



Overview
Fuente: Perchet Thibaut1,2,3, Meunier Sylvain1,2,3, Sophie Novault4, Rachel Golub1,2,3
1 Unidad de Linfopoyesis, Departamento de Inmunología, Instituto Pasteur, París, Francia
2 INSERM U1223, París, Francia
3 Université Paris Diderot, Sorbonne Paris Cité, Cellule Pasteur, París, Francia
4 Flow Cytometry Platfrom, Citometría y Biomarcadores UtechS, Centro de Ciencias Traslacionales, Instituto Pasteur, París, Francia
La función general del sistema inmunológico es defender el cuerpo contra organismos infecciosos y otros invasores. Los glóbulos blancos, o leucocitos, son los actores clave del sistema inmunitario. Tras la infección, se activan e inician una respuesta inmunitaria. Los leucocitos se pueden dividir en varias subpoblaciones (por ejemplo, células mieloides, linfocitos, células dendríticas) basándose en diferentes parámetros que pueden ser biológicos, físicos y/o funcionales (por ejemplo, tamaño, granularidad y secreción). Una forma de caracterizar a los leucocitos es a través de sus proteínas superficiales, que son principalmente receptores. Cada población de leucocitos expresa una combinación específica de receptores (por ejemplo, citotóxicos, activadores, receptores de migración) que pueden definir subconjuntos entre las poblaciones. Como el sistema inmunitario abarca una amplia gama de poblaciones celulares, es esencial caracterizarlas para descifrar su participación en la respuesta inmunitaria.
La citometría de flujo (FC o FCM) es un método ampliamente utilizado para analizar la expresión de la superficie celular y las moléculas intracelulares, caracterizando y definiendo diferentes tipos de células en una mezcla de células heterogéneas. Los citometros de flujo se componen de tres subsistemas principales: fluidos, ópticos y electrónicos. El sistema de fluidos transporta las células en una corriente de tal manera que pasan delante de un láser uno por uno. El sistema óptico consta de fuentes de luz (láser) para iluminar las partículas, filtros ópticos para dirigir la luz resultante y señales fluorescentes a los detectores adecuados. Por último, el sistema electrónico convierte las señales de luz detectadas en señales electrónicas que pueden ser procesadas por el ordenador. A medida que una célula individual pasa delante del rayo láser, dispersa la luz. Un detector delante del haz mide la dispersión hacia adelante (FS) y varios detectores hacia el lado de la dispersión lateral (SC). El FS se correlaciona con el tamaño de celda y SC es proporcional a la granularidad de las células. De esta manera, las poblaciones celulares a menudo se pueden distinguir en función de las diferencias en su tamaño y granularidad solamente.
Además de analizar el tamaño, la forma y la complejidad de una célula, la citometría de flujo se utiliza ampliamente para detectar la expresión de los receptores de superficie celular (1). Esto se logra mediante el uso de anticuerpos monoclonales etiquetados con fluorocromo que se unen a receptores específicos de células conocidos. Tras la excitación, estos fluorocromos enlazados emiten una luz de longitud de onda específica, llamada longitud de onda de emisión, que puede ser detectada y puntuada. Las mediciones de fluorescencia proporcionan datos cuantitativos y cualitativos sobre los receptores de superficie celular etiquetados con fluorocromo. Los hematólogos utilizaron por primera vez la FC para el seguimiento terapéutico de las poblaciones de células inmunitarias (2). Ahora, se utiliza para una amplia gama de aplicaciones tales como inmunofenotipado, viabilidad celular, expresión génica, recuento celular y análisis de GFP.
FACS (Fluorescent Activated Cell Sorter) es un tipo especializado de citometría de flujo, que ordena una población de células en subpoblación utilizando etiquetado fluorescente. Al igual que la citometría de flujo convencional, se recopilan los primeros datos FS, SC y fluorescentes. A continuación, la máquina aplica una carga (negativa o positiva) y un sistema de desviación electrostática (electroimanes) facilita la recolección de gotas cargadas que contienen células en tubos apropiados.
