



Overview
Quelle: Dominique R. Bollino1, Eric A. Legenzov2, Tonya J. Webb1
1 Department of Microbiology and Immunology, University of Maryland School of Medicine and the Marlene and Stewart Greenebaum Comprehensive Cancer Center, Baltimore, Maryland 21201
2 Center for Biomedical Engineering and Technology, University of Maryland School of Medicine, Baltimore, Maryland 21201
Die konfokale Fluoreszenzmikroskopie ist eine bildgebende Technik, die eine höhere optische Auflösung im Vergleich zur herkömmlichen "Weitfeld"-Epifluoreszenzmikroskopie ermöglicht. Konfokale Mikroskope sind in der Lage, eine verbesserte x-y optische Auflösung durch "Laserscanning" zu erreichen - typischerweise eine Reihe von spannungsgesteuerten Spiegeln (Galvanometer oder "Galvo"-Spiegel), die Laserbeleuchtung auf jedes Pixel der Probe zu einem Zeitpunkt leiten. Noch wichtiger ist, dass konfokale Mikroskope eine überlegene z-axiale Auflösung erreichen, indem sie ein Loch verwenden, um aus fokussiertem Licht zu entfernen, das von Orten stammt, die sich nicht in der zu scannenden Z-Ebene befinden, wodurch der Detektor Daten von einer bestimmten Z-Ebene sammeln kann. Durch die hohe Z-Auflösung, die in der konfokalen Mikroskopie erreichbar ist, ist es möglich, Bilder aus einer Reihe von Z-Ebenen (auch z-stack genannt) zu sammeln und ein 3D-Bild durch Software zu konstruieren.
Bevor Sie den Mechanismus eines konfokalen Mikroskops diskutieren, ist es wichtig zu überlegen, wie eine Probe mit Licht interagiert. Licht besteht aus Photonen, Paketen elektromagnetischer Energie. Ein Photon, das auf eine biologische Probe einwirkt, kann mit den Molekülen interagieren, die die Probe umfassen, auf eine von vier Arten: 1) das Photon interagiert nicht und durchläuft die Probe; 2) das Photon reflektiert/gestreut wird; 3) das Photon wird von einem Molekül absorbiert und die absorbierte Energie wird als Wärme durch Prozesse freigesetzt, die kollektiv als nicht-radioativer Zerfall bekannt sind; und 4) das Photon wird absorbiert und die Energie wird dann schnell als sekundäres Photon durch den als Fluoreszenz bekannten Prozess übersendet. Ein Molekül, dessen Struktur die Fluoreszenzemission zulässt, wird als Fluorophor bezeichnet. Die meisten biologischen Proben enthalten vernachlässigbare endogene Fluorophore; daher müssen exogene Fluorophore verwendet werden, um Merkmale hervorzuheben, die für die Probe von Interesse sind. Während der Fluoreszenzmikroskopie wird die Probe mit Licht der entsprechenden Wellenlänge für die Absorption durch das Fluorophor beleuchtet. Bei der Aufnahme eines Photons wird ein Fluorophor als "erregt" bezeichnet und der Absorptionsprozess wird als "Erregung" bezeichnet. Wenn ein Fluorophor Energie in Form eines Photons aufgibt, wird das Verfahren als "Emission" bezeichnet, und das emittierte Photon wird Fluoreszenz genannt.
Der Lichtstrahl, der zur Erregung eines Fluorophors verwendet wird, wird durch die Objektivlinse eines Mikroskops fokussiert und konvergiert an einem "Fokalpunkt", wo er maximal fokussiert ist. Jenseits des Brennpunkts weicht das Licht wieder ab. Die ein- und austretenden Balken können als kegelndes Kegelpaar visualisiert werden, das sich am Brennpunkt berührt (siehe Abbildung 1, linkes Panel). Das Phänomen der Beugung setzt eine Grenze, wie eng ein Lichtstrahl fokussiert werden kann - der Strahl konzentriert sich tatsächlich auf einen Punkt von endlicher Größe. Zwei Faktoren bestimmen die Größe des Brennpunkts: 1) die Wellenlänge des Lichts und 2) die Lichtaufnahmefähigkeit der Objektivlinse, die sich durch ihre numerische Blende (NA)auszeichnet. Der brennpunktige "Spot" erstreckt sich nicht nur in der x-y-Ebene, sondern auch in z-Richtung und ist in Wirklichkeit ein Brennvolumen. Die Abmessungen dieses Brennvolumens definieren die maximale Auflösung, die durch optische Bildgebung erreicht wird. Obwohl die Anzahl der Photonen innerhalb des Brennvolumens am größten ist, enthalten die konischen Lichtpfade oberhalb und unterhalb des Fokus auch eine geringere Dichte von Photonen. Jedes Fluorophor im Lichtpfad kann so angeregt werden. In der konventionellen (Weitfeld-)Epifluoreszenzmikroskopie tragen die Emissionen von Fluorophoren oberhalb und unterhalb der Brennebene zur außerfokussierenden Fluoreszenz bei (ein "hazy background"), was die Bildauflösung und den Kontrast reduziert, wie in Abbildung 1 der rote Würfel, der die Fluorophoremission über der Brennebene (roter Stern) darstellt, die zu einer nicht fokussierten Fluoreszenz führt (oben rechts). Dieses Problem wird in der konfokalen Mikroskopie durch die Verwendung eines Lochs gemildert. (Abbildung 2, unten rechts). Wie in Abbildung 3 dargestellt, ermöglicht das Loch Emissionen, die vom Brennpunkt ausgehen, den Detektor (links) zu erreichen, während die nicht fokussierte Fluoreszenz (rechts) am Erreichen des Detektors gehindert wird, wodurch sowohl die Auflösung als auch der Kontrast verbessert werden.
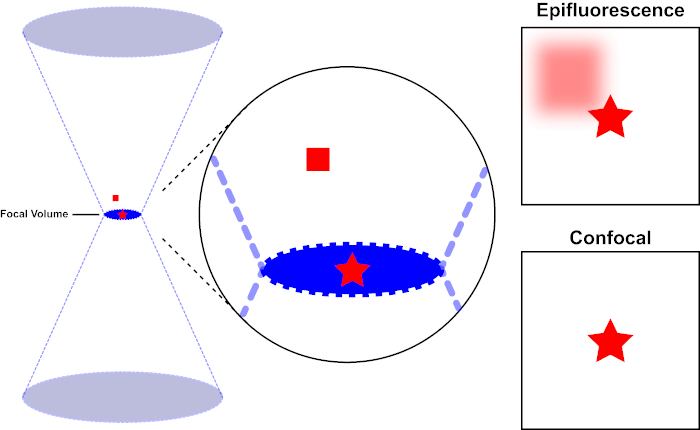
Abbildung 1. Optische Auflösung der Epifluoreszenz versus konfokale Mikroskopie. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Der Lichtstrahl, der zur Anregung eines Fluorophors verwendet wird, wird durch die Objektivlinse eines Mikroskops fokussiert und konvergiert bei einem Brennvolumen und weicht dann ab (links). Der rote Stern stellt die Brennebene einer Probe dar, die abgebildet wird, während das rote Quadrat die Fluorophoremission über der Brennebene darstellt. Bei der Aufnahme eines Bildes dieser Probe mit einem Epifluoreszenzmikroskop wird die Emission des roten Quadrats a-fokus sichtbar und trägt zu einem "hazy background" (oben rechts) bei. Konfokale Mikroskope haben ein Loch, das die Erkennung von Licht verhindert, das außerhalb der Brennebene emittiert wird, wodurch der "hazy background" (unten rechts) eliminiert wird.
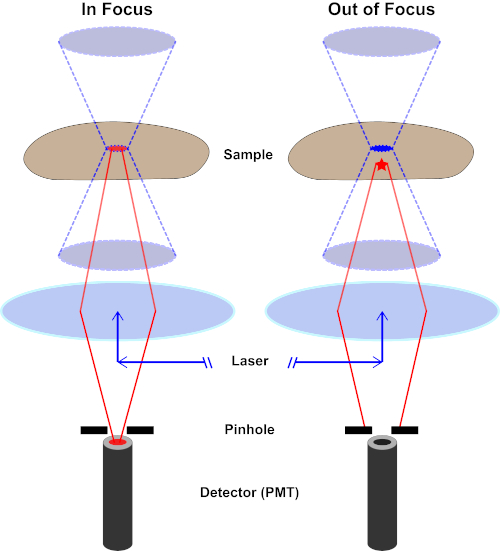
Abbildung 2. Locheffekt in der konfokalen Mikroskopie. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Obwohl die höchste Intensität des Anregungslichts am Brennpunkt der Linse (links, rot oval) liegt, erhalten andere Teile der Probe, die sich nicht im Brennpunkt befindet (rechts, roter Stern), Licht und Fluoreszenz. Um zu verhindern, dass Licht aus diesen nicht fokussierten Bereichen zum Detektor gelangt, befindet sich ein Bildschirm mit einem Loch vor dem Detektor. Nur das von der Brennebene emittende Infokuslicht (links) kann durch das Loch reisen und den Detektor erreichen. Das nicht fokussierte Licht (rechts) wird mit dem Loch blockiert und erreicht den Detektor nicht.
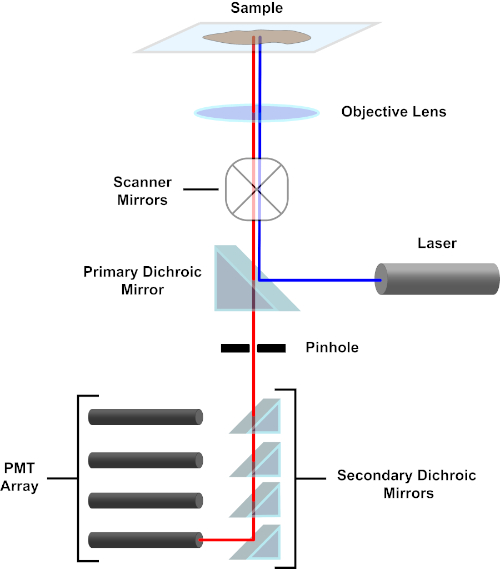
Abbildung 3. Hauptkomponenten eines konfokalen Laserscanmikroskops. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Der Einfachheit halber wird sich die mechanistische Beschreibung eines konfokalen Mikroskops auf die des Nikon Eclipse Ti A1R beschränken. Obwohl es geringfügige technische Unterschiede zwischen verschiedenen konfokalen Mikroskopen geben kann, dient das A1R sowohl als gutes Modell für die Beschreibung der konfokalen Mikroskopfunktion. Der anregungslichtstrahl, der durch eine Reihe von Diodenlasern erzeugt wird, wird vom primären dichroitischen Spiegel in das Objektiv reflektiert, der das Licht auf die abgebildete Probe fokussiert. Der primäre dichroitische Spiegel reflektiert selektiv das Anregungslicht und lässt gleichzeitig Licht bei anderen Wellenlängen passieren. Das Licht trifft dann auf die Scanspiegel, die den Lichtstrahl x-y x-y über die Probe fegen und gleichzeitig ein einzelnes (x,y) Pixel beleuchten. Fluoreszenz, die von Fluorophoren am beleuchteten Pixel emittiert wird, wird von der Objektivlinse erfasst und durchläuft den primären dichroitischen Spiegel, um eine Reihe von Photomultiplierröhren (PMTs) zu erreichen. Sekundäre dichroitische Spiegel leiten das Emissionslicht auf das entsprechende PMT. Von der Probe in das Objektiv gestreutes Anregungslicht wird durch den primären dichroitischen Spiegel zurück zur Probe reflektiert und somit am Eindringen in den Nachweis gehindert. Lichtpfad und erreichen die PMTs (siehe Abbildung 3). Dadurch kann die relativ schwache Fluoreszenz ohne Kontamination durch Licht quantifiziert werden, das vom Anregungslichtstrahl gestreut wird, der typischerweise um Größenordnungen intensiver ist als die Fluoreszenz. Da das Loch Licht von außerhalb des Brennvolumens blockiert, kommt das Licht, das beim Detektor ankommt, von einer schmalen, ausgewählten Z-Ebene. Daher können Bilder aus einer Reihe benachbarter z-Ebenengesammelt werden; diese Bilderserie wird oft als "Z-Stack" bezeichnet. Mit der entsprechenden Software kann ein z-Stackverarbeitet werden, um ein 3D-Bild der Probe zu erzeugen. Ein besonderer Vorteil der konfokalen Mikroskopie ist die Fähigkeit, die subzelluläre Lokalisation der Färbung zu unterscheiden. Zum Beispiel die Unterscheidung zwischen Membranfärbung und intrazellulärer Färbung, die bei der konventionellen Epifluoreszenzmikroskopie (1, 2, 3) sehr anspruchsvoll ist.
Die Probenvorbereitung ist eine wichtige Facette der konfokalen Bildgebung. Eine Stärke der optischen Mikroskopie-Techniken ist die Flexibilität, lebende oder feste Zellen abzubilden. Beim Versuch, 3D-Bilder zu erzeugen, ist die Verwendung fester Zellen aufgrund der Anzahl der Bilder, die für einen Z-Stack erworben werden müssen, der Schwierigkeit, die Zellgesundheit zu erhalten, und der Bewegung von lebenden Zellen und ihren Organellen typisch. Das Verfahren zur Fixierung und Färbung von Zellen zur konfokalen Fluoreszenz ähnelt dem, das konventionell bei der Immunfluoreszenz verwendet wird. Nach der Kultur in Kammerrutschen oder auf Abdeckungen werden Zellen mit Paraformaldehyd fixiert, um die zelluläre Morphologie zu erhalten. Die unspezifische Antikörperbindung wird mit Rinderserumalbumin, Milch oder normalem Serum blockiert. Um die Spezifität der sekundären Antikörper aufrechtzuerhalten, sollte die verwendete Lösung nicht von derselben Art stammen, bei der die primären Antikörper erzeugt wurden. Die Zellen werden mit primären Antikörpern inkubiert, die das Antigen von Interesse binden. Bei der Kennzeichnung mehrerer zellulärer Ziele müssen die primären Antikörper jeweils von einer anderen Spezies abgeleitet werden. Antikörper, die ein Antigen kennzeichnen, werden dann durch fluorophorkonjugierte sekundäre Antikörper gebunden. Fluorophorkonjugierte Sekundärantikörper sollten so ausgewählt werden, dass sie mit den Wellenlängen der Lasererregung im konfokalen Mikroskop kompatibel sind. Bei der Visualisierung mehrerer Antigene sollten sich die Anregungs-/Emissionsspektren der Fluorophore so weit unterscheiden, dass ihre Signale durch mikroskopische Analysen diskriminiert werden können. Die gebeizte Probe wird dann zur Bildgebung auf einem Dia montiert. Ein Montagemedium wird verwendet, um Photobleichungen und Probenaustrocknung zu verhindern. Auf Wunsch kann ein Montagemedium verwendet werden, das einen nuklearen Gegenfleck enthält (z.B. DAPI oder Hoechst) (4).
Im folgenden Protokoll wurden Maus-Fibroblasten, die auf CD1d (LCD1) transfiziert wurden, mit Antikörpern gefärbt, die CD1d und CD107a (LAMP-1) erkennen. CD1d ist ein wichtiger Histokompatibilitätskomplex 1 (MHC 1)-ähnlicher Rezeptor, der auf der Oberfläche von Antigen-Präsentierenden Zellen vorhanden ist, die Lipidantigene darstellen. LAMP-1 (lysosomal assoziiertes Membranprotein-1) ist ein Transmembranprotein, das hauptsächlich in lysosomalen Membranen vorkommt. Für die richtige Antigen-Präsentation wird CD1d durch das niedrige pH-Lysosomalfach geschmuggelt, so dass LAMP-1 als Marker des lysosomalen Fachs für dieses Protokoll verwendet wird. Durch die Untersuchung der LCD1-Zellen mit Anti-CD1d und Anti-LAMP-1, die in verschiedenen Arten hergestellt wurden, können sekundäre Antikörper mit einzigartigen Fluorophoren verwendet werden, um die Lokalisation jedes Proteins in der Zelle zu bestimmen und ob CD1d in der LAMP-1-positiven lysosomalen Fächern.
