

Summary
Serial Block-Face Scanning Electron Microscopy (SBEM) wordt toegepast op beeld en analyseren van dendritische stekels in de muriene hippocampus.
Abstract
Driedimensionale elektronenmicroscopie (3D EM) geeft de mogelijkheid om morfologische parameters van dendritische stekels met nanoschaalresolutie te analyseren. Bovendien kunnen sommige kenmerken van dendritische stekels, zoals volume van de wervelkolom en post-synaptische dichtheid (PSD) (die post-synaptisch deel van de synaps vertegenwoordigt), aanwezigheid van presynaptische terminal, en glad endoplasmatisch reticulum of atypische vorm van PSD (bijv. multi-innervated stekels), alleen worden waargenomen met 3D EM. Door gebruik te maken van seriële block-face scanning elektronenmicroscopie (SBEM) is het mogelijk om 3D EM-gegevens gemakkelijker en reproduceerbaarder te verkrijgen dan bij het uitvoeren van traditionele seriële doorsneden. Hier laten we zien hoe we hippocampale monsters van muizen kunnen voorbereiden voor SBEM-analyse en hoe dit protocol kan worden gecombineerd met immunofluorescentiestudie van dendritische stekels. Milde fixatieperfusie stelt ons in staat om immunofluorescentiestudies uit te voeren met lichte microscopie op de ene helft van de hersenen, terwijl de andere helft was voorbereid op SBEM. Deze aanpak vermindert het aantal dieren dat voor het onderzoek moet worden gebruikt.
Introduction
De meeste excitatory synapsen in het centrale zenuwstelsel bevinden zich op dendritische stekels - kleine uitsteeksels van een neuronaal membraan. Deze uitsteeksels vormen beperkte biochemische compartimenten die de intracellulaire signaaltransductie regelen. Structurele plasticiteit van dendritische stekels en synapsen hangt nauw samen met de functionele veranderingen in synaptische werkzaamheid die ten grondslag liggen aan belangrijke processen zoals leren en geheugen1,2. Het is belangrijk op te merken dat elektronenmicroscopie (EM) de enige techniek is die het mogelijk maakt om te bepalen of een dendritische wervelkolom een presynaptische input heeft. EM-resolutie is ook nodig om ultrastructurele details te bestuderen, zoals de vorm van een postsynaptische dichtheid (PSD), die een postsynaptisch deel van een synaps vertegenwoordigt, of afmetingen van een dendritische wervelkolom, evenals de grootte en vorm van een axonale bouton. Bovendien is het met EM mogelijk om synapsen en hun omgeving te visualiseren.
Dankzij de vooruitgang in beeldvormings- en computertechnologieën is het mogelijk om hele neurale circuits te reconstrueren. Volume-elektronenmicroscopietechnieken, zoals seriële sectietransmissie-elektronenmicroscopie (ssTEM), seriële blok-gezicht scanning elektronenmicroscopie (SBEM) en gerichte ionenbundel scanning elektronenmicroscopie (FIB-SEM) worden vaak gebruikt voor neuronale circuitreconstructies3.
In onze studies wordt de SBEM-methode met succes gebruikt om de structurele plasticiteit van dendritische stekels en PSD's te onderzoeken in monsters van de muis hippocampus en organotypische hersenschijfjes 4,5. De SBEM is gebaseerd op de installatie van een miniatuur ultramicrotome in de scanning elektronenmicroscoopkamer6,7,8,9. De bovenkant van het monsterblok wordt afgebeeld en vervolgens wordt het monster op een opgegeven diepte door de ultramicrotome gesneden, waardoor een nieuw blokvlak wordt onthuld, dat opnieuw wordt afgebeeld en vervolgens wordt het proces herhaald8. Als gevolg hiervan blijft alleen het beeld van een blokvlak over, terwijl het gesneden plakje verloren gaat als puin. Daarom wordt SBEM een destructieve techniek genoemd, wat betekent dat het niet mogelijk is om dezelfde plaats opnieuw in beeld te brengen. Het voordeel van de destructieve on-block-methoden is echter dat ze geen last hebben van warpingproblemen en sectieverlies die de gegevenskwaliteit en de gegevensanalyse aanzienlijk kunnen beïnvloeden3. Bovendien biedt SBEM de mogelijkheid om een relatief groot gezichtsveld (> 0,5 mm × 0,5 mm) in hoge resolutie 3 in beeld tebrengen.
Om SBEM te gebruiken, moeten monsters worden voorbereid volgens een speciaal, zeer contrasterend protocol vanwege de backscattered elektronendetector die wordt gebruikt voor het verkrijgen van afbeeldingen. We laten hier zien hoe u monstervoorbereiding uitvoert volgens het protocol op basis van een procedure ontwikkeld door Deerinck10 (National Center for Microscopy and Imaging Research (NCMIR) methode), met behulp van verminderde osmium-thiocarbohydrazide-osmium (rOTO) vlekken ontwikkeld in de jaren 19808,11. Daarnaast introduceren we een fixatiebenadering in twee stappen, met milde fixatieperfusie die het mogelijk maakt om dezelfde hersenen te gebruiken, zowel voor immunofluorescentiestudies met lichte microscopie als SBEM.
In het protocol wordt een muisbrein voornamelijk gefixeerd met een mild fixatief en vervolgens in tweeën gesneden, en de ene hemisfeer wordt gepostfixeerd en voorbereid op immunofluorescentie (IF), terwijl de andere voor EM-studies (Figuur 1).

Figuur 1. Schematische weergave van de workflow voor de dendritische stekels voorbereiding voor de analyse met SBEM. Muizen werden geofferd en doordrenkt met een mild primair fixatief. De hersenen werden in tweeën gesneden en de ene hemisfeer werd gepostfixeerd met immunofluorescentie (IF)-specifiek fixatief, cryoprotected, gesneden met behulp van een cryostaat en verwerkt voor IF-studies, terwijl de andere hemisfeer werd gepostfixeerd met EM-fixatief, gesneden met het vibratoom en voorbereid op EM-studies. Hersenschijfjes voor SBEM-studies waren contrast, plat ingebed in hars, waarna een CA1-gebied van de hippocampus aan de pin werd gemonteerd en afgebeeld met SBEM (figuur 1). Het deel van het protocol dat in een geel vak is gemarkeerd, is in de video opgenomen. Klik hier om een grotere versie van deze afbeelding te bekijken.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
Het onderzoek is uitgevoerd in overeenstemming met de richtlijnen van het Nencki Instituut en toestemming van de Local Ethical Committee. De studies zijn uitgevoerd overeenkomstig de richtlijn van de Raad van de Europese Gemeenschappen van 24 november 1986 (86/609/EEG), animal protection act van Polen en goedgekeurd door de eerste plaatselijke ethische commissie in Warschau. Alle inspanningen werden gedaan om het aantal gebruikte dieren en hun lijden tot een minimum te beperken.
LET OP: Alle hieronder beschreven procedures moeten worden uitgevoerd in een laboratorium afzuigkap. Vanwege de gevaarlijke aard van de gebruikte reagentia. Persoonlijke veiligheidsmaatregelen zoals handschoenen, labjas, veiligheidsbril en een mondkapje zijn vereist.
1. Bereiding van het fixatief voor perfusie (2% wt/vol paraformaldehyde (PFA) en 0,5% vol/vol glutaraldehyde (GA) in 0,1 M fosfaatbuffer (PB), pH 7,4)
OPMERKING: Bereid de fixatieve oplossing voor op dezelfde dag dat deze wordt gebruikt en bewaar deze niet langer dan 3 uur. In geval van tijdstekort, bereid 2% PFA in 0,1 M PB de dag ervoor, bewaar het bij 4 °C en voeg kort voor de perfusie verse GA toe.
- Neem 400 ml steriel dubbel gedestilleerd water (ddH2O) en verwarm het tot 60 °C met behulp van een roerende kookplaat. Voeg vervolgens 20 g PFA toe. Voeg druppels van 1 M NaOH toe totdat PFA volledig is opgelost en laat het mengsel afkoelen.
- Voeg 500 ml 0,2 M PB (pH 7,4) toe.
- Filter de oplossing om eventuele afzetting te verwijderen en af te koelen tot 4 °C.
- Voeg vlak voor de perfusie 20 ml 25% GA toe aan de oplossing en vul het volume vervolgens aan met ddH2O tot 1 L.
2. Bereiding van postperfusiefixatief voor SBEM (2% wt/vol PFA en 2,5% vol/vol GA in 0,1 M PB, pH 7,4)
- Neem 50 ml fixatief voor perfusie (2% PFA en 0,5% GA in 0,1 M PB).
- Voeg 5 ml 25% GA toe.
3. Bereiding van postperfusiefixatief voor IF-kleuring (4% PFA in fosfaatgebufferde zoutoplossing (PBS))
- Los een tablet van 1x PBS (pH 7.4) op in een gezuiverd en gedeioneerd water (H2O) volgens de instructies van de fabrikant.
- Gebruik een roerhete plaat om de oplossing tot 60 °C te verwarmen en voeg 40 g PFA toe.
- Voeg druppels van 1 M NaOH toe totdat de PFA volledig is opgelost en laat het mengsel afkoelen.
- Stel de pH van de oplossing in op 7,5 met 1 M HCl en vul het volume vervolgens aan met H2O tot 1 L.
- Filter de oplossing om eventuele afzettingen te verwijderen.
4. Transcardiale perfusie van dieren
OPMERKING: Alle PFA- en GA-afvalstoffen moeten worden ingezameld en opgeslagen voor verwijdering volgens de lokale voorschriften. Anesthesie en perfusie moeten de lokale voorschriften volgen. In het beschreven protocol werden volwassen 3 maanden oude en 20±1 maanden oude vrouwelijke Thy1-GFP(M) muizen (Thy1-GFP +/-)12 die groene fluorescentie-eiwit (GFP) uitdrukken in een schaars verdeelde populatie glutamatergische neuronen gebruikt, maar elke andere kan ook worden gebruikt. Dieren werden gefokt als heterozygoten met de C57BL/6J-achtergrond in het Dierenhuis van het Nencki Institute of Experimental Biology.
- Vóór perfusie een muis verdoven door toediening van een ketamine/xylazinemengsel (tot 90 mg/kg lichaamsgewicht ketamine en 10 mg/kg lichaamsgewicht xylazine) via intraperitoneale injectie (27-gauge naald).
- Beoordeel of de diepte van anesthesie voldoende is door de reflex op pijnprikkels (knijpen) en de hoornvliesreflex (knijpen) te controleren.
- Voer na 20 minuten een intraperitoneale injectie (27-gauge naald) van natriumpentobarbital (50 mg/kg lichaamsgewicht) uit.
- Perfuseer een muis volgens een perfusiechirurgieprotocol beschreven door Gage et al.13 (zie punt 4; Figuur 5-6). Gebruik van een perfusiepomp begin met een fosfaatbuffer van 0,1 M, pH 7,4 (30 ml) gedurende 3 minuten en ga verder met 2% PFA en 0,5% GA in 0,1 M PB, pH 7,4 gedurende 6 minuten (80 ml).
- Ontleed voorzichtig de hersenen van de schedel en deel deze doormidden (zie figuur 9-10 in Gage et al., 201213). Plaats een stuk in een flacon met fixatief voor SBEM en het tweede in de flacon met 4% PFA/PBS voor IF-kleuring.
- Houd de hemisferen 's nachts bij 4 °C in het fixatief.
5. Hersenschijfjes voorbereiding voor elektronenmicroscopie
- Kies de vibratoominstellingen (veersnelheid blad: 0,075 mm/s, snijfrequentie: 80 Hz).
- Plaats de snijkamer in de houder, bevestig deze aan het vibratoom en omring deze met ijs. Plaats vervolgens een scheermesje in de vibratome meshouder.
- Gebruik een lepel of een soortgelijk voorwerp en plaats de gekoelde hersenen (dorsaal oppervlak omhoog) op een taai snijoppervlak (bijv. een glazen petrischaaldeksel). Om een coronale plak van de hippocampus te bereiden, maak je een loodrechte snede tussen de hersenhelft en het cerebellum met een scheermesje of scalpel, waardoor het cerebellum wordt verwijderd. De olfactorische lamp kan ook worden verwijderd.
- Breng cyanoacrylaatlijm aan op het droge platform van het vibratoom.
- Pak de hersenen op met een tang en droog deze voorzichtig op filterpapier.
- Lijm de hemisfeer op het platform dicht bij het snijblad met de rostrale punt naar boven. Bevestig het platform aan de houder en vul het onmiddellijk met ijskoud 0,1 M PB, pH 7,4. Als de loodrechte snede goed is gemaakt, zal de hemisfeer recht omhoog staan en een hoek van 90° bieden die nodig is om een symmetrische coronale snede te maken die de hippocampus bevat. Zorg ervoor dat de hersenen bedekt zijn met PB.
- Plaats het vibratoomblad voor de hemisfeer en laat het zakken naar de coronale kant van de hemisfeer. Laat het mes zakken tot 400 μm verder in de staartrichting en begin met snijden. Blijf snijden totdat de eerste twee plakjes volledig gescheiden zijn van het weefselblok.
- Trek het mes terug en laat nog eens 100 μm zakken en snijd vervolgens opnieuw.
- Wanneer de hippocampus zichtbaar wordt (gebruik de muishersenatlas Paxinos en Franklin, 200414) verzamel je de plakjes met de kleine penseel of verbrede plastic Pasteur pipet.
- Breng de plakjes over in een 12-putplaat gevuld met koude 0,1 M PB, pH 7,4.
- Ontleed de hippocampus in een glazen petrischaal gevuld met 0,1 M PB, pH 7,4 met behulp van een scheermesje of scalpel, en doe het in glazen flacons met dezelfde fosfaatbuffer.
OPMERKING: Voor langdurige opslag van de plakjes vult u 0,1 M PB aan met 0,05% natriumaziide (NaN3).
6. Voorbereiding van hersenmonsters voor immunostaining
- Na een nachtelijke fixatie in 4% PFA/PBS-oplossing in een zuurkast, plaatst u het hersenweefsel in de cryopreservatieoplossing (30% sacharose in PBS met 0,05% NaN3)en houdt u het gedurende 2 dagen op 4 °C (tot het is gezonken).
- Bereid een antivriesoplossing (15% sacharose/30% ethyleenglycol/0,05% NaN3/PBS).
- Stel de kasttemperatuur van de cryostaat in op -19 °C en zorg ervoor dat de temperatuur wordt bereikt voordat u verder gaat. Zorg er tijdens het doorsnijden voor dat de kasttemperatuur tussen -18 °C en -20 °C blijft.
- Gebruik een lepel of een soortgelijk voorwerp en plaats de gekoelde hersenen (dorsaal oppervlak omhoog) op een taai snijoppervlak (bijv. een glazen petrischaaldeksel). Om een coronale plak van de hippocampus te bereiden, maak je een loodrechte snede tussen de hersenhelft en het cerebellum met een scheermesje of scalpel, waardoor het cerebellum wordt verwijderd.
- Selecteer een voorgekoelde monsterschijf, bedek deze met een medium om in te vriezen op een losplank en bevestig met een tang de hemisfeer aan de schijf met een rostrale punt naar boven en wacht tot het monster volledig bevroren is. Plaats de monsterschijf in een monsterkop.
- Plaats een mes in een meshouder in de cryokamer en snijd 40 μm dikke plakjes.
- Breng plakjes met een kleine kwast over op de 96-well plaat gevuld met koude antivriesoplossing en rol de secties voorzichtig uit (verzamel een plakje na elke ronde snijden om te voorkomen dat ze verloren gaan in de plakkamer).
OPMERKING: Hersenen of secties kunnen lange tijd in een antivriesoplossing bij -20 °C worden bewaard.
7. Immunostaining van hersenschijfjes
OPMERKING: Alle kleuringsstappen werden uitgevoerd in een 24-put plaat op een platform shaker.
- Was de plakjes drie keer met PBS, telkens gedurende 6 minuten.
- Incubeer plakjes in 300 μL blokkerende oplossing (5% normaal ezelsserum (NDS)/0,3% Triton X-100) gedurende 1 uur met zachtjes schudden op een rotator.
- Incubeer de plakjes met het primaire antilichaam tegen PSD-95 verdund 1:500 in 5% NDS/0,3% Triton X-100/PBS (300 μL per put) 's nachts bij 4 °C. De uiteindelijke concentratie van het primaire antilichaam is 2 μg/ml.
- Was de plakjes met 0,3% Triton X-100/PBS driemaal op kamertemperatuur (RT), telkens gedurende 6 minuten.
- Incubeer plakjes met het secundaire antilichaam verdund 1:500 in 300 μL van 0,3% Triton X-100/PBS gedurende 90 minuten. De uiteindelijke concentratie van het secundaire antilichaam is 4 μg /ml.
- Was de plakjes drie keer met PBS, telkens gedurende 6 minuten.
- Monteer de plakjes op dia's met behulp van een bevestigingsmedium en sluit ze vervolgens met een afdekschuif.
- Onderzoek de monsters met behulp van een confocale microscoop (63 × oliedoelstelling, NA 1.4, pixelgrootte 0,13 μm × 0,13 μm).
OPMERKING: Voor langdurige opslag: bewaar de monsters op 4 °C, bescherm ze tegen licht.
8. SBEM monstervoorbereiding
LET OP: Vanwege de gevaarlijke aard van de gebruikte reagentia moeten alle hieronder beschreven procedures in een afzuigkap van het laboratorium worden uitgevoerd. Lees voor het gebruik van deze chemicaliën zorgvuldig de materiaalveiligheidsinformatiebladen van de fabrikanten en vraag de veiligheidsfunctionaris naar de lokale regels om een veilige behandeling en afvalverwijdering te garanderen.
- Voorbeeldcontrast
OPMERKING: Snijdt wassen en incubatie op kamertemperatuur moet worden uitgevoerd met licht schudden (bijv. op een platform shaker). Autoclaaf ontgast water werd gebruikt.- Was de plakjes met koude 0,1 M PB, pH 7,4 vijf keer, telkens gedurende 3 minuten.
- Bereid een 1:1 mengsel van 4% waterig osmium tetroxide en 3% kalium ferrocyanide (1:1 bij vol). Het eindproduct wordt bruin. Dompel de monsters onder in dit mengsel, leg ze op ijs en vanaf dit stadium is het belangrijk om ze te beschermen tegen licht. Incubeer ze met zachtjes schudden gedurende 1 uur.
- Bereid ondertussen een thiocarbohydrazide (TCH) oplossing voor. Meng 10 ml dubbel gedestilleerde H2O (ddH2O) en 0,1 g TCH en zet deze gedurende 1 uur in een oven op 60 °C. Het is belangrijk om de oplossing van tijd tot tijd te wervelen (bijv. elke 10 minuten). Als het klaar is, koel het dan af tot kamertemperatuur.
- Was de plakjes vijf keer met ddH2O, telkens gedurende 3 minuten.
- Filter de TCH-oplossing met een spuitfilter van 0,22 μm en dompel het monster onder in een gefilterde oplossing. De plakjes worden zwart. Incubeer ze gedurende 20 minuten op kamertemperatuur.
- Was de monsters vijf keer met ddH2O, telkens gedurende 3 minuten.
- Incubeer de monsters met een waterige oplossing van 2% OsO4 gedurende 30 minuten bij kamertemperatuur.
- Was de monsters vijf keer met ddH2O, telkens gedurende 3 minuten.
- Plaats de monsters in gefilterd 1% waterig uranylacetaat en incubeer ze 's nachts bij 4 °C. Gebruik een spuitfilter van 0,22 μm voor filtratie.
- Bereid L-asparaginezuuroplossing door 0,4 g L-asparaginezuur en 80 ml ddH2O te mengen, stel de pH in op 3,8 om gemakkelijker op te lossen en vul vervolgens aan met water tot 100 ml.
- Begin de volgende dag met Walton's leidende aspartate15 voorbereiding. Meng 0,066 g loodnitraat met 10 ml L-asparaginezuuroplossing (punt 8.1.10) voorverwarmd tot 60 °C en stel de pH in op 5,5 (gemeten bij 60 °C) met 1 M NaOH. Sluit de flacon met lood aspartaat en laat deze 30 minuten op 60 °C staan in een waterbad. De oplossing moet duidelijk zijn. Als het bewolkt wordt, moet het worden weggegooid en moet er een nieuwe worden voorbereid.
- Was ondertussen de monsters vijf keer met ontgaste ddH2O, telkens gedurende 3 minuten. Bewaar ze vervolgens 30 minuten in de oven op 60 °C.
- Dompel de monsters onder in de vers bereide lood aspartaatoplossing en incubeer ze gedurende 20 minuten in de oven op 60 °C.
- Was de monsters vijf keer met ontgaste ddH2O, telkens gedurende 3 minuten.
- Uitdroging en harsinbedding
- Bereid epoxyhars voor. Weeg de ingrediënten (33,3 g component A/M, 33,3 g component B en 1 g component D) en meng de hars goed (schud deze bijvoorbeeld op een roterende shaker in een buis van 15 ml) gedurende ten minste 30 minuten voordat u 16 druppels versneller DMP 30 toevoegt. Roer nog 10 minuten.
OPMERKING: Weeg de ingrediënten af onder de zuurkast. Deze hoeveelheid componenten geeft ongeveer 60 ml hars wat genoeg is voor 15 flacons met monsters. U kunt de rest van de hars minder bereiden of opslaan in een spuit bij 4 °C en de volgende dag gebruiken. Vergeet niet om de punt van de spuit af te dichten. - Bereid flacons met gesorteerde verdunningen van ethanol (30%, 50%, 70%, 90%, 100%, 100% ethanol in water en 100% gedroogd op een moleculaire zeef).
- Meng de hars met 100% ethanol in 1:1 verhouding om 50% hars te verkrijgen. Meng het goed door elkaar.
- Dehydrateer de monsters gedurende 5 minuten in elke verdunning ethanol vanaf 30% en eindigend met 100% watervrije ethanol gedroogd op moleculaire zeef. Vergeet niet dat de monsters nooit volledig mogen drogen.
- Infiltreer de monsters eerst in 50% hars gedurende 30 minuten, vervolgens in 100% hars gedurende een uur en vervolgens 's nachts opnieuw in 100% hars. Voer alle infiltratiestappen uit met constant langzaam schudden.
- Plaats de volgende dag de monsters een uur in verse 100% hars en sluit ze vervolgens in tussen fluoropolymeer insluitbladen. Ontvetten stukken inbeddingsplaat met ethanol en de monsters plat insluiten tussen twee lagen ervan, met behulp van twee glazen dia's als ondersteuning. Probeer luchtbellen in hars op of in de buurt van het monster te vermijden.
- Hard de monsters minstens 48 uur uit in de oven bij 70 °C.
- Bereid epoxyhars voor. Weeg de ingrediënten (33,3 g component A/M, 33,3 g component B en 1 g component D) en meng de hars goed (schud deze bijvoorbeeld op een roterende shaker in een buis van 15 ml) gedurende ten minste 30 minuten voordat u 16 druppels versneller DMP 30 toevoegt. Roer nog 10 minuten.
- Trimmen en monteren
- Scheid inbeddingsbladen en snijd een stuk van het ingebedde monster (ongeveer 1 mm x 1 mm) met een scheermesje. Breng het over naar een parafilm. Dit minimaliseert het risico van verlies van het monster als gevolg van elektrostatica.
- Neem een aluminium pin die is ontvet met ethanol. Meng een geleidende epoxy goed en gebruik er een beetje van om het monster aan de pin te bevestigen. Hard geleidende epoxy uit bij 70 °C gedurende 10 minuten.
- Knip elke kant van het monsterblok af met het diamantmes en polijst vervolgens het gezicht van het blok totdat het weefsel wordt blootgesteld.
OPMERKING: Bevestig bij deze stap de aanwezigheid van een interessegebied door enkele secties te verzamelen en toluidineblauwe kleuring16 uit te voeren of onder de elektronenmicroscoop te controleren. - Verklein het monster zoveel mogelijk. Aard het monster vervolgens met geleidende verf tot de pin en hard het uit (gedurende 24 uur bij kamertemperatuur of in de oven bij 65 °C gedurende 40 minuten).
- Om het opladen van artefacten te minimaliseren, sputtert sputter de monsters met een dunne laag goud of goud / palladium.
9. SBEM beeldvorming
- Plaats de pin met een monster in de kamer van de seriële blok-gezicht scanning elektronenmicroscoop. Lijn het monster uit op het mes. Sluit de kamer en stel de parameters in.
- Verzamel een stapel afbeeldingen met de gewenste vergroting, pixelgrootte, plakdikte, versnelde spanning (EHT), diafragma, druk, enz.
OPMERKING: De parameters zijn afhankelijk van het monster en het doel van het experiment. Voorbeeldige initiële beeldvormingsinstellingen zijn opgenomen in de tabel met materialen.
10.3D reconstructies
OPMERKING: Voor de onderstaande stappen gebruiken we open-access software zoals FijiJ17 (ImageJ versie 1.49b), Microscopy Image Browser (MIB)18 en Reconstruct19, maar er kunnen verschillende andere software worden gebruikt.
- Converteer digitale micrograafbestanden (dm) naar TIFF-formaat. Importeer eerst de afbeeldingsvolgorde (in FijiJ: > Importeer > afbeeldingsvolgorde > Kies 8-bits indeling)en sla vervolgens op als TIFF (in FijiJ: Bestand > Opslaan als > afbeeldingsvolgorde > Kies Tiff).
- Pas de helderheid en het contrast van de stapel afbeeldingen aan (in FijiJ: Image > Adjust > Brightness/Contrast) en, indien nodig, denoise it (gebruik DenoisEM plugin voor FijJ20 : Plugins > DenoiseEM > Denoise).
- De stapel uitlijnen (in FijiJ: Plug-ins > StagReg of MIB: Dataset > Alignment Tools).
- Segment dendritische stekels en synapsen (In MIB- of Reconstruct-software zijn uitgebreide tutorials beschikbaar (zie tabel met materialen voor meer informatie).
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Met behulp van de hierboven beschreven methode met hoog contrast kunnen afbeeldingen met een goede resolutie van het hersenweefsel van de muis worden verkregen. Een groot gezichtsveld van de SBEM-techniek vergemakkelijkt een nauwkeurige selectie van de interesseregio. Het grote beeld van het CA1-gebied van de hippocampus werd genomen om de lengte van stratum radiatum (SR) te meten (figuur 2A) en om de beeldvorming precies in het midden in te stellen (figuur 2B). Vervolgens werd de stapel afbeeldingen verworven en werden de interessante objecten, zoals dendrieten, dendritische stekels, postsynaptische dichtheden van de synapsen gesegmenteerd (Figuur 2C,D).
Het gebruik van de SBEM-techniek geeft de mogelijkheid om een relatief groot volume weefsel te analyseren en zeldzame gebeurtenissen te herkennen, zoals een multi-innervated dendritische wervelkolom (Figuur 2E) - een bepaald type synapsen dat wordt gekenmerkt door verschillende presynaptische terminals die in contact komen met dezelfde postsynaptischewervelkolom 21.

Figuur 2. Dendritische stekels in muis hippocampus CA1 regio. Representatieve afbeelding van muis hippocampal CA1 regio. Stratum radiatum (SR) wordt weergegeven met een witte pijl (A). Het gebied in het midden van SR werd gekozen voor beeldvorming (B). Reconstructie van een stuk dendriet met dendritische stekels (C). Reconstructie van dendritische stekels in een volume weefsel (D). Reconstructie van een multi-innervated dendritische wervelkolom (E). Twee presynaptische terminals die contact maken met dezelfde postsynaptische wervelkolom worden weergegeven (in magenta en groen). Klik hier om een grotere versie van deze afbeelding te bekijken.
Het gepresenteerde protocol voor SBEM-monstervoorbereiding stelde ons in staat om hoogwaardige beelden van het hersenweefsel met sterk contrasterende membranen te verkrijgen, wat een gemakkelijkere segmentatie en reconstructie van membraanomringsstructuren mogelijk maakt. De standaardfixatie met de hogere concentratie PFA (figuur 3B) en de tweestapsfixatiebenadering resulteerden in vergelijkbare weefselmorfologie (figuur 3A).

Figuur 3. Morfologische details. Met het gebruik van het hier gepresenteerde protocol werden afbeeldingen van goede kwaliteit verkregen die de morfologie van synaptische blaasjes, PSD en mitochondriën lieten zien. Hersenweefsel gefixeerd met tweestapsbenadering (A) of met een standaard fixatief (B). Schaalbalk 2 μm. Klik hier om een grotere versie van deze afbeelding te bekijken.
De resultaten zijn reproduceerbaar, wat betekent dat elk specimen dat met behulp van dit protocol werd bereid, niet alleen hersenweefsel, maar ook de monolaag van gekweekte cellen, goed contrasteerde.
Het gebruik van milde primaire fixatie biedt de mogelijkheid om het weefsel ook te gebruiken voor immunofluorescentiestudies met een lichtmicroscoop. In het geval van het PSD-95-antilichaam hebben we het verschil in immunostainingresultaten niet waargenomen tussen de monsters die zijn gefixeerd met tweestapsfixatie (mild primair en vervolgens secundair met een traditioneel fixatief) en de monsters die zijn verkregen met traditionele fixatie met alleen 4% PFA (respectievelijkfiguur 4A en 4B).

Figuur 4. Vergelijking van PSD-95 immunofluorescentie in monsters gefixeerd met verschillende protocollen. Representatieve microfoto's van PSD-95 immunostaining in de stratum radiatum laag van CA1 gebied in de dorsale hippocampus. (A) Confocale afbeelding na milde primaire fixatie gevolgd door postfixatie met een standaard fixatief. (B) Confocale afbeelding na standaard, sterkere fixatie. Schaalbalken: 5 μm. Klik hier om een grotere versie van deze afbeelding te bekijken.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Er zijn veel variaties van de primaire NCMIR-methode beschreven door Deerinck in 201010. De basisprincipes blijven hetzelfde, maar afhankelijk van het soort bestudeerd materiaal worden kleine veranderingen doorgevoerd. Eerder werd beschreven dat verschillende harsen kunnen worden gebruikt om monsters voor SBEM in te bedden en bijvoorbeeld in het geval van planten is Spurr's de hars bij uitstek vanwege de lage viscositeit die een betere infiltratie door de celwanden mogelijk maakt22,23. Bovendien kunnen verschillende buffers worden toegepast voor fixatie (bijv. cacodylaatbuffer, HEPES of fosfaatbuffer, evenals een andere concentratie glutaraldehyde (2-3%) en paraformaldehyde (2-4%)23,24,25,26,27,28). Het gebruik van looizuur om de kleuring van monsters die rijk zijn aan extracellulaire matrix29 of rutheniumrood te verbeteren om de kleuring van plantaardig materiaal te verbeteren23,30 zijn andere wijzigingen die kunnen worden gebruikt. Wat uitdroging betreft, wordt gewoonlijk ethanol en/of aceton aanbevolen voor epoxyhars24,28.
Hetzelfde protocol zoals hier beschreven, of zeer vergelijkbaar, is eerder met succes gebruikt door ons team en andere groepen4,5,24. Monsters die volgens deze methode worden verwerkt, zijn compatibel met TEM imaging31. In vergelijking met het oorspronkelijke NCMIR-protocol gepubliceerd in 201010 introduceren we enkele kleine wijzigingen, zoals vervanging van toxische cacodylaatbuffer tijdens fixatie en osmium postfixatiestap door een fosfaatbuffer. Zoals hierboven vermeld, kunnen diverse buffers worden gebruikt tijdens em-voorbereiding, en volgens onze ervaring geeft niet-toxische PB bevredigende resultaten.
Een andere verandering is een vervanging van aceton door minder door de gebruiker schadelijke ethanol tijdens de laatste stap van uitdroging. Ethanol werd ook gebruikt als harsoplosmiddel tijdens het inbeddingsproces. Bovendien werd DMP-30 gebruikt in plaats van epoxyharscomponent C, zoals gepubliceerd vóór32,33.
Vanwege de noodzaak om dezelfde hersenen te gebruiken voor immunofluorescentiestudies werd de tweestapsfixatiebenadering geïntroduceerd. Ten eerste werden de dieren doordrenkt met een buffer met een lagere concentratie glutaraldehyde (milde primaire fixatie door perfusie met een buffer die 2% PFA en 0,5% GA bevat) en daarna werd slechts de helft van de hersenen gewijd aan EM verder gepostfixeerd met een hogere concentratie glutaraldehyde (2% PFA en 2,5% GA), terwijl de andere helft van de hersenen werd gepostfixeerd met een standaard fixatief voor immunostaining (4%). Het gebruik van EM-fixatief met een lagere concentratie PFA werd eerder gepubliceerd door anderen en onze groep4,5,31,34. De hier gepresenteerde tweestapsfixatie is even voldoende als een traditionele eenstapsbenadering (figuur 3 en figuur 4). Omdat we echter voorstellen dat elke helft van de hersenen afzonderlijk kan worden verwerkt, maakt ons protocol het mogelijk om het aantal gebruikte dieren te verminderen. Milde primaire fixatie handhaaft met succes weefselantigeniciteit, waardoor verdere immunofluorescentie mogelijk is, zoals aangetoond voor het antilichaam tegen PSD-95-eiwit. Er moet echter worden benadrukt dat het nut van onze aanpak moet worden gevalideerd voor andere antilichamen.
De gepresenteerde methode resulteert in contrastrijke beelden van het hersenweefsel met duidelijk zichtbare membranen. Het is echter vermeldenswaard dat er dezelfde stappen zijn die vatbaar zijn voor mislukking, zoals in het geval van elke andere methode voor de bereiding van SBEM-monsters, en er moet bijzondere aandacht aan worden besteed. De kwaliteit van de weefselmorfologie zelf hangt vooral af van de perfusie. Het is een cruciale stap, omdat alleen met succes vaste exemplaren bevredigende resultaten geven. Helaas wordt fixatieperfusie beïnvloed door individuele variabiliteit tussen dieren, dus de kwaliteit van beelden kan van dier tot dier verschillen. Bovendien moet men oppassen dat het monster niet droogt tijdens uitdroging; anders wordt het monster niet goed geïnfiltreerd met de hars. Een ander belangrijk aspect is de monstermontage aan de pin. De epoxyharsen zijn niet-geleidend en daarom accumuleren ze elektronen tijdens beeldvorming, waardoor artefacten worden opgeladen. Om het laadeffect bij SBEM te verminderen, is het noodzakelijk om het teveel aan hars aan elke kant van het monster zorgvuldig te verwijderen en het monster nauwkeurig te aarden met zilververf en sputtercoating.
Het huidige protocol richt zich op hersenweefsel; het kan echter worden aangepast voor celmonolayer, organotypische plakjes of andere weefsels. Net als het originele NCMIR-protocol resulteert het hier gepresenteerde protocol in zeer contrasterende monsters die niet alleen geschikt zijn voor SBEM, maar ook voor FIB-SEM- en ATUM-SEM-experimenten.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
De auteurs hebben niets bekend te maken.
Acknowledgments
SBEM-beeldvorming, lichte microscopiebeeldvorming en elektronenmicroscopiemonstervoorbereiding werden uitgevoerd met behulp van de apparatuur van laboratory of Imaging Tissue and Function, die dient als een imaging-kernfaciliteit van het Nencki Institute of Experimental Biology.
Voor de voorbereiding van figuur 1 werd de afbeelding van een muis (Souris_02) en een flacon van de https://smart.servier.com/ gebruikt.
Dit werk werd ondersteund door het National Science Centre (Polen) Grant Opus (UMO-2018/31/B/NZ4/01603) toegekend aan KR.
Materials
| Name | Company | Catalog Number | Comments |
| Anesthetic: | |||
| Ketamine/xylazine mixture (Ketamina/Sedazin) | Biowet Pulawy, Pulawy, Poland | ||
| Sodium pentobarbital (Morbital) | Biowet Pulawy, Pulawy, Poland | ||
| Fixatives: | |||
| Glutaraldehyde (GA) | Sigma-Aldrich,St. Louis, MI, USA | G5882 | Grade I, 25% in H2O, specially purified for use as an electron microscopy fixative |
| Hydrochloric acid (HCl) | POCH, Gliwice, Poland | 575283115 | pure p.a. |
| Paraformaldehyde (PFA) | Sigma-Aldrich,St. Louis, MI, USA | 441244 | prilled, 95% |
| Phosphate buffered saline (PBS), pH 7.4 | Sigma-Aldrich,St. Louis, MI, USA | P4417-50TAB | tablets |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade, 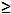 98%, pellets (anhydrous) 98%, pellets (anhydrous) |
| Sodium phosphate dibasic (Na2HPO4) | Sigma-Aldrich,St. Louis, MI, USA | S3264 | |
| Sodium phosphate monobasic (NaH2PO4) | Sigma-Aldrich,St. Louis, MI, USA | S3139 | |
| Perfusion: | |||
| Large blunt/blunt curved scissors (~14.5 cm) | Fine Science Tools, Foster City, CA, USA | 14519-14 | |
| Micro-spatula (double 2" flat ends, one rounded, one tapered to 1/8") | Fine Science Tools, Foster City, CA, USA | 10091-12 | |
| Needle tip, 15 GA, blunt (perfusion needle) | KD Medical GmbH Hospital Products, Berlin, Germany | KD-FINE 900413 | 1.80 x 40 mm |
| Pair of fine (Graefe) tweezers | Fine Science Tools, Foster City, CA, USA | 11050-10 | |
| Perfusion pump | Lead Fluid | BQ80S | |
| Plastic vials | Profilab, Warsaw, Poland | 534.02 | plastic vials with blue cap for tissue storage, 20 ml, 31 x 48 mm |
| Straight iris scissors (~9 cm) | Fine Science Tools, Foster City, CA, USA | 14058-11 | |
| Brain slices preparation for EM: | |||
| 12-well plate | NEST, Rahway, NJ, USA | 712001 | |
| Cyanoacrylic glue | Fenedur, Montevideo, Uruguay | ||
| Glass vials | Electron Microscopy Sciences, Hatfield, PA, USA | 72632 | 20 ml Scintillation Vial, a pack of 100 |
| Pasteur pipette | VWR, Radnor, PA, USA | 612-4545 | LDPE, disposable, 7.5 ml |
| Razor blade | Wilkinson Sword, London, UK | Classic double edge safety razor blades | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Vibratome | Leica Microsystems, Vienna, Austria | Leica VT1000 S | |
| Brain slices preparation for IF: | |||
| 96-well plate | NEST, Rahway, NJ, USA | 701101 | |
| Criostat | Leica Microsystems, Vienna, Austria | Leica CM 1950 | |
| Ethylene glycol | Bioshop, Burlington, Canada | ETH001 | |
| Low-profile disposable blade 819 | Leica Biosystems Inc., USA | 14035838925 | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Sodium azide (NaN3) | POCH, Gliwice, Poland | 792770426 | |
| Sucrose | POCH, Gliwice, Poland | 772090110 | |
| Tissue freezing medium for cryosectioning, OCT-Compound | Leica Biosystems, Switzerland | 14020108926 | |
| Immunostaining: | |||
| 24-well plate | NEST, Rahway, NJ, USA | 702001 | |
| Anti-Post Synaptic Density Protein 95 Antibody | Merck-Millipore, Burlington, MA, USA | MAB1598 | |
| Confocal microscope | Zeiss, Göttingen, Germany | Zeiss Spinning Disc microscope (63 × oil objective, NA 1.4, pixel size 0.13 µm × 0.13 µm) | |
| Cover slide | Menzel Glaser, Braunschweig, Germany | B-1231 | 24 x 60 mm |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Invitrogen, Carlsbad, CA, USA | A31570 | |
| Fluoromount-G Mounting Medium, with DAPI | Invitrogen, Carlsbad, CA, USA | 00-4959-52 | |
| Microscope slide | Thermo Scientific, Waltham, MA, USA | AGAA00008 | SuperFrost |
| Normal donkey serum (NDS) | Jackson ImmunoResearch Laboratories, West Grove, PA, USA | 017-000-121 | |
| Shaker | JWElectronic, Warsaw, Poland | KL-942 | |
| TritonT X-100 Reagent Grade | Bioshop, Burlington, Canada | TRX506 | |
| Electron microsocpy sample preparation | |||
| Potassium hexacyanoferrate(II) trihydrate | POCH, Gliwice, Poland | 746980113 | |
| Aclar 33C Film | Electron Microscopy Sciences, Hatfield, PA, USA | 50425 | Fluoropolymer Film embedding sheet |
| DMP-30, 2,4,6-Tris(dimethylaminomethyl)phenol | Sigma-Aldrich,St. Louis, MI, USA | T58203 | Epoxy embedding medium accelerator |
| Durcupan ACM single component A, M | Sigma-Aldrich,St. Louis, MI, USA | 44611 | Durcupan ACM single component A, M epoxy resin |
| Durcupan ACM single component B | Sigma-Aldrich,St. Louis, MI, USA | 44612 | Durcupan ACM single component B, hardener 964 |
| Durcupan ACM single component D | Sigma-Aldrich,St. Louis, MI, USA | 44614 | Durcupan ACM single component D , plasticizer |
| Ethyl alcohol absolute | POCH, Gliwice, Poland | 64-17-5 | Ethyl alcohol absolute 99.8 % pure P.A.-BASIC |
| Genlab laboratory oven | Wolflabs, York, UK | Mino/18/SS | Oven Genlab MINO/18/SS 18l volume, no fan circulation, no digital display, standard temperature gradient, standard recovery rate, no timer, 250°C maximum temperature, 240V electrical supply |
| L-Aspartic acid | Sigma-Aldrich,St. Louis, MI, USA | A-9256 | reagent grade, 98% (HPLC) |
| Lead (II) nitrate | Sigma-Aldrich,St. Louis, MI, USA | 467790 | 99.95% trace metals basis |
| Osmium tetroxide | Sigma-Aldrich,St. Louis, MI, USA | 75632 | for electron microscopy, 4% in H2O |
| pH meter | Elmetron, Zabrze, Poland | CP-5-5 | |
| Rotator | BioSan, Józefów, Poland | Multi Bio RS-24 | rotator Multi Bio RS-24 |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade, 98%, pellets (anhydrous) |
| Sunflower mini shaker | Grant bio, Shepreth,UK | PD-3D | |
| Syringe filter | Millipore, Burlington, MA, USA | SLGP033NB | 0,22 µm pore size |
| Thiocarbohydrazide | Sigma-Aldrich,St. Louis, MI, USA | 88535 | purum p.a., for electron microscopy, 99.0% (N) |
| Uranyl acetate | Serva, Heidelberg, Germany | 77870 | Uranyl acetate·2H2O, research grade |
| Water bath | WSL, Swietochlowice, Poland | LWT | |
| Specimen mounting for SBEM | |||
| 96-well culture plate | VWR, Radnor, PA, USA | 734-2782 | 96-well plates, round bottom, non treated |
| AM Gatan 3View stub handling tweezers | Micro to Nano, Haarlem, Netherlands Netherlands |
50-001521 | |
| Binocular | OPTA-TECH, Warsaw, Poland | X2000 | |
| Conductive glue | Chemtronics, Georgia, USA | CW2400 | conductive eopxy |
| Gatan 3View sample pin stubs | Micro to Nano, Haarlem, Netherlands Netherlands |
10-006003 | |
| Parafilm | Sigma-Aldrich,St. Louis, MI, USA | P7793 | roll size 20 in. × 50 ft |
| Pelco conductive silver paint | Ted Pella, Redding, CA, USA | 16062-15 | PELCO® Conductive Silver Paint, 15g |
| Razor blades double edge | Electron Microscopy Sciences, Hatfield, PA, USA | 72000 | Stainless Steel "PTFE" coated. PERSONNA brand .004" thick, wrapped individually, 250 blades in a box. |
| Scanning Electron Microscope | Zeiss, Oberkochen, Germany | Sigma VP with Gatan 3View2 chamber, acceleration voltage 2.5 kV, variable pressure 5 Pa, aperture 20 µm, dwell time 6 µs, slice thickness 60 nm, magnification 15 000 x, image resolution 2048 x 2048 pixels, pixel size 7.3 nm | |
| trim 90° diamond knife | Diatome Ltd., Nidau, Switzerland | DTB90 | |
| Ultramicrotome | Leica Microsystems, Vienna, Austria | Leica ultracutR | |
| Software | webpage | tutorials | |
| FijiJ | https://fiji.sc/ | ||
| Microscopy Image Browser | http://mib.helsinki.fi/ | http://mib.helsinki.fi/tutorials.html | |
| Reconstruct | https://synapseweb.clm.utexas.edu/software-0 | https://synapseweb.clm.utexas.edu/software-0) | |
| Animals | |||
| Mice | Adult 3-month old and 20±1 month old female Thy1-GFP(M) mice (Thy1-GFP +/-) (Feng et al.,2000) which express GFP in a sparsely distributed population of glutamatergic neurons. Animals were bred as heterozygotes with the C57BL/6J background in the Animal House of the Nencki Institute of Experimental Biology. |
References
- Bosch, M., Hayashi, Y. Structural plasticity dendritic spines. Current Opinion in Neurobiology. 22 (3), 383-388 (2012).
- Borczyk, M., Radwanska, K., Giese, K. P. The importance of ultrastructural analysis of memory. Brain Research Bulletin. 173, 28-36 (2021).
- Wanner, A. A., Kirschmann, M. A., Genoud, C. Challenges of microtome-based serial block-face scanning electron microscopy in neuroscience: challenges of SBEM in neuroscience. Journal of Microscopy. 259 (2), 137-142 (2015).
- Śliwińska, M. A., et al. Long-term Memory Upscales Volume of Postsynaptic Densities in the Process that Requires Autophosphorylation of αCaMKII. Cerebral Cortex. 30 (4), 2573-2585 (2020).
- Borczyk, M., Śliwińska, M. A., Caly, A., Bernas, T., Radwanska, K. Neuronal plasticity affects correlation between the size of dendritic spine and its postsynaptic density. Scientific Reports. 9 (1), 1693 (2019).
- Leighton, S. B. SEM images of block faces, cut by a miniature microtome within the SEM - a technical note. Scanning Electron Microscopy. , Pt 2 73-76 (1981).
- Denk, W., Horstmann, H. Serial Block-Face Scanning Electron Microscopy to Reconstruct Three-Dimensional Tissue Nanostructure. PLoS Biology. 2 (11), 329 (2004).
- Smith, D., Starborg, T. Serial block face scanning electron microscopy in cell biology: Applications and technology. Tissue and Cell. 57, 111-122 (2019).
- Titze, B., Genoud, C. Volume scanning electron microscopy for imaging biological ultrastructure: Volume scanning electron microscopy. Biology of the Cell. 108 (11), 307-323 (2016).
- Deerinck, T. J., Bushong, E. A., Thor, A., Ellisman, M. H. NCMIR methods for 3D EM: A new protocol for preparation of biological specimens for serial block face scanning electron microscopy. , 6-8 (2010).
- Willingham, M. C., Rutherford, A. V. The use of osmium-thiocarbohydrazide-osmium (OTO) and ferrocyanide-reduced osmium methods to enhance membrane contrast and preservation in cultured cells. Journal of Histochemistry & Cytochemistry. 32 (4), 455-460 (1984).
- Feng, G., et al. Imaging Neuronal Subsets in Transgenic Mice Expressing Multiple Spectral Variants of GFP. Neuron. 28 (1), 41-51 (2000).
- Gage, G. J., Kipke, D. R., Shain, W. Whole Animal Perfusion Fixation for Rodents. Journal of Visualized Experiments. (65), e3564 (2012).
- Paxinos, G., Franklin, K. B. J. The mouse brain in stereotaxic coordinates. , Acad. Press. San Diego, Calif. (2004).
- Walton, J. Lead aspartate, an en bloc contrast stain particularly useful for ultrastructural enzymology. Journal of Histochemistry & Cytochemistry. 27 (10), 1337-1342 (1979).
- Mercer, E. H. a scheme for section staining in electron microscopy. Journal of the Royal Microscopical Society. 81 (3-4), 179-186 (1963).
- Schindelin, J., et al. Fiji: an open-source platform for biological-image analysis. Nature Methods. 9 (7), 676-682 (2012).
- Belevich, I., Joensuu, M., Kumar, D., Vihinen, H., Jokitalo, E. Microscopy Image Browser: A Platform for Segmentation and Analysis of Multidimensional Datasets. PLOS Biology. 14 (1), 1002340 (2016).
- Fiala, J. C. Reconstruct: a free editor for serial section microscopy. Journal of Microscopy. 218 (1), 52-61 (2005).
- Roels, J., et al. An interactive ImageJ plugin for semi-automated image denoising in electron microscopy. Nature Communications. 11 (1), 771 (2020).
- Radwanska, K., et al. Mechanism for long-term memory formation when synaptic strengthening is impaired. Proceedings of the National Academy of Sciences. 108 (45), 18471-18475 (2011).
- Kittelmann, M., Hawes, C., Hughes, L. Serial block face scanning electron microscopy and the reconstruction of plant cell membrane systems: SBFSEM Methods for Plant Cells. Journal of Microscopy. 263 (2), 200-211 (2016).
- Fendrych, M., et al. Programmed Cell Death Controlled by ANAC033/SOMBRERO Determines Root Cap Organ Size in Arabidopsis. Current Biology. 24 (9), 931-940 (2014).
- Russell, M. R. G., et al. 3D correlative light and electron microscopy of cultured cells using serial blockface scanning electron microscopy. Journal of Cell Science. 130 (1), 278-291 (2017).
- Płachno, B. J., Świątek, P., Jobson, R. W., Małota, K., Brutkowski, W. Serial block face SEM visualization of unusual plant nuclear tubular extensions in a carnivorous plant (Utricularia, Lentibulariaceae). Annals of Botany. 120 (5), 673-680 (2017).
- Genoud, C., Titze, B., Graff-Meyer, A., Friedrich, R. W. Fast Homogeneous En Bloc Staining of Large Tissue Samples for Volume Electron Microscopy. Frontiers in Neuroanatomy. 12, (2018).
- Puhka, M., Joensuu, M., Vihinen, H., Belevich, I., Jokitalo, E. Progressive sheet-to-tubule transformation is a general mechanism for endoplasmic reticulum partitioning in dividing mammalian cells. Molecular Biology of the Cell. 23 (13), 2424-2432 (2012).
- Gluenz, E., Wheeler, R. J., Hughes, L., Vaughan, S. Scanning and three-dimensional electron microscopy methods for the study of Trypanosoma brucei and Leishmania mexicana flagella. Methods in Cell Biology. 127, 509-542 (2015).
- Starborg, T., et al. Using transmission electron microscopy and 3View to determine collagen fibril size and three-dimensional organization. Nature Protocols. 8 (7), 1433-1448 (2013).
- Hughes, L., Borrett, S., Towers, K., Starborg, T., Vaughan, S. Patterns of organelle ontogeny through a cell cycle revealed by whole-cell reconstructions using 3D electron microscopy. Journal of Cell Science. 130 (3), 637-647 (2017).
- Bojko, A., et al. Improved Autophagic Flux in Escapers from Doxorubicin-Induced Senescence/Polyploidy of Breast Cancer Cells. International Journal of Molecular Sciences. 21 (17), 6084 (2020).
- Knott, G. W., Holtmaat, A., Trachtenberg, J. T., Svoboda, K., Welker, E. A protocol for preparing GFP-labeled neurons previously imaged in vivo and in slice preparations for light and electron microscopic analysis. Nature Protocols. 4 (8), 1145-1156 (2009).
- Glauert, A. M., Lewis, P. R. Biological specimen preparation for transmission electron microscopy. , Princeton University Press. Princeton. (2014).
- Genoud, C. Altered Synapse Formation in the Adult Somatosensory Cortex of Brain-Derived Neurotrophic Factor Heterozygote Mice. Journal of Neuroscience. 24 (10), 2394-2400 (2004).