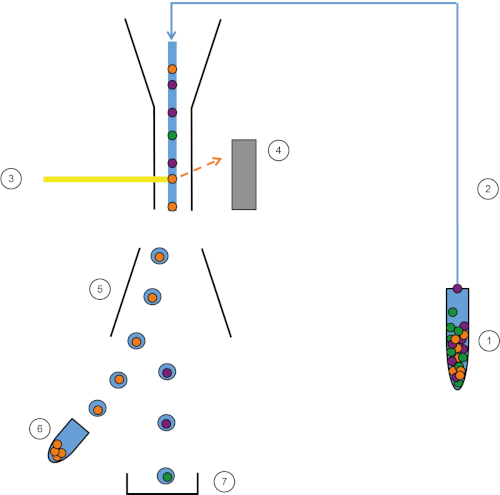
Figura 1: Representación esquemática de FACS. La muestra (1) se aspira en el FACS (2) y se pasa delante del láser (3). La fluorescencia celular es sintensada por detectores de fluorescencia (4). Finalmente, las células se incorporan en gotas y las células de interés son desviadas por placas de desviación (5) y recogidas en un tubo de recolección (6). Las celdas restantes entran en la basura (7). Haga clic aquí para ver una versión más grande de esta figura.
El aspecto de clasificación del FACS presenta muchas ventajas. Muchas pruebas pueden ayudar a entender el papel de las células específicas en el sistema inmunitario, como los análisis de la expresión génica como RT-qPCR, ciclo celular o secreción de citoquinas. Sin embargo, las células deben purificarse aguas arriba para obtener resultados claros y específicos. Aquí, FACS viene en útil y las células deseadas se pueden clasificar con gran pureza, produciendo resultados altamente confiables y reproducibles. FacS también se puede utilizar para clasificar las células en función de la tinción nuclear u otra intracelular y de acuerdo con la presencia, ausencia y densidad de los receptores de superficie. FACS es ahora una técnica estándar para la purificación de subpoblaciones de células y tiene la capacidad de ordenar hasta cuatro poblaciones simultáneamente.
Este ejercicio de laboratorio demuestra cómo aislar leucocitos esplénicos y luego cómo clasificar específicamente las células linfoides B de la mezcla de células de leucocitos esplénicos utilizando FACS.
Procedure
1. Preparación
- Antes de comenzar, ponte guantes de laboratorio y la ropa protectora adecuada.
- Esterilice todas las herramientas de disección, primero con un detergente y luego con 70% de etanol y luego seque bien.
- Preparar 50 ml de la solución salina equilibrada de Hank (HBSS) que contiene un 2% de suero de becerro fetal (FCS).
2. Disección
- Usando un sistema de administración de dióxido de carbono, eutanasia el ratón por hipoxia. Asegure el ratón eutanasiado en una placa de disección en posición supina y realice una laparotomía longitudinal utilizando tijeras y fórceps.
- Usando fórceps, mueva los intestinos y el estómago en el lado derecho del abdomen para exponer el estómago y el bazo. El bazo está unido al estómago.
- Usando fórceps, sepáre cuidadosamente el bazo del estómago y colóquelo en la placa Petri que contiene 5 ml de HBSS 2% FCS.
3. Aislamiento celular inmune
- Coloque el bazo en un colador de células de 40 m sobre la misma placa De Petri. Aplastar el bazo con un émbolo para disociarlo en el mismo plato.
- Transfiera el bazo disociado y el líquido a un tubo centrífugo de 15 ml.
- Centrifugar el tubo a 370 x g durante 7 min a 10oC y desechar el sobrenadante, evitando el pellet.
- Resuspenda el pellet en 2 ml de acetato de potasio para lisar los eritrocitos. Espere 2 minutos y luego suba el volumen hasta 15 ml usando HBSS 2% FCS.
- Centrifugar el tubo de nuevo a 370 x g durante 7 min a 10oC. Deseche el sobrenadante y resuspenda el pellet en 5 ml de HBSS 2% FCS.
- Cuente las células usando un ensayo de tinción azul trypan y ajuste la concentración celular final a 107 celdas/ml usando el volumen apropiado de HBSS 2% FCS.
4. Tinción celular
- Transfiera 200 l de la suspensión celular a seis tubos FACS, etiquetados del 1 al 6.
- Centrifugar los tubos a 370 x g durante 7 min a 10oC y desechar el sobrenadante evitando el pellet.
- A continuación, prepare seis nuevas mezclas de anticuerpos añadiendo la cantidad adecuada de anticuerpos a 200-L HBSS 2% FCS de acuerdo con la Tabla 1.
- A continuación, transfiera estas mezclas de anticuerpos a los tubos FACS numerados correspondientes.
- Incubar las suspensiones celulares mezcladas con los anticuerpos durante 20 minutos sobre hielo en la oscuridad.
- Añadir 1 ml de HBSS 2% FCS a cada tubo y luego centrifugar de nuevo a 370 x g durante 3 min a 10oC.
- Deseche el sobrenadante y resuspenda el pellet en 200 s de HBSS 2% FCS.
- Transfiera los pellets resuspendidos a los nuevos tubos FACS.

Tabla 1: Los anticuerpos mezclan composición. Para el experimento se prepararon seis mezclas de 200 l de HBSS + anticuerpos. Mix 1 es para el ajuste PMT, las mezclas 2 a 5 son para los ajustes de compensación, y la mezcla 6 es para la clasificación de celdas.
5. Calibración FACS
- En primer lugar, encienda el clasificador y realice"Fluidics Startup."
- Encienda la secuencia y espere 15 minutos para que la secuencia se estabilice.
- Ajuste la amplitud de la corriente para obtener la formación de gotas separadas. A continuación, haga clic en"Punto dulce"para completar el ajuste de amplitud.
- Ponga el filtro de densidad neutra (N.D.) - 1.0 y abra la interfaz "CST" que significa configuración y seguimiento del citometro.
- Para realizar un control diario de calidad, primero diluir las perlas CST con el medio FACS siguiendo las instrucciones del fabricante y, a continuación, realizar el control CST.
- Una vez que el control CST esté completo, substituya el filtro N.D. 1.0 por el filtro N.D. 2.0 en el catómetro.
- A continuación, diluir las perlas de retardo de caída en el medio FACS siguiendo las instrucciones del fabricante y luego cargar en el FACS.
- Para garantizar una clasificación adecuada, realice Drop Delay-
- Primero haga clic en "Voltaje" y luego "Filtro óptico". El cuadrante derecho debe ser igual al 100%. Si es necesario, ajuste el tornillo láser rojo en el citómetro izquierdo o derecho para obtener el 100% en el cuadrante derecho.
- A continuación, realice una ordenación de prueba para asegurarse de que la secuencia cae en el tubo de recogida. Para ello, haga clic en"Cajónde residuos" y comience "Ordenar prueba". Compruebe que las corrientes laterales caigan en tubos de recolección. Si no lo hacen, ajuste el voltaje hasta que lo hagan.
- Vaya a la plantilla experimental. A continuación, abra el experimento "Accudrop Drop Delay" y haga clic en el"Diseño de clasificación".
- Cambie el caudal para obtener de 1000 a 3000 eventos por segundo.
- Haga clic en "Voltaje" y luego "Filtro óptico". El cuadrante izquierdo debe ser igual a 0 y el cuadrante derecho a 100.
- En la ventana"Ordenar diseño"haga clic en "Ordenar" y luego haga clic en "Cancelar". El cuadrante izquierdo debe ser igual a 100 y el cuadrante derecho a 0. Si el cuadrante izquierdo es inferior a 95, haga clic en "Retardo automático"para ajustarlo automáticamente.
6. Citometría de flujo y control de pureza
- Comience la citometría de flujo comenzando con el tubo 1 (células no manchadas) para definir la morfología celular y los picos negativos de los fluorocromos. Configure la dispersión hacia delante y hacia los lados y defina los voltajes de cada parámetro fluorescente. Coloque la población negativa en la primera década utilizando las cuadrículas en cada parcela de puntos.
- A continuación, el tubo de carga 2 (control de un solo color) en el catómetro. Ajuste la superposición espectral hasta que las medianas de población negativas y positivas estén alineadas o utilice el software de cálculo automático. Es importante mantener la señal a escala. Los controles de compensación deben coincidir con la configuración experimental de fluorocromos y detectores. Graba 10.000 eventos.
- Repita estos pasos con los tubos 3, 4 y 5 (otros controles de color único).
- A continuación, cargue el tubo 6 (células de varios puntos) y defina las poblaciones celulares de interés mediante una estrategia de gating específica.
- En la ventana Diseño de ordenación, seleccione el rellenado de celdas de interés. Seleccione el umbral de celda en el "Eventos de destino" y el nivel de precisión en el "Precisión". Aquí sólo se está ordenando una población, sin embargo, cuatro poblaciones diferentes se pueden clasificar al mismo tiempo.
- Una vez listo, haga clic en "Ordenar" y"Aceptar",a continuación, espere a la ordenación de celdas.
- Una vez completada la clasificación celular, realice un control de pureza pipeteando por primera vez 10 l de células clasificadas en un nuevo tubo FACS con 90 microlitros de HBSS 2% FCS.
- A continuación, cargue el tubo del catómetro. Registre y analice los fenotipos de las células para verificar que la estrategia de gating funcionó según lo previsto.
7. Análisis de datos
- Abra el software 'FlowJo' y arrastre los archivos para cada tubo en la ventana"Todos los muestras".
- Haga doble clic en un archivo para abrirlo en una nueva ventana.
- Haga clic en el"Polígono"y vuelva a crear la estrategia de gating utilizada anteriormente.
- Repita los pasos con todos los demás archivos.
- Para visualizar las gráficas de puntos, haga clic en " Editor dediseño" y arrastre las poblaciones de interés desde el tubo 6 y el control de pureza en la pestaña editor de diseño.
- Para comprobar la pureza de los linfocitos B en las células ordenadas, haga clic en"Editorde tablas". Arrastre la población de linfocitos B del tubo 6 y el control de pureza en la tabla.
- En el menú"Estadística"seleccione la frecuencia de las celdas CD45+ para probar la pureza de esta población de células, luego haga clic en"Crear tabla".
- Los valores de parámetro aparecen en una nueva tabla. En la ventana de control de pureza, compruebe la frecuencia de los linfocitos B dentro de las células CD45+, que debe ser superior al 98% (ver figura 2, panel inferior).
El sistema inmunitario protege al cuerpo de los patógenos invasores mediante la generación de leucocitos, también llamados glóbulos blancos. Cuando un patógeno infecta con éxito un organismo, se activa una amplia variedad de leucocitos y esta reacción coordinada se denomina respuesta inmunitaria.
Con frecuencia, es útil para los investigadores ser capaces de identificar el tipo específico y el número de células inmunitarias que se han activado en respuesta a un patógeno. La citometría de flujo es una técnica que permite a los investigadores separar las células en función de epítopos específicos expresados en sus superficies. Esto se logra utilizando anticuerpos monoclonales etiquetados con fluorocromo que se unen a epítopos específicos de células inmunitarias conocidos, y tras la excitación, estos fluorocromos enlazados emiten una longitud de onda de luz que puede ser detectada y puntuada por un citómetro de flujo.
Los citometros de flujo se componen de tres sistemas. El sistema fluido transporta las células en una corriente de tal manera que pasan delante de un láser uno por uno. El sistema óptico está compuesto por láseres y detectores que reconocen la presencia o ausencia de los fluoróforos. Por último, el sistema electrónico convierte los datos ópticos recogidos en archivos electrónicos para su análisis.
Una extensión de la citometría de flujo es el Clasificador de Células Activado por Fluorescencia, o FACS, que permite el enriquecimiento de poblaciones celulares específicas para que puedan ser estudiadas de forma independiente. La clasificación celular se realiza utilizando una boquilla vibratoria dentro de la corriente fluida que forma microgotas, cada una de las cuales contiene una sola célula. Luego, un detector determina si la luz fluorescente se emite o no de cada gota, y en base a esa información, un electroimán da a cada célula una carga negativa o positiva. A continuación, un campo eléctrico fuerte ordena las gotas cargadas de manera diferente en contenedores separados. En última instancia, uno de los contenedores contendrá una población homogénea de células basada en la expresión de una molécula de superficie celular específica.
En este video, aprenderás a usar citometría de flujo para aislar leucocitos del tejido del bazo del ratón y FACS para seleccionar linfocitos B.
Para empezar, ponte guantes de laboratorio y la ropa protectora adecuada. A continuación, lave un par de tijeras dissección y fórceps primero con detergente y luego con 70% de etanol y luego séquelos con una toalla de papel limpia.
Luego, agregue 49 mililitros de solución de sal equilibrada de Hank, o HBSS, a un tubo de 50 mililitros. Agregue un mililitro de suero de becerro fetal, o FCS, para crear una solución de FCS HBSS 2% y mezcle pipeteando suavemente hacia arriba y hacia abajo aproximadamente 10 veces.
A continuación, coloque un ratón eutanasiado en la posición supina en una placa de disección. Con las tijeras y fórceps, realice una laparotomía longitudinal para acceder a la cavidad abdominal. Use los fórceps para mover los intestinos del lado derecho del abdomen a un lado para exponer el estómago y el bazo. El bazo está unido al estómago. Luego, con una pipeta, coloque cinco mililitros del HBSS 2% FCS en una placa Petri. Con fórceps, separa cuidadosamente el bazo del estómago y coloca el bazo en la placa De Petri.
Para aislar las células inmunitarias, coloque primero el bazo en un colador de células de 40 micras en un plato de Petri. Aplastar el bazo con un émbolo para disociarlo en el plato. Luego, pipetee el bazo disociado y el líquido de la placa Petri en un tubo centrífugo de 15 mililitros. Centrifugar el tubo a 370 veces g durante siete minutos a 10 grados Centígrados y luego recuperar el tubo con cuidado para no molestar el pellet.
Ahora, retire el sobrenadante, evitando el pellet, y deseche el líquido en un contenedor de residuos apropiado. A continuación, agregue dos mililitros de tampón de levas ACK en el tubo de centrífuga para resuspender y lise los eritrocitos. Espere dos minutos y luego agregue HBSS 2% FCS para obtener un volumen total de 15 mililitros. Repite la centrifugación. Recupere el tubo con cuidado y deseche el sobrenadante. Resuspenda el pellet de nuevo en cinco mililitros de HBSS 2% FCS.
Para contar las células resuspendidas, diluya cinco microlitros de la suspensión celular con cinco microlitros de Trypan Blue. A continuación, deposite suavemente una gota de cinco microlitros de esta suspensión de celda diluida entre el vidrio de cubierta y la corredera Malassez. Ahora, bajo un microscopio con aumento 40X, cuente el número de células presentes. Luego, ajuste la concentración celular a 10 a las séptimas células por mililitro agregando el volumen apropiado de HBSS 2% FCS.
Para manchar las células inmunitarias, comience por etiquetar seis tubos FACS del uno al seis. A continuación, transfiera 200 microlitros de la solución celular a cada uno de los seis tubos. Centrifugar estos tubos a 370 veces g durante siete minutos a 10 grados Centígrados y retirar el sobrenadante.
Luego, etiquete seis nuevos tubos FACS como uno a seis y pipetee 200 microlitros de HBSS 2% FCS en cada uno. Preparar las seis mezclas de anticuerpos nuevos añadiendo la cantidad adecuada de anticuerpos a cada tubo de acuerdo con la tabla uno. Mezclar uno es para células no manchadas sin adición de anticuerpos. Las mezclas de dos a cinco contienen un anticuerpo único diferente para los ajustes de compensación. Mix six contiene los cuatro anticuerpos para las células multi-teñidas que se utilizarán para la clasificación.
A continuación, transfiera estas mezclas de anticuerpos a los tubos FACS numerados correspondientes. Incubar estas soluciones durante 20 minutos a cuatro grados centígrados o sobre hielo en la oscuridad. A continuación, agregue un mililitro de HBSS 2% FCS a cada tubo y luego centrifugar de nuevo. Deseche el sobrenadante y luego resuspenda los pellets en 200 microlitros de HBSS 2% FCS. Finalmente, transfiera los pellets resuspendidos a los nuevos tubos FACS etiquetados.
Para realizar FACS, primero encienda el clasificador. A continuación, seleccione el menú del citómetro y haga clic en inicio de fluidos. Siga las instrucciones que aparecen en pantalla.
En la pestaña stream, haga clic en la cruz roja para activar la secuencia y, a continuación, espere 15 minutos para que la secuencia se estabilice. Ajuste la amplitud de la secuencia hasta que vea que aparece una caída separada clara en la pestaña de la secuencia. A continuación, haga clic en punto dulce para completar el ajuste de amplitud. Inserte el filtro Densidad neutra, o ND, 1.0 delante del láser.
Abra el menú del citómetro en la parte superior de la pantalla y seleccione CST, que significa Cytometer Setup and Tracking. Para realizar un control de calidad diario, primero diluir las perlas CST con medio FACS en un tubo FACS siguiendo las instrucciones del fabricante. A continuación, cargue el tubo en la máquina y realice el control CST haciendo clic en Ejecutar en la pestaña CST.
Cuando se complete el control CST, reemplace el filtro ND 1.0 por el filtro ND 2.0 en el catómetro. A continuación, diluir las perlas de retardo de gota en el medio FACS siguiendo las instrucciones del fabricante y luego cargar el tubo en el FACS. Para garantizar una clasificación adecuada, realice el retardo de caída haciendo clic primero en el voltaje y luego en el filtro óptico. El cuadrante derecho del filtro óptico debe ser igual al 100%, lo que indica que el 100% de las gotas están registradas por la máquina. Si es necesario, ajuste el tornillo láser rojo en el citómetro izquierdo o derecho para obtener el 100% en el cuadrante derecho. Es importante asegurarse de que la corriente cae en el tubo de recogida. Para ello, realice una clasificación de prueba haciendo clic en el cajón de residuos y, a continuación, pruebe la ordenación. Compruebe que las corrientes laterales caigan en los tubos de recogida. Si no lo hacen, ajuste la tensión debajo de la pestaña de clasificación hasta que lo hagan.
Vaya a la plantilla experimental seleccionando la pestaña del explorador y haciendo clic en Vista compartida. A continuación, abra el experimento Accudrop_DROP DELAY y haga clic en el botón de diseño de ordenación. Ahora, cambie la velocidad de umbral en el panel de adquisición manipulando el caudal hasta que alcance los 3.000 eventos por segundo. Haga clic en voltaje y, a continuación, haga clic en filtro óptico. El cuadrante izquierdo debe ser igual a cero y el cuadrante derecho igual a 100.
Por último, en la ventana de diseño de ordenación, haga clic en ordenar y, a continuación, haga clic en cancelar. El cuadrante izquierdo debe ser igual a 100 y el cuadrante derecho igual a cero. Si el cuadrante izquierdo es menor que 95, haga clic en retardo automático para indicar al software que aumente automáticamente el voltaje para obtener el 100% de las caídas en el cuadrante izquierdo.
Para comenzar la citometría de flujo, primero usaremos células no manchadas para definir la morfología celular y los picos negativos de los fluorocromos. Para ello, coloque el tubo uno que contenga celdas no retenidas en la máquina y, debajo de la pestaña del panel de adquisición, haga clic en cargar. En la pestaña del citómetro, ajuste los voltajes de dispersión hacia adelante y hacia los lados hasta que vea su población celular como una densa concentración de puntos en la pantalla. Los linfocitos son células pequeñas, por lo que tendrán una dispersión hacia adelante baja y dispersión lateral baja.
A continuación, elimine la fluorescencia de fondo ajustando el voltaje de los fluorocromos en la pestaña del citómetro hasta que las poblaciones celulares a un nivel negativo estén en la primera década en la pestaña de la hoja de trabajo global. En el menú del citómetro, haga clic en la configuración de la vista y verifique que todos los fluorocromos estén presentes. A continuación, coloque el tubo dos en el citómetro y haga clic en cargar. Ajuste la superposición espectral en la pestaña del citómetro hasta que las medianas de población negativas y positivas estén alineadas en la pestaña de la hoja de cálculo global. En la pestaña adquisición, establezca los eventos para registrar el parámetro en 10.000 y haga clic en registro. Repita estos pasos con los tubos tres, cuatro y cinco.
A continuación, cargue el tubo seis que contiene las células de varios manchas. Para aislar los linfocitos B, primero configure los parámetros para ordenar las células en función de su morfología. En la primera ventana, trace el área de dispersión hacia adelante FSC-A en el eje Y y el área de dispersión del lado SSC-A en el eje X. En el gráfico de dispersión, cada punto representa una celda. Haga clic en la puerta poligonal en la hoja de trabajo global y luego seleccione la población con una dispersión hacia adelante baja y una dispersión lateral intermedia. En una nueva ventana de trazado de puntos, haga clic con el botón derecho en la ventana y seleccione Mostrar poblaciones en el menú y haga clic en P1.
Luego, en la nueva ventana, agarre las celdas positivas viables CD45 trazando la viabilidad en el eje Y y CD45 en el eje X. Utilice la puerta poligonal para rodear las celdas con una baja viabilidad y una señal CD45 alta y seleccione P2 para mostrar las celdas seleccionadas en una nueva ventana. En la siguiente ventana, puerta para leucocitos positivos CD45, excluyendo linfocitos T. Con CD45 en el eje X y CD3 en el eje Y, circule a la población con una señal CD45 alta y una señal CD3 negativa baja y seleccione P3. Por último, puerta para células positivas CD19 que identifican los linfocitos B. Con CD19 en el eje Y y CD3 en el eje X, circule a la población con una señal CD19 alta y una señal CD3 negativa baja y seleccione P4.
Ahora se establecen todos los parámetros de ordenación. A continuación, en la ventana de diseño de clasificación, seleccione su población celular de interés- P4, que es la cuarta población que estaba cerrada, y le dice a la máquina que ordene sólo los linfocitos B. Establezca los eventos de destino en 10.000 celdas y establezca la precisión en pureza. Sólo estamos clasificando una población. Sin embargo, se pueden clasificar hasta cuatro poblaciones diferentes al mismo tiempo. Una vez listo, haga clic en ordenar y Aceptar. Luego, espere a la clasificación celular.
Una vez completada la clasificación celular, realice un control de pureza pipeteando 10 microlitros de las células clasificadas en un nuevo tubo FACS con 90 microlitros de HBSS 2% FCS. Coloque el tubo en el citómetro, haga clic en cargar y luego haga clic en registro para analizar los fenotipos de las celdas para verificar que la estrategia de gating funcionó según lo previsto.
Ahora, analizaremos las células ordenadas para determinar el porcentaje de linfocitos B entre los leucocitos que fueron aislados del bazo del ratón. Para empezar, haga doble clic en el icono FlowJo y arrastre los archivos de cada tubo a la ventana de muestra.
Haga clic en polígono y vuelva a crear las estrategias de gating que se utilizaron en la sección anterior. A continuación, haga clic en el editor de diseño y arrastre las poblaciones de linfocitos B de interés desde el tubo seis y el control de pureza a la pestaña del editor de diseño. En este ejemplo, la gráfica en la parte superior derecha representa los linfocitos B ordenados de la suspensión total de la célula del bazo y la gráfica en la parte inferior derecha es el control de pureza. Las células sólo deben aparecer en la población de interés en el control de pureza.
Para comprobar la pureza de los linfocitos B en las células ordenadas, haga clic en el editor de tablas. Arrastre la población de linfocitos B del tubo seis y el control de pureza en la tabla. En el menú estadístico, seleccione la frecuencia de las células positivas CD45 para probar la pureza de esta población celular. A continuación, haga clic en crear tabla. Los valores de parámetro aparecen en una nueva tabla. En la ventana de control de pureza, compruebe la frecuencia de los linfocitos B dentro de las células positivas CD45, que debe ser superior al 98%.
Subscription Required. Please recommend JoVE to your librarian.
Results
En este protocolo, purificamos linfocitos B esplénicos utilizando la tecnología FACS. Primero aislamos leucocitos del bazo y los manchamos. Usando una combinación de marcadores de superficie de celda B, creamos una estrategia de gating para ordenarlos (Figura 2, panel superior). Al final del experimento verificamos si las células en el tubo de recolección eran células B a través de una "prueba de pureza". Mantuvimos la misma estrategia de gating y observamos que más del 98% de las células eran de hecho células B (Figura 2, panel inferior). Por lo tanto, FACS es un protocolo eficaz para aislar las poblaciones de células inmunitarias con un alto grado de pureza. Las celdas recopiladas se pueden utilizar para experimentos posteriores, como el cultivo celular, RT-qPCR y los ensayos de citotoxicidad.

Figura 2: Estrategia de gating y pruebas de pureza post-clasificación. (A) Las células primero fueron cerradas en función de su morfología (izquierda: FSC-A, SSC-A), luego sólo vivas (izquierda media: viabilidad, CD45), CD45+ células (CD45, CD3) se trazaron contra CD19 y CD3. Solo se ordenaron las celdas CD19+. (B) Resultados de la prueba de pureza de una fracción de las células obtenidas después de la clasificación celular. Haga clic aquí para ver una versión más grande de esta figura.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
La citometría de flujo es una técnica de primera mano para caracterizar y clasificar las poblaciones de células inmunitarias con un alto grado de pureza. Es una herramienta primordial en el campo de la investigación, ya que permite el enriquecimiento de poblaciones celulares específicas y para descifrar la respuesta inmune a los patógenos. Con el aumento en el número de fluorocromos y citómetros disponibles, el número de parámetros detectables se incrementa considerablemente. Como resultado, el análisis bioinformático de los datos FACS ha comenzado a surgir y ha abierto nuevos horizontes para fluir la citometría (3). La citometría de flujo ofrece otras aplicaciones en hematología y oncología (4) donde se utiliza para el desarrollo de herramientas de diagnóstico.
Subscription Required. Please recommend JoVE to your librarian.
References
- Lanier, L. L. Just the FACS. The Journal of Immunology, 193 (5), 2043-2044 (2014).
- Walker, J. M. Epiblast Stem Cells IN Series Editor.
- Tung, J. W., Heydari, K., Tirouvanziam, R., Sahaf, B., Parks. D. R., Herzenberg, L. A., and Herzenberg. L. A. Modern Flow Cytometry: A Practical Approach. Clinics in Laboratory Medicine. 27 (3), 453-468 (2007).
- Walker, J. M. Tumor Angiogenesis Assays IN Series Editor.