Procedure
1. Materialien
Puffer
- Waschpuffer: 1 X sterile Phosphat-gepufferte Saline (PBS) ohne Calcium oder Magnesium
- Fixationspuffer: 1% Paraformaldehyd in PBS
- Permeabilisationspuffer: 0,1% Triton X-100 in PBS
- Sperrpuffer: 1% Rinderserumalbumin in PBS
- Zellwachstumsmedium: DMEM ergänzt mit 10% fetalem Rinderserum (FBS), Penicillin/Streptomycin und L-Glutamin
ausrüstung
- Laminare Durchflusshaube
- Befeuchtter Inkubator (37°C, 5%CO2)
- Konfokales Laser-Scanning-Mikroskop; hier, Nikon Eclipse Ti Laser
Materialien und Reagenzien
- Kammerzellenkultur-Dias
- Anti-Fade-Montagemedium mit DAPI (zur Färbung von Kernen)
- Mikroskopabdeckung Glas
- Zarte Aufgabentücher
- Pipettierer und Spitzen
Assay Spezifische Reagenzien
- Anhimierbare Zellen (primäre Zellen oder Zelllinien); hier wurden Maus-Fibroblasten verwendet, die mit CD1d transfiziert wurden (LCD1).
- Primäre Antikörper zum Nachweis zellulärer Ziele; hier wurden Ratten-Anti-Maus-CD107a (LAMP-1) und Maus-Anti-Maus-CD1d verwendet.
- Fluorophorkonjugierte sekundäre Antikörper, die für primäre Antikörper-Isotypen spezifisch sind; hier wurden Ratten-Anti-Ratten-IgG-konjugierte Alexafluor 488 und Anti-Maus-IgG-konjugierte Alexafluor 647 verwendet.
2. Protokoll
Vorbereitung auf Antikörperfärbung
Säzellen
- Setzen Sie die Zellen des Interesses an Wachstumsmedien aus.
- Dann säen Sie 500 l der Zellsuspension/pro Brunnen in die Brunnen einer 4-Well-Kammerrutsche. (Hier wurden LCD1-Zellen mit 2,5x105 Zellen/Kammer in 500 l Wachstumsmedien gesät. Die Saatdichte kann zwischen den Zelllinien variieren).
- Inkubieren Sie die Kammer über Nacht in einem 5% CO2-Inkubator bei 37°C, damit die Zellen am Glas haften können.
- Am nächsten Tag die Medien aus jedem Brunnen aspirieren und dann die Zellen 1X mit 500 L PBS waschen.
fixierung
- Um die Zellen zu fixieren, fügen Sie 500 l 1% Paraformaldehydlösung in jeden Brunnen und inkubieren für 15 min bei Raumtemperatur.
- Nach der Inkubation das Paraformaldehyd in einen geeigneten Behälter für gefährliche Flüssigkeitsabfälle einsammeln.
- Waschen Sie dann die Zellen 3 mal mit 500 L PBS, um alle Reste des Fixiermittels zu entfernen.
Permeabilisierung
- Permeabilisieren Sie die Zellen, indem Sie bei Raumtemperatur 15 Minuten lang mit 500 L Permeabilisationspuffer/Well inkubieren.
- Dann waschen Sie die Zellen kurz 3 mal mit 500 l PBS.
Blockieren
- Inkubieren Sie die Zellen in jedem Brunnen mit 500 l Blockierpuffer für 1 Stunde bei 4°C, um die unspezifische Antikörperbindung zu blockieren.
Primäre Antikörper-Inkubation
- Saugen Sie den Sperrpuffer aus den Schiebekammern.
- Fügen Sie dann den Zellen 500 l verdünnte Primärantikörