tumorali che si basano meno sui fattori di crescita per la progressione del ciclo cellulare rispetto alle linee cellulari non trasformate. Il sistema FUCCI (Fluorescence Ubiquitin Cell Cycle Indicator) è uno strumento particolarmente utile per studiare il ciclo cellulare, ma ha un limite fondamentale quando si differenzia tra le fasi del ciclo cellulare S e G23.
Qui è dimostrato che l'uso di PCNA con tag fluorescenti come marcatore non invasivo per la fase S limita gli svantaggi dei metodi di sincronizzazione chimica del ciclo cellulare, consentendo al contempo una maggiore specificità e flessibilità rispetto al sistema FUCCI. Come singolo marcatore, non solo il PCNA può evidenziare le cellule della fase S in una popolazione asincrona, ma può anche mostrare l'esatta progressione delle cellule all'interno della fase S(cioèfase S iniziale, media o tardiva) 4 . Bassi livelli di espressione di PCNA esogeno e marcato assicurano interferenze minime sia con la progressione del ciclo cellulare che con i processi di riparazione del DNA. È importante sottolineare che il PCNA funge anche da controllo interno per una corretta induzione del danno al DNA in quanto è coinvolto nella riparazione di diverse lesioni del DNA e viene reclutato nei siti di danno al DNA indotti localmente1,4.
Gli esperimenti qui presentati dimostrano come misurare le dinamiche di reclutamento di EXO1b in fase S e come questo è influenzato dal consolidato inibitore PARP, olaparib. L'attività della nucleasi EXO1b è rilevante per una vasta gamma di percorsi di riparazione del DNA, tra cui la riparazione del mismatch (MMR), la riparazione dell'escissione nucleotidica (NER) e la riparazione della rottura a doppio filamento (DSB). Nella fase S, EXO1b svolge un ruolo importante nella ricombinazione omologa (HR) attraverso la formazione di sporgenze di 3' ssDNA durante la resezione del DNA5. EXO1b è stato ulteriormente implicato nella replicazione del DNA con ruoli nell'attivazione del checkpoint per riavviare le forchette del DNA in stallo, nonché nella rimozione del primer e nella maturazione del frammento di Okazaki nel filamento in ritardo durante lo spostamento del filamento nella replicazione5. Il reclutamento di EXO1b in siti di DNA danneggiati è regolato dall'interazione diretta con il poli (ADP-ribosio) (PAR)6,7. A causa delle numerose implicazioni specifiche del ciclo cellulare di EXO1b, è una scelta eccellente per gli studi di reclutamento specifici della fase S che utilizzano PCNA.
Protocol
1. Coltivazione di cellule derivate dall'osteosarcoma umano (U-2 OS)
NOTA: le cellule OS U-2 sono ideali per questi studi in quanto hanno una morfologia piatta, un nucleo grande e fortemente attaccate a diverse superfici, incluso il vetro. Potrebbero essere utilizzate anche altre linee cellulari con caratteristiche simili.
- Per la coltivazione di linee cellulari U-2 OS, utilizzare il mezzo 5A di McCoy integrato con siero bovino fetale al 10% (FBS) e antibiotici (100 U / mL di penicillina e 100 μg / mL di streptomicina). Incubare le cellule a 37 °C in atmosfera umidificata contenente il 5% di CO2. Per gli studi di microscopia, mantenere la coltura cellulare in un piatto di 10 cm per fornire un numero sufficiente di cellule.
- Quando le cellule si avvicinano al 90% di confluenza (~ 7 x 106 celle / piatto di 10 cm), dividere le cellule.
- Risciacquare le cellule con PBS per lavare via gli inibitori della tripsina contenuti nel siero.
- Aggiungere 1 mL di tripsina-EDTA e assicurarsi che lo strato cellulare sia ugualmente coperto.
- Incubare a 37 °C fino a quando lo strato cellulare non viene sollevato dalla piastra (circa 6 min).
- Rinsoppiare le cellule tripsinizzate nel siero contenente mezzi per inattivare la tripsina e aggiungere 1/10 del volume (~ 0,7 x 106 cellule) in una nuova piastra da 10 cm contenente 10 ml di terreno di crescita integrato.
- Prima della sperimentazione, testare regolarmente le cellule per la contaminazione da micoplasma utilizzando il kit universale di rilevamento del micoplasma seguendo la raccomandazione del produttore.
2. Infezione retrovirale
NOTA: Per le misure di sicurezza BSL-2 e mentre si lavora con virus ricombinanti, fare riferimento a: Linee guida NIH, Sezione III-D-3: Virus ricombinanti in coltura tissutale.
- Semina 4 x 106 celle HEK293T per ottenere una confluenza di ~ 60% entro 24 ore dopo la placcatura in una piattaforma di coltura di 10 cm.
- Per coltivare HEK293T si prega di seguire le fasi di coltivazione di U-2 OS descritte in 1.1-1.3 di questo protocollo. Per HEK293T sostituire il mezzo 5A di McCoy per DMEM. Assicurati di lavare sempre delicatamente le cellule HEK293T mentre si attaccano debolmente alle piastre di coltura tissutale.
- Trasfetturare le cellule HEK293T utilizzando un reagente di trasfezione a base lipidica per il confezionamento virale di plasmidi.
- Per i vettori retrovirali, combinare 1,5 μg di VSV-G (Addgene #8454) e 1,5 μg di vettori di imballaggio pUMVC (Addgene #8449) insieme a 3 μg del vettore contenente il gene di interesse (in una dorsale vettoriale retrovirale con resistenza alla puromicina) in 250 μL di mezzi sierici ridotti Opti-MEM in un tubo di microcentrifuga. Aggiungere 1 μL di reagente P3000 per ogni μg di DNA aggiunto nella miscela Opti-MEM/DNA (in questo caso 6 μL) e mescolare delicatamente picchiettando. Non vortice o pipetta su e giù.
- In un altro tubo microcentrifuga, combinare 2 μL per μg di DNA (in questo caso 12 μL) di reagente di trasfezione con 250 μL di mezzi sierici ridotti Opti-MEM.
- Unire le due miscele (500 μL combinate, non vortice, mescolare solo picchiettando delicatamente) e lasciarlo incubare per 15 minuti a temperatura ambiente.
- Con attenzione, aggiungere la miscela a goccia alle cellule HEK293T seminate senza staccare le cellule. Ruotare delicatamente i piatti.
- Infezione virale per generare linee cellulari stabili.
- Rimuovere il virus contenente surnatante dalle cellule HEK293T 72 ore dopo la trasfezione. Filtrare accuratamente la soluzione con un filtro da 0,45 μm per rimuovere i detriti cellulari e le cellule staccate. Facoltativamente, aggiungere 8 μg / mL di polibrene al surnatante virale per facilitare l'infezione virale.
- Aggiungere virus contenente surnatante alle cellule U-2 OS a ~ 50% di confluenza in un piatto di 10 cm (~ 3 x 106 cellule). Semina le cellule del sistema operativo U-2 il giorno prima.
- Infettare per 6-16 ore prima di rimuovere e scartare il surnatante contenente virus.
NOTA: Per ottenere la quantità desiderata di sovraespressione per il gene di interesse, incubare una serie di diluizioni virali per un determinato periodo di tempo. Controllare i livelli di espressione del transgene in ogni linea cellulare appena stabilita con Western blot confrontandolo con i livelli endogeni. - Consentire alle cellule di selezionare in presenza di antibiotici appropriati (per 3-4 giorni in caso di puromicina a 2 μg/mL di concentrazione finale) e verificare al microscopio l'espressione del gene di interesse marcato con proteina fluorescente.
- Ripetere questi passaggi per generare linee di cella con doppia etichetta. Negli esperimenti qui presentati mPlum-PCNA è stato espresso da un vettore retrovirale (pBABE) combinato con EXO1B-AcGFP, espresso anche da un vettore retrovirale (pRetroQ-AcGFP1-N1).
3. Preparazione delle cellule per il microirraggiamento
- Celle di placcatura: 24 ore prima dell'esperimento, placcare un totale di 8,0 x10 4 celle in un volume compreso tra 500 μL-1 mL di mezzo (per circa il 70% di confluenza) su un vetro di copertura a quattro camere con un fondo di vetro borosilicato n. 1,5 che offre risultati ideali per la microscopia confocale ad alto ingrandimento e la microirraggiamento laser. Una maggiore confluenza cellulare consente di misurare più celle in un singolo campo visivo (FOV); tuttavia i vetrini completamente confluenti introdurranno irregolarità del ciclo cellulare.
- Mezzi di imaging: un'ora prima della microirraggiamento, scambiare un mezzo di crescita regolare per FluoroBrite DMEM integrato con 10% FBS, 100 U / mL penicillina e 100 μg / mL streptomicina, 15 mM HEPES (pH = 7,4) e 1 mM sodio-piruvato. Questo supporto di imaging aiuta a massimizzare il rapporto segnale-rumore consentendo il rilevamento di una fluorescenza molto fioca. Poiché contiene HEPES, stabilizza anche il pH in assenza di un'atmosfera di CO2 al 5%.
- Applicare qualsiasi trattamento aggiuntivo prima dell'imaging in questa fase. Negli esperimenti qui presentati, le cellule sono state pre-trattate un'ora prima dell'imaging con olaparib (inibitore PARP, a concentrazione finale di 1 μM) o un controllo del veicolo (DMSO)1,8,9.
4. Preparare il microscopio e selezionare le cellule della fase S per l'imaging.
- Utilizzare un sistema confocale che abbia le proprietà simili al sistema descritto qui per ottenere i migliori risultati. Gli esperimenti qui presentati sono stati eseguiti utilizzando un microscopio confocale montato su un supporto per microscopio invertito (vedi Tabella dei materiali).
NOTA: Il microscopio utilizzato qui era dotato di un modulo laser FRAP da 50 mW 405 nm e di un obiettivo a piano di olio da 60x 1,4 NA. La testina di scansione confocale aveva due opzioni di scanner: uno scanner galvano (per alta risoluzione) e uno scanner risonante (per immagini ad alta velocità).- Introdurre il recupero di fluorescenza dopo il laser FRAP (Photobleaching) nel campione tramite un dispositivo galvano XY controllato da software. Utilizzare una linea laser a 488 nm per eccitare AcGFP e una linea laser a 561 nm o 594 nm per eccitare mPlum.
NOTA: La seguente combinazione di filtri fornisce risultati ottimali: utilizzando un filtro a passaggio lungo da 560 nm, la luce di emissione con una lunghezza d'onda inferiore a 560 nm è stata fatta passare attraverso un filtro di emissione a 525/50 nm per AcGFP, mentre la luce di emissione con una lunghezza d'onda superiore a 560 nm è stata fatta passare attraverso un filtro di emissione a 595/50 nm per mPlum. È possibile utilizzare qualsiasi set di filtri appropriato (ad esempio, FITC/ TRITC, GFP/mCherry, FITC/TxRed) che garantisca un minimo spurgo di fluorescenza.
- Introdurre il recupero di fluorescenza dopo il laser FRAP (Photobleaching) nel campione tramite un dispositivo galvano XY controllato da software. Utilizzare una linea laser a 488 nm per eccitare AcGFP e una linea laser a 561 nm o 594 nm per eccitare mPlum.
- Accendere la camera ambientale e i componenti del microscopio.
- Accendere il riscaldamento (stadio, obiettivo e camera ambientale quando possibile), l'alimentazione di CO2 e il regolatore di umidità almeno 4 ore prima dell'inizio dell'esperimento per garantire l'equilibrio termico per un'acquisizione stabile dell'immagine.
- Inizializzare le sorgenti luminose insieme alle linee laser almeno 1 ora prima del trasferimento delle cellule al microscopio.
- Selezionare le celle della fase S in una popolazione asincrona utilizzando pcNA con tag fluorescenti come marcatore. Fallo seguendo i passaggi seguenti.
- Cerca il modello di localizzazione unico del PCNA con tag mPlum in fase S rendendo possibile l'identificazione di questa fase del ciclo cellulare. Il PCNA ha una distribuzione completamente omogenea nel nucleo nelle fasi G1 e G2 del ciclo cellulare, pur essendo escluso dai nucleoli. Nella fase S, pcna forma focolai nella posizione dei rettili nel nucleo. La Figura 1 mostra i diversi modelli di focolai PCNA durante la fase S, il che rende possibile persino differenziare la fase S iniziale, media e tardiva.
- Guarda attraverso l'oculare per selezionare un FOV che abbia abbastanza cellule in fase S per la micro-irradiazione. Le cellule asincrone U-2 OS di solito hanno il 30-40% della loro popolazione in fase S.
- Cerca di evitare estremi nei livelli di espressione (cellule luminose e fioche allo stesso modo) sia per il PCNA che per la proteina di interesse (POI), in questo caso EXO1b-AcGFP, che potrebbe portare a artefatti sperimentali.
- Quando trovi un FOV adatto, cerca di evitare di scansionare il campo per un lungo periodo di tempo per ridurre al minimo il fotosciviazione e i danni indesiderati al DNA.
- Impostare la regione di interesse (ROI) desiderata per la microirraggiamento. Utilizzando il software associato (vedi Tabella dei materiali),imposta il ROI desiderato inserendo prima le linee binarie (imposta il numero desiderato di linee e la spaziatura). Fare clic su Binario, quindi su Inserisci riga | Cerchio | Ellisse per disegnare il numero desiderato di linee.
- Converti queste linee binarie in ROI e infine converti questi ROI in ROI di stimolazione. Per fare ciò, fai prima clic su ROI, quindi fai clic su Sposta binarioin ROI , quindi fai clic con il pulsante destro del mouse su uno qualsiasi dei ROI e seleziona Usa come ROI di stimolazione: S1. Posizionare queste linee nel FOV per passare attraverso il nucleo delle cellule. I ROI con una lunghezza di 1024 pixel che si estendevano sull'intero FOV sono stati utilizzati in tutto il protocollo.
5. Microir irradiazione per colorazione a immunofluorescenza o imaging time lapse.
- Determinazione delle impostazioni ottimali di microirraggiamento.
- Prima della micro-irradiazione delle cellule, prendere un'immagine ad alta risoluzione del FOV per identificare i focolai di PCNA per un'analisi successiva. Invece della scansione sequenziale, registrare contemporaneamente entrambi i canali ottici utilizzati (verde e rosso), per evitare il movimento cellulare tra la scansione alle due lunghezze d'onda. Per una corretta risoluzione dei fuochi utilizzare almeno 1024 x 1024 pixel / risoluzione di campo con zoom 1x (dimensione dei pixel di 0,29 μm sul sistema di imaging utilizzato qui), con velocità di scansione di 1/8 frame / s (4,85 μs / pixel) con media 2x. Una volta impostati questi parametri nella GUI compatta LFOV A1 e nelle finestre Area di scansione LFOV A1, premere il pulsante Acquisizione per registrare il FOV.
NOTA: è importante mantenere la stessa dimensione in pixel durante gli esperimenti per garantire risultati comparabili. - Per impostare la microirraggiamento, aprire la scheda Stimolazione ND nel software di imaging per accedere alla finestra Time schedule (A1 LFOV / Galvano Device). Questo utilizza gli scanner galvano per acquisire una serie di immagini di pre-stimolazione, stimolare (utilizzando il laser FRAP LUN-F 50 mW 405 nm) e quindi acquisire nuovamente una serie di immagini post-stimolazione utilizzando gli scanner galvano. Impostare innanzitutto tre fasi nella finestra Pianificazione temporale. Nella colonna Acq/Stim selezionare Acquisizione | | sbiancante Acquisizione rispettivamente per le tre fasi. Per la fase di sbiancamento, impostare S1 come ROI.
NOTA: Nell'esperimento qui presentato, nessuna immagine è stata acquisita durante la fase di stimolazione. - Nella finestra galvano XY,impostare i fattori chiave per la microirraggiamento: potenza laser di uscita a 405 nm, tempo di permanenza (l'iterazione è 1 per impostazione predefinita su questo sistema). Negli esperimenti qui presentati, le cellule sono state irradiate con il laser FRAP a 405 nm (50 mW sulla punta della fibra) al 100% di potenza con un tempo di permanenza di 1000-3000 μs.
NOTA: poiché il tempo di permanenza del laser è su base per pixel, finché la dimensione dei pixel rimane la stessa, la relazione tra il tempo di permanenza e la densità di potenza sarà comparabile tra diversi FOV. La Figura 2A mostra l'uso di proteine specifiche del percorso di risposta al danno del DNA (DDR) (FBXL10 per DSB e NTHL1 per danno ossidativo alla base) per ottimizzare le impostazioni di potenza del laser per l'induzione del danno specifico. Queste linee cellulari stabili sono state generate con infezione virale dopo la sezione 2 del protocollo.
- Prima della micro-irradiazione delle cellule, prendere un'immagine ad alta risoluzione del FOV per identificare i focolai di PCNA per un'analisi successiva. Invece della scansione sequenziale, registrare contemporaneamente entrambi i canali ottici utilizzati (verde e rosso), per evitare il movimento cellulare tra la scansione alle due lunghezze d'onda. Per una corretta risoluzione dei fuochi utilizzare almeno 1024 x 1024 pixel / risoluzione di campo con zoom 1x (dimensione dei pixel di 0,29 μm sul sistema di imaging utilizzato qui), con velocità di scansione di 1/8 frame / s (4,85 μs / pixel) con media 2x. Una volta impostati questi parametri nella GUI compatta LFOV A1 e nelle finestre Area di scansione LFOV A1, premere il pulsante Acquisizione per registrare il FOV.
- Imaging time lapse.
- Imposta l'imaging time lapse per la finestra temporale e gli intervalli desiderati utilizzando la pianificazione temporale, la GUI compatta A1 LFOV e le finestre A1 LFOV Scan Area. Negli esperimenti qui presentati, il reclutamento di EXO1b e PCNA è stato ripreso per 12 minuti, scansionando il FOV ogni 5 secondi, a 1024 x 1024 pixel / campo, utilizzando lo zoom 1x (con conseguente dimensione dei pixel di 0,29 μm sul sistema di imaging utilizzato qui) con velocità di scansione di 0,35 fotogrammi / s (1,45 μs / pixel) senza media per ridurre il foto-sbiancamento.
- Ottimizza le impostazioni di potenza del laser %, guadagno e offset per ridurre il foto-sbiancamento durante l'imaging nella finestra A1 LFOV Compact GUI. Se si mira a misurare sia il POI che il PCNA, utilizzare la scansione simultanea anziché la scansione sequenziale per evitare il movimento cellulare tra la scansione del campo per i due fluorofori separati.
- Il sistema di imaging è stato utilizzato con le seguenti impostazioni. Per la linea laser 488 nm (20 mW): 7% di potenza laser, guadagno: 45 (rilevatore GaAsP) con e offset di 2, per la linea laser 561 nm (20 mW): 4% di potenza laser, guadagno 40 (rilevatore GaAsP) con e offset di 2.
- A seconda della cinetica della proteina, estendere o abbreviare l'intervallo tra le immagini o la durata del time lapse totale. Nella finestra Pianificazione temporale, impostare l'intervallo e la durata desiderati per la riga Acquisizione della terza fase.
- Premere Esegui ora per eseguire la microirraggiamento e il successivo imaging time lapse.
- Al termine dell'imaging time lapse, salvare i ROI di stimolazione come immagini separate, che saranno un utile aiuto per identificare le coordinate di microirraggiamento in qualsiasi software a valle utilizzato per l'analisi.
- Colorazione immunofluorescenza.
NOTA: La fase 5.1.3 e la figura 2A dimostrano l'uso di proteine di riparazione del DNA note per valutare i tipi di lesioni del DNA introdotte dalla microirraggiamento. Alcune lesioni del DNA possono anche essere rilevate utilizzando anticorpi specifici dopo aver fissato le cellule. È anche possibile rilevare il reclutamento del POI mediante il rilevamento anticorpale della proteina endogena. La visualizzazione di γH2A.X per verificare la disponibilità di DSB è illustrata di seguito (Figura 2B). La Figura 3 mostra la coerenza della localizzazione e del reclutamento di PCNA durante tutto il ciclo cellulare sia per il PCNA endogeno che per quello esogeno.- Dopo il passaggio 5.1.3, prendere una sola immagine dopo la micro-irradiazione per garantire un evento FRAP corretto basato sul reclutamento di mPlum-PCNA. Prendi nota delle coordinate esatte del FOV per trovare il campo in seguito dopo l'etichettatura immunofluorescente.
- Togliere la camera di coltura cellulare dal microscopio e incubare le cellule a 37 °C in un'atmosfera umidificata contenente il 5% di CO2 per 5-10 minuti.
NOTA: la paraformaldeide (PFA) è tossica e il lavoro deve essere svolto in un'area ben ventilata o in una cappa aspirante. Tutti i successivi lavaggi e incubazione saranno eseguiti con volumi di 0,5 ml nella slitta a 4 camere di pozzo. Dopo il tempo di incubazione, lavare le cellule con 0,5 mL di PBS (137 mM NaCl, 2,7 mM KCl, 8 mM Na2HPO4e 2 mM KH2PO4) e fissare con 0,5 mL di PFA al 4% in PBS per 10 minuti a temperatura ambiente (RT). - Lavare le cellule una volta con PBS, quindi lavarle con 50 mM NH4Cl per estinguere il PFA residuo.
- Permeabilizzare le cellule per 15 minuti a RT con lo 0,1% di Triton X-100 in PBS.
- Bloccare i campioni per 1 ora con buffer di blocco (5% FBS, 3% BSA, 0,05% Triton X-100 in PBS).
- Rimuovere la soluzione bloccante e aggiungere l'anticorpo primario diluito (anti-γH2A.X, 1:2000) nel tampone di blocco per 1 ora a RT.
- Lavare i pozzi con tampone di blocco 3 x 10 min.
- Aggiungere anticorpi secondari diluiti (coniugato anti-mouse Alexa 488 Plus, 1:2000) nel tampone di blocco per 1 ora a RT.
- Lavare i pozzi con tampone di blocco 3 x 10 min.
- Controbattere il nucleo con 1 μg/mL di soluzione DAPI in PBS per 15 min.
- Lavare le cellule una volta con PBS. L'imaging può essere eseguito direttamente in PBS o in una soluzione PBS con reagenti antifade (ad esempio, AFR3) per ridurre il fotosciviazione.
6. Analisi del reclutamento
NOTA: La Figura 4A mostra immagini rappresentative del reclutamento di Exo1b e PCNA in presenza di DMSO o olaparib. La Figura 4B mostra un'immagine rappresentativa per l'analisi dei dati. I valori medi di fluorescenza sono stati calcolati misurando le intensità medie acGFP utilizzando un rettangolo lungo la traccia laser evidenziato dal mPlum-PCNA (A, rettangoli gialli) su diversi punti temporali utilizzando Fiji. PCNA può servire come controllo interno per evidenziare l'irradiazione di successo lungo le coordinate ROI. Allo stesso modo, i valori medi di fluorescenza AcGFP sono stati calcolati anche per le regioni non danneggiate del nucleo (B, rettangoli blu). L'intensità del segnale di fondo è stata misurata in aree non popolate (C, rettangoli rossi) ed è stata sottratta dai valori fluorescenti medi (Figura A e B). Pertanto, l'unità fluorescente media relativa (RFU) per ciascun punto di raccolta dati è stata calcolata dall'equazione RFU = (A − C)/ (B − C)8,9. I valori RFU risultanti della regione microirraggiata vengono normalizzati ai valori RFU prima della microirraggiamento.
- Per definire la regione A del sito microisolato, escludere dalla misurazione le regioni nucleolari, i focolai di replicazione e le regioni nucleari irregolari della cellula. Tieni premuto il tasto shift tra il disegno di due ROI nelle Figi per raggruppare due regioni separate come una sola.
NOTA: Il reclutamento delle proteine varierà tra i diversi geni e le condizioni di irradiazione; pertanto, la dimensione della regione A deve essere determinata individualmente. Una volta determinata la larghezza in pixel della regione A, dovrebbe rimanere costante per eventuali assunzioni comparative. Negli esperimenti qui presentati, sono stati utilizzati rettangoli di larghezza di 7 pixel. - Escludere dall'analisi le celle che si sono spostate durante la durata dei video registrati. Per includere celle altamente mobili, l'analisi descritta deve essere eseguita fotogramma per fotogramma.
- Per visualizzare il profilo di reclutamento, tracciare i valori RFU normalizzati rispetto al tempo utilizzando un software statistico.
- Calcolare la differenza in un punto temporale indicato tra il trattamento con DMSO e olaparib (n = 31) utilizzando un test di Mann-Whitney.
Representative Results
Le cellule affrontano ogni tipo di lesione del DNA in un modo specifico che dipende anche dalla fase del ciclo cellulare in cui si trovano. Ad esempio, dopo la microirraggiamento, le rotture a doppio filamento (DSB) verranno elaborate mediante unione finale non omologa (NHEJ) o HR a seconda della fase del ciclo cellulare. Le nucleasi che agiscono più estesamente durante le fasi S e G2 del ciclo cellulare creano sporgenze di DNA che sono cruciali per una corretta HR. Per promuovere la valutazione delle cellule in fase S, il PCNA è stato impiegato come marcatore del ciclo cellulare a colore singolo. La Figura 1A mostra il profilo di localizzazione di mPlum-PCNA durante la progressione del ciclo cellulare. Il PCNA ha una distribuzione completamente omogenea nel nucleo in fase G1 e G2 (pur essendo per lo più escluso dai nucleoli). Nella fase S, PCNA si localizza in siti di replicazione del DNA, che possono essere visualizzati come punti luminosi nel nucleo. Nelle prime cellule della fase S, le macchie sono relativamente piccole e equamente distribuite in tutto il nucleo della cellula. Progredendo nella fase S media, le macchie diventano sfocate e si localizzano maggiormente verso il perimetro del nucleo e dei nucleoli. Nella fase S tardiva, le macchie si riducono in numero ma diventano sempre più grandi man mano che il PCNA si concentra nei siti di replicazione tardiva (Figura 1B). È importante sottolineare che l'espressione esogena di PCNA dalla spina dorsale del vettore pBABE era inferiore ai livelli endogeni, ma era sufficiente per il rilevamento al microscopio che riduce al minimo i potenziali artefatti nella progressione del ciclo cellulare e nella DDR. La Figura 1C mostra l'estensione della sovraespressione di PCNA rispetto ai livelli endogeni. Si noti che la banda corrispondente a mPlum-PCNA migra più lentamente a causa delle sue dimensioni maggiori.
Abbiamo mirato a introdurre DSB durante la microirraggiamento per studiare il reclutamento PARP1/2-dipendente di EXO1b a queste lesioni in fase S. La Figura 2A mostra che basse dosi di energia (1000 μs di tempo di permanenza) non inducono il reclutamento di EGFP-FBXL10, un responder DSB (componente del complesso FRUCC 8),mentre è stato sufficiente indurre il reclutamento di NTHL1-mCherry, una proteina pathway di riparazione dell'escissione di base (BER), reclutando in siti di danno ossidativo al DNA10,11,12. A 3000 μs di tempo di permanenza, sia EGFP-FBXL10 che NTHL1-mCherry reclutano, dimostrando un'uscita laser che genera sia lesioni ossidative che DSB. Rafforzando questi risultati, la Figura 2B mostra la colorazione di immunofluorescenza contro γH2A.X (marcatore DSB), che è chiaramente più evidente quando si utilizzano dosi di energia più elevate. PcNA funge sia da marcatore del ciclo cellulare che da marcatore per il successo della microirraggiamento, in quanto recluta adeguatamente con entrambe le impostazioni del tempo di permanenza laser. È importante sottolineare che sia il PCNA esogeno che/o endogeno con tag proteico fluorescente può essere utilizzato per questa funzione reporter in quanto si comportano in modo simile (Figura 3). Il PCNA etichettato in modo endogeno è stato progettato inserendo mRuby nel telaio con il primo esone in un allele del locusPCNA 13 (la linea cellulare era un dono gentile di Jörg Mansfeld).
La Figura 4A e la Figura 4C mostrano il reclutamento di EXO1b con tag AcGFP nelle celle della fase S. EXO1b raggiunge il massimo livello di accumulo nei siti di microirraggiamento intorno a 1 minuto e poi inizia lentamente a disimpegnarsi dalle lesioni del DNA in seguito. Gli arricchimenti nei siti di microirraggiamento sono indicati da un'unità di fluorescenza relativa > 1 sul grafico. In presenza di olaparib, l'accumulo di EXO1b sulla striscia laser a 1 minuto è significativamente inferiore rispetto al controllo del veicolo. Questi risultati sono in accordo con la letteratura6,7. La figura 4B illustra le regioni rappresentative per la quantificazione (aree A, B e C) come descritto al punto 6 del protocollo. La Figura 4D mostra i livelli di espressione comparabili di EXO1b endogeno e EXO1b-AcGFP esogeno nelle cellule utilizzate per la microirraggiamento.
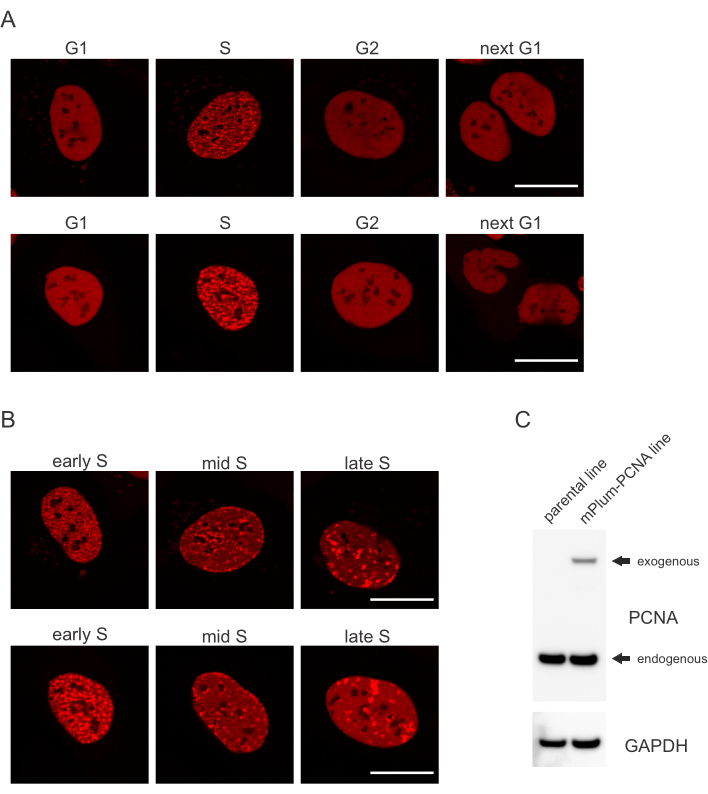
Figura 1: Modello di localizzazione di PCNA. (A) Le immagini mostrano il modello di localizzazione di PCNA esogeno stabilmente integrato durante tutto il ciclo cellulare in cellule U-2 OS. (B) Le immagini mostrano modelli di focolai PCNA in diverse fasi della fase S (iniziale, media e tardiva) nelle cellule OS U-2. (C) Western blot che mostra livelli endogeni ed esogeni di PCNA nelle cellule U-2 OS utilizzate per l'imaging. La barra della scala rappresenta 20 μm. Fare clic qui per visualizzare una versione più grande di questa figura.

Figura 2: Induzione di DSB attraverso l'uscita di potenza laser ottimizzata. (A) Le impostazioni laser possono essere ottimizzate per indurre diverse forme di danno al DNA. Le cellule U-2 OS che esprimono stabilmente sia EGFP-FBXL10 che NTHL1-mCherry sono state utilizzate per identificare rispettivamente DSB e siti di lesioni ossidative. La microirraggiamento con una linea laser a 405 nm è stata effettuata su celle asincrone U-2 OS con tempo di permanenza di 1000 μs o 3000 μs. La barra di scala rappresenta 20 μm. (B) La colorazione immunofluorescente contro γH2A.X è stata eseguita su cellule epiteliali pigmentate retiniche umane (hTERT RPE-1) con PCNA endogeno marcato con mRuby. Le cellule sono state fissate ed elaborate 5 minuti dopo la microirraggiamento con un tempo di permanenza di 1000 μs o 3000 μs. La barra della scala rappresenta 20 μm. Fare clic qui per visualizzare una versione più grande di questa figura.

Figura 3: Reclutamento comparabile di mRuby-PCNA endogeno e mPlum-PCNA esogeno in siti di microirraggiamento a 1000 μs o 3000 μs di tempo di permanenza laser. Sia il PCNA endogeno che l'esogeno marcata formano focolai di replicazione durante la fase S. Fare clic qui per visualizzare una versione più grande di questa figura.

Figura 4: Reclutamento PARP1/2-dipendente di EXO1b in fase S. Le cellule U-2 OS che esprimono stabilmente EXO1b-AcGFP e mPlum-PCNA sono state micro-irradiate con una linea laser FRAP a 405 nm utilizzando un tempo di permanenza di 3000 μs. (A) Immagini rappresentative di cellule microirraggiate nei punti temporali indicati dopo il pretrattamento con controllo del veicolo (DMSO) o olaparib (1 μM). La barra di scala rappresenta 20 μm. (B) Immagini rappresentative di regioni definite di aree A, B e C per l'analisi del reclutamento. La barra della scala rappresenta 20 μm. (C) La dinamica di reclutamento del danno al DNA è stata catturata dall'imaging di cellule vive. I valori medi di fluorescenza relativa e le immagini sono stati acquisiti ogni 5 s per 12 min. Per ogni condizione, sono state valutate ≥30 cellule. I valori medi di fluorescenza relativa (linee nere solide) e l'errore standard (intervallo visualizzato da un'area ombreggiata) sono stati tracciati rispetto al tempo. La linea tratteggiata mostra i valori di reclutamento a 1 minuto dopo la microirraggiamento. La differenza tra il trattamento con DMSO (n=32) e olaparib (n=31) è stata calcolata utilizzando un test di Mann-Whitney. Asterix indica p<0.0001. (D) Western blot confronta i livelli di espressione di EXO1b endogeno e EXO1b-AcGFP esogeno in cellule utilizzate per la microirraggiamento. Fare clic qui per visualizzare una versione più grande di questa figura.
Discussion
Passaggi critici e potenziali problemi/modifiche del protocollo
Un adeguato vaso di coltura tissutale per la micro-irradiazione è fondamentale per il successo. La maggior parte dei sistemi di imaging ad alta risoluzione sono ottimizzati per uno spessore del vetro di copertura di 0,17 mm. L'utilizzo di camere di imaging di spessore superiore o inferiore o di quelle realizzate con polimeri plastici (non ottimizzate per l'imaging a 405 nm) può ridurre significativamente la qualità dell'immagine. Quando si utilizzano superfici di vetro, assicurarsi che siano trattate con colture tissutali per migliorare l'adesione cellulare. Se non sono trattate con coltura tissutale, queste camere dovranno essere rivestite, ad esempio, con poli-D-lisina prima di seminare le cellule. Quando si placcano le celle nel vetro di copertura camerato, la densità cellulare ideale è fondamentale per evitare irregolarità del ciclo cellulare e stress aggiuntivo per le cellule. Un corretto equilibrio termico dei componenti del microscopio prima della sperimentazione per mantenere una temperatura stabile è fondamentale sia per mantenere la messa a fuoco durante l'imaging time lapse sia per garantire una DDR omogenea nel tempo e nei campioni.
È fondamentale che le cellule siano in condizioni sane prima della micro-irradiazione per ridurre i dati artefatti. Se le cellule hanno una morfologia irregolare post-infezione / selezione, consentire alle cellule di progredire attraverso più passaggi fino a quando la morfologia non ritorna alla normalità. Assicurarsi sempre che le linee di cellule utilizzate siano prive di contaminazione da micoplasma. Tra i molti effetti avversi dell'infezione da micoplasma, provoca anche danni al DNA delle cellule ospiti e potrebbe influenzare le loro vie DDR14,15. Il modo più sensibile per rilevare il micoplasma nella coltura cellulare è attraverso la PCR (versus. rilevamento con DAPI o Hoechst).
La sovraespressione ottimale della proteina di riparazione di interesse dovrebbe essere paragonabile ai livelli endogeni, tuttavia, abbastanza alta per il rilevamento. Il promotore utilizzato sui vettori virali, il titolo virale durante l'infezione e la durata del tempo di infezione possono essere regolati per i livelli di espressione ideali. Per risultati coerenti, isolare i singoli cloni cellulari per garantire livelli di espressione omogenei e una normale morfologia cellulare. Si raccomanda di utilizzare costrutti vettoriali che non sovraesprimano il PCNA marcato a livelli superiori a quelli endogeni per una corretta funzione di marcatore di riparazione del DNA e del ciclo cellulare. Anche bassi livelli di sovraespressione di PCNA sono sufficienti per discriminare le cellule in fase S. I vettori pBABE retrovirali sono stati utilizzati con successo per questo scopo (Addgene #1764, #1765, #1766, #1767). PcNA può essere etichettato con qualsiasi proteina fluorescente monomerica rossa(ad esempio, mPlum, mCherry, mRuby, ecc.) o monomerica verde fluorescente (ad esempio, mEGFP, AcGFP, mWasabi, mNeonGreen, mEmerald, ecc.) che potrebbe quindi essere combinata con un POI alternativamente taggato. La sovraespressione di un POI con tag fluorescenti presenta alcune limitazioni e considerazioni. I tag fluorescenti possono interrompere la normale funzione e localizzazione delle proteine. Pertanto, la posizione del tag (N o C-terminale) deve essere considerata. Utilizzare sempre proteine fluorescenti monomeriche, poiché l'oligomerizzazione di varianti non monomeriche può influenzare la funzione del POI.
Le impostazioni laser devono essere determinate per ciascun sistema di imaging poiché molti componenti del percorso ottico influenzeranno la potenza effettiva erogata nelle celle. La microirraggiamento laser può causare diversi tipi di lesioni del DNA a seconda della lunghezza d'onda di eccitazione, della potenza erogata dal laser FRAP e se sono stati utilizzati agenti pre-sensibilizzanti (come Bromodeoxyuridine o Hoechst). I laser a 405 nm possono causare danni ossidativi al DNA, rotture a singolo e doppio filamento16,17. Utilizzando impostazioni di uscita laser più elevate, la quantità di DSB aumenta. In questo protocollo non sono stati utilizzati metodi di pre-sensibilizzazione, ma queste tecniche sono ampiamente trattate in letteratura e ricapitole nella discussione seguente. A nostro parere, il modo migliore per verificare se la lesione desiderata viene generata è testare il reclutamento di geni specifici del percorso di danno al DNA noti. Il reclutamento di NTHL1 o OGG1, componenti della via BER, suggerisce l'induzione delle basi di DNA ossidato10,11,17,18,19,mentre FBXL10 o XRCC5 indicano la presenza di DSB8,20,21. Il reclutamento di XRCC1 può indicare sia la presenza di basi di DNA ossidate che di rotture a singolo filamento (SSB)22,23. XPC (cioè RAD4) è un buon indicatore di NER che rimuove gli addotti di DNA voluminosi generati dalla luce ultravioletta (UV)17,24. Poiché il reclutamento di proteine esogene può introdurre alcune irregolarità, la colorazione immunofluorescente di proteine o marcatori di riparazione del DNA endogeno (come γH2A.X per le rotture a doppio filamento) può confermare la presenza di specifiche lesioni del DNA. In alternativa, potrebbero essere utilizzati anche anticorpi sollevati contro specifici tipi di lesioni del DNA. Per regolare la potenza del laser erogata, è possibile modificare sia il tempo di permanenza che la potenza del laser.
Con l'aiuto della modellazione matematica, è stato possibile eseguire un'analisi cinetica dettagliata in grado di fornire preziose informazioni sulle proprietà di reclutamento del POI (ad esempio, contributo di più domini di legame del DNA, sensibilità verso diversi eventi di segnalazione, ecc.). La valutazione automatizzata del reclutamento e il monitoraggio delle cellule potrebbero essere combinati per creare flussi di lavoro robusti 1,25.
Vantaggi e limiti della pre-sensibilizzazione del DNA
La pre-sensibilizzazione del DNA prima della micro-irradiazione è uno strumento comunemente usato per il reclutamento delle proteine di riparazione del DNA16,17. La sensibilizzazione del DNA prima della micro-irradiazione lo rende più suscettibile ai DSB. I due metodi più comuni per la pre-sensibilizzazione del DNA sono il pre-trattamento delle cellule con bromodeossiuridina (BrdU) o colorante Hoechst. Per i sistemi non in grado di microirraggiare ad alte potenze laser, questi metodi possono essere necessari per indurre lesioni del DNA come i DSB. Inoltre, in assenza di un rivelatore di luce trasmessa o di un segnale fluorescente che evidenzi il nucleo cellulare (ad esempio, quando si studia il reclutamento di proteine di riparazione del DNA endogene senza tag), Hoechst agisce sia come strumento di presensibilizzante che come macchia nucleare fluorescente. Tuttavia, la pre-sensibilizzazione del DNA può introdurre complicazioni significative. BrdU (utilizzato ad una concentrazione finale di 10 μM) deve essere aggiunto alle cellule 24 ore (o tempo equivalente a un ciclo cellulare completo nella linea cellulare utilizzata) per incorporarsi correttamente nel DNA e può causare interferenze del ciclo cellulare26. Hoechst 33342 (utilizzato ad una concentrazione finale di 1 μg/mL) è citotossico dopo lunghi periodi di incubazione, ma richiede tempo sufficiente per saturare il nucleo con il colorante. Pertanto, dovrebbe essere applicato solo 15-20 minuti prima della microirraggiamento; in caso contrario, i dati di reclutamento non saranno coerenti. Le cellule macchiate in questo modo non possono essere tenute in coltura per più di qualche ora27,28. Assicurarsi di non utilizzare Hoechst 33358, che non è permeabile alle cellule come il colorante Hoechst 33342. La pre-sensibilizzazione può anche introdurre inutili variazioni tra gli esperimenti e rende l'esperimento ancora più sensibile alle differenze nella densità cellulare (poiché ciò influenzerà la quantità di colorante / cellula incorporato).
Vantaggi e limiti della microscopia confocale
La velocità di imaging della microscopia confocale può essere limitante rispetto alla microscopia a campo largo. Tuttavia, un microscopio confocale dotato di uno scanner risonante può migliorare enormemente la velocità di imaging (a scapito della risoluzione) avvicinandosi alle velocità della microscopia a disco rotante. Tre caratteristiche rendono il sistema confocale A1R HD25 una scelta eccellente per il protocollo qui presentato. In primo luogo, il FOV di 25 mm del sistema consente di eseguire l'immagine tra 15-20 celle in un singolo campo scansionato (rispetto alle 5-10 celle nelle configurazioni regolari), limitando il numero di acquisizioni necessarie per ottenere abbastanza celle per l'analisi statistica. In secondo luogo, il modulo FRAP e due teste di scansione consentono di image e micro-irradiare le cellule contemporaneamente, non solo in sequenza. Infine, la flessibilità di avere sia gli scanner risonanti che galvano offre la possibilità di passare facilmente tra l'imaging ad alta risoluzione temporale con una velocità eccezionale che riduce al minimo la tempra dei fluorofori e l'imaging ad alta risoluzione spaziale che utilizza velocità di scansione più lente per produrre immagini con un rapporto segnale/rumore più elevato. Mentre il sistema utilizzato consentiva la suddetta flessibilità, per assomigliare a configurazioni di microscopio confocale più ampiamente disponibili, solo lo scanner galvano è stato utilizzato negli esperimenti presentati (sia per la micro-irradiazione che per l'imaging successivo).
Vantaggi e limiti della microirraggiamento
Mentre la microirraggiamento fornisce una risoluzione spaziale e temporale senza rivali, non è senza limitazioni. Il danno al DNA da microirraggiamento laser è altamente raggruppato in parti specifiche del nucleo rispetto agli agenti dannosi presenti in natura. Pertanto, la risposta alla cromatina dovuta alla microirraggiamento può differire rispetto al danno distribuito in modo omogeneo. Inoltre, la microirraggiamento richiede molto tempo e può essere condotta solo su poche dozzine di cellule, mentre i metodi biochimici basati su grandi popolazioni (frazionamento della cromatina, immunoprecipitazione, ChIP) possono fornire una maggiore robustezza studiando migliaia di cellule alla volta. Verificare le osservazioni fatte dalla microirraggiamento con tecniche biochimiche tradizionali è una strategia efficace per conclusioni affidabili. Sebbene sia possibile la microirraggiamento simultaneo di molte cellule in un determinato FOV, il sistema di imaging avrà bisogno di più tempo per eseguire l'attività. Pertanto, misurare la dinamica delle proteine che reclutano molto rapidamente le lesioni del DNA limita il numero di possibili ROI per la microirraggiamento utilizzate contemporaneamente. Sul sistema di imaging utilizzato per questo protocollo, la microirraggiamento di un singolo ROI lungo 1024 pixel richiede 1032 ms utilizzando un tempo di permanenza di 1000 μs e 3088 ms utilizzando un tempo di permanenza di 3000 μs per essere completato. L'utilizzo di più linee di ROI aumenterà significativamente il tempo necessario per completare la microirraggiamento (ad esempio, il ROI lungo 7 x 1024 pixel richiede 14402 ms utilizzando il tempo di permanenza di 1000 μs e 21598 ms utilizzando il tempo di permanenza di 3000 μs). Questo tempo è perso dall'acquisizione di immagini e deve essere preso in considerazione. Quando si visualizzano eventi di reclutamento rapido, utilizzare il ROI più breve possibile e micro-irradiare solo una cella alla volta.
Vantaggi e limitazioni rispetto ai metodi di sincronizzazione
Per gli studi specifici del ciclo cellulare, i metodi esistenti prevedono la sincronizzazione delle cellule in specifiche fasi del ciclo cellulare o l'utilizzo di reporter fluorescenti per identificare la specifica fase del ciclo cellulare della cellula. Tuttavia, ognuno di questi metodi fornisce le proprie sfide e limitazioni.
Il sistema FUCCI3 (che si basa su proteine fluorescenti marcate forme troncate di CDT1 e Geminina) è uno strumento particolarmente utile per gli studi sul ciclo cellulare, ma ha dei limiti quando si tratta di differenziare tra le fasi S e G2 del ciclo cellulare. I livelli di Geminina sono già alti dalla fase S media e rimangono alti fino alla fase M, rendendo queste fasi difficili da separare. L'utilizzo del sistema FUCCI significa anche che due canali ottici del microscopio non possono essere utilizzati per l'imaging del POI.
Le linee cellulari non tumorali potrebbero essere sincronizzate in G0 mediante la rimozione dei fattori di crescita trovati nel siero (fame sierica) causando poco o nessun danno al DNA alle cellule. Tuttavia, la maggior parte delle linee cellulari tumorali continuerà parzialmente a progredire attraverso il ciclo cellulare anche senza adeguate quantità di siero nei loro mezzi. Inoltre, le cellule iniziano parzialmente a perdere la sincronizzazione entro la fine di G1, la fase S iniziale. Oltre alla fame di siero, ci sono numerosi metodi chimici per ottenere la sincronizzazione del ciclo cellulare. I blocchi di idrossiurea, afildicolina e timidina sono metodi per fermare la replicazione del DNA per sincronizzare le cellule nella fase S iniziale. Mentre questi metodi sono economici e semplici, introducono lo stress di replicazione che provoca danni al DNA. Questi inibitori della replicazione del DNA hanno dimostrato di indurre la fosforilazione di H2A. X, un noto marcatore di DSB2,29. Il metodo di utilizzo del PCNA marcato come marcatore per le cellule in fase S riduce il potenziale di artefatti causati dalla sincronizzazione chimica e può essere applicato a una vasta gamma di linee cellulari rispetto alla fame sierica.
Conclusione
Il danno al DNA è una forza trainante per le malattie genetiche in cui le lesioni mutagene possono portare alla trasformazione maligna delle cellule. Il targeting del meccanismo di sintesi del DNA è una strategia terapeutica fondamentale nel trattamento di malattie iperproliferative come il cancro. Per trattare queste malattie in modo più mirato, abbiamo bisogno di una migliore comprensione delle proteine che riparano le lesioni del DNA. Il protocollo qui descritto aiuta gli studi basati sulla microirraggiamento in fase S riducendo al minimo le sfide presentate dai metodi di sincronizzazione tradizionali per ridurre i possibili artefatti e aumentare la riproducibilità degli esperimenti.
Disclosures
Gli autori affermano che la pubblicazione del lavoro presentato è stata sponsorizzata da Nikon Corporation. Gli autori dichiarano che non esistono interessi concorrenti.
Acknowledgments
Gli autori ringraziano M. Pagano per il suo continuo supporto così come D. Simoneschi, A. Marzio e G. Tang per la loro revisione critica del manoscritto. B. Miwatani-Minter ringrazia R. Miwatani e B. Minter per il loro continuo supporto. G. Rona ringrazia K. Ronane Jurasz e G. Rona per il loro continuo supporto.
Materials
| Name | Company | Catalog Number | Comments |
| Ammonium chloride | Sigma-Aldrich | A9434-500G | For quenching formaldehyde |
| Anti-EXO1 Rabbit Polyclonal Antibody | Proteintech | 16253-1-AP | primary antibody |
| Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301 | Millipore | 05-636 | primary antibody |
| Bovine Serum Albumin | Sigma-Aldrich | 3117332001 | BSA for blocking |
| BrdU (5-Bromo-2'-deoxyuridine) | Merck | 19-160 | pre-sensitizing agent |
| Citifluor™ Mountant Solution AFR3 | Electron Microscopy Sciences | 17973-10 | antifade containing PBS solution for imaging |
| DAPI | Sigma-Aldrich | D9542-1MG | nucleic acid stain |
| DMEM Medium | Thermo Fisher Scientific | 10569010 | Cell culture medium for HEK293T cells |
| DMSO | Sigma-Aldrich | D2650-100ML | Vehichle control and dissolution solvent |
| EGFP-FBXL10 | Addgene | #126542 | viral expression vector for EGFP-FBXL10 |
| EXO1b-AcGFP (in pRetroQ) | custom cloning | na | EXO1b cDNA was cloned in the NheI, BamHI sites of pRetroQ-AcGFP1-N1 vector. |
| Fetal Bovine Serum | Gibco | 16140071 | Media supplement |
| FluoroBrite DMEM | Thermo Fisher Scientific | A1896701 | Phenol red free medium for microscopy |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | A32723 | secondary antibody |
| HEK293T cells | ATCC | ATCC CRL-3216 | Cell line for viral packaging |
| HEPES | Sigma-Aldrich | H0887-100ML | Buffering agent to supplement live cell imaging medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | pre-sensitizing agent |
| Lipofectamine 3000 | Thermo Fisher Scientific | L3000015 | Transfection reagent |
| McCoy’s 5A (Modified) Medium | Life Technologies | 16600-108 | Cell culture medium for U-2 OS cells |
| mCherry-PCNA | Addgene | #55117 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA | Addgene | #55994 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA (in pBABE) | custom cloning | na | mPlum-PCNA cDNA was cloned from Addgene #55994 in the BamHI, SalI sites of pBABE (puro) |
| Nikon A1R-HD25 Confocal Scanhead and Controller | Nikon | na | confocal imaging system |
| Nikon LUN4 laser unit | Nikon | na | excitation system |
| Nikon LUN-F 50 mW 405 nm FRAP laser unit | Nikon | na | FRAP laser unit |
| Nikon NIS Elements Confocal Controller Software | Nikon | na | Confocal controlling software |
| Nikon Ti2-E Inverted Microscope | Nikon | na | inverted epifluorescent microscope base |
| Nikon Ti2-LAPP Modular Illumination System | Nikon | na | illumination system |
| NTHL1-mCherry (in pRetroQ) | custom cloning | na | NTHL1 cDNA was cloned in the NheI, SalI sites of pRetroQ-mCherry-N1 vector. |
| Nunc Lab-Tek II Chambered Coverglass (4 well) | Thermo Fisher Scientific | 155382PK | Live cell microscopy cell culture chamber |
| Olaparib | Selleck Chemicals | S1060 | PARP inhibitor |
| Opti-MEM reduced serum media | Thermo Fisher Scientific | 31985062 | Dilution medium for transient transfection |
| Paraformaldehyde aqueous solution (32%) | Thermo Fisher Scientific | 50-980-494 | Fixative |
| pBABE (hygro) | Addgene | #1765 | retroviral expression vector (for low expression levels) |
| pBABE (neo) | Addgene | #1767 | retroviral expression vector (for low expression levels) |
| pBABE (puro) | Addgene | #1764 | retroviral expression vector (for low expression levels) |
| pBABE (zeo) | Addgene | #1766 | retroviral expression vector (for low expression levels) |
| PCNA Antibody (PC10) | Santa Cruz | sc-56 | primary antibody |
| Penicillin-Streptomycin-Glutamine (100x) | Gibco | 10378016 | Media supplement |
| polybrene | Sigma-Aldrich | TR-1003 | Increase viral infection efficiency |
| pRetroQ-AcGFP-C1 | Takara | 632506 | retroviral expression vector |
| pRetroQ-AcGFP-N1 | Takara | 632505 | retroviral expression vector |
| pRetroQ-mCherry-C1 | Takara | 632567 | retroviral expression vector |
| pRetroQ-mCherry-N1 | Takara | 632568 | retroviral expression vector |
| pUMVC | Addgene | #8449 | Viral packaging vector |
| Sodium-pyruvate | Thermo Fisher Scientific | 11360070 | Supplement for live cell imaging medium |
| Triton X-100 aqueous solution (10%) | Sigma-Aldrich | 11332481001 | Dilute in PBS for cell permeabilization buffer |
| Trypsin-EDTA Solution 10X | Sigma-Aldrich | 59418C-100ML | Dilute in PBS to split cells |
| U-2 OS Cells | ATCC | HTB-96 | Optimal cell line for microscopy experiments |
| Universal Mycoplasma Detection Kit | ATCC | 30-1012K | PCR based Mycoplasma detection kit |
| VSV-G | Addgene | #8454 | Viral protein envelope vector |
References
- Aleksandrov, R., et al. Protein dynamics in complex DNA lesions. Molecular Cell. 69 (6), 1046-1061 (2018).
- Darzynkiewicz, Z., Halicka, H. D., Zhao, H., Podhorecka, M. Cell synchronization by inhibitors of DNA replication induces replication stress and DNA damage response: Analysis by flow cytometry. Methods in Molecular Biology. 761, 85-96 (2011).
- Sakaue-Sawano, A., et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 132 (3), 487-498 (2008).
- Herce, H. D., Rajan, M., Lattig-Tunnemann, G., Fillies, M., Cardoso, M. C. A novel cell permeable DNA replication and repair marker. Nucleus. 5 (6), 590-600 (2014).
- Keijzers, G., et al. Human exonuclease 1 (EXO1) regulatory functions in dna replication with putative roles in cancer. International Journal of Molecular Sciences. 20 (1), (2018).
- Cheruiyot, A., et al. Poly(ADP-ribose)-binding promotes Exo1 damage recruitment and suppresses its nuclease activities. DNA Repair (Amsterdam). 35, 106-115 (2015).
- Zhang, F., Shi, J., Chen, S. H., Bian, C., Yu, X. The PIN domain of EXO1 recognizes poly(ADP-ribose) in DNA damage response. Nucleic Acids Research. 43 (22), 10782-10794 (2015).
- Rona, G., et al. PARP1-dependent recruitment of the FBXL10-RNF68-RNF2 ubiquitin ligase to sites of DNA damage controls H2A.Z loading. elife. 7, (2018).
- Young, L. M., et al. TIMELESS forms a complex with PARP1 distinct from its complex with TIPIN and plays a role in the dna damage response. Cell Reports. 13 (3), 451-459 (2015).
- Kong, X., et al. Laser microirradiation to study in vivo cellular responses to simple and complex dna damage. Journal of Visualized Experiments. (131), e56213 (2018).
- Kong, X., et al. Condensin I recruitment to base damage-enriched DNA lesions is modulated by PARP1. PLoS One. 6 (8), 23548 (2011).
- Lan, L., et al. Novel method for site-specific induction of oxidative DNA damage reveals differences in recruitment of repair proteins to heterochromatin and euchromatin. Nucleic Acids Research. 42 (4), 2330-2345 (2014).
- Zerjatke, T., et al. Quantitative cell cycle analysis based on an endogenous all-in-one reporter for cell tracking and classification. Cell Reports. 19 (9), 1953-1966 (2017).
- Ji, Y., Karbaschi, M., Cooke, M. S. Mycoplasma infection of cultured cells induces oxidative stress and attenuates cellular base excision repair activity. Mutation Research. 845, 403054 (2019).
- Sun, G., et al. Mycoplasma pneumoniae infection induces reactive oxygen species and DNA damage in A549 human lung carcinoma cells. Infection and Immunity. 76 (10), 4405-4413 (2008).
- Gassman, N. R., Wilson, S. H. Micro-irradiation tools to visualize base excision repair and single-strand break repair. DNA Repair (Amsterdam). 31, 52-63 (2015).
- Muster, B., Rapp, A., Cardoso, M. C. Systematic analysis of DNA damage induction and DNA repair pathway activation by continuous wave visible light laser micro-irradiation. AIMS Genetics. 4 (1), 47-68 (2017).
- Ikeda, S., et al. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. Journal of Biological Chemistry. 273 (34), 21585-21593 (1998).
- Rosenquist, T. A., Zharkov, D. O., Grollman, A. P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proceedings of the National Academy of Science U. S. A. 94 (14), 7429-7434 (1997).
- Reid, D. A., et al. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proceedings of the National Academy of Science U. S. A. 112 (20), 2575-2584 (2015).
- Taccioli, G. E., et al. Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science. 265 (5177), 1442-1445 (1994).
- Marsin, S., et al. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. Journal of Biological Chemistry. 278 (45), 44068-44074 (2003).
- Thompson, L. H., Brookman, K. W., Jones, N. J., Allen, S. A., Carrano, A. V. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Molecular and Cell Biology. 10 (12), 6160-6171 (1990).
- Scharer, O. D. Nucleotide excision repair in eukaryotes. Cold Spring Harbor Perspective Biology. 5 (10), 012609 (2013).
- Oeck, S., et al. High-throughput evaluation of protein migration and localization after laser micro-irradiation. Science Reports. 9 (1), 3148 (2019).
- Mistrik, M., et al. Cells and stripes: A novel quantitative photo-manipulation technique. Science Reports. 6, 19567 (2016).
- Durand, R. E., Olive, P. L. Cytotoxicity, mutagenicity and dna damage by hoechst 33342. Journal of Histochemistry and Cytochemistry. 30 (2), 111-116 (1982).
- Tobey, R. A., Oishi, N., Crissman, H. A. Cell cycle synchronization: reversible induction of G2 synchrony in cultured rodent and human diploid fibroblasts. Proceedings of the National Academy of Science U. S. A. 87 (13), 5104-5108 (1990).
- Podhorecka, M., Skladanowski, A., Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. Journal of Nucleic Acids. 2010, (2010).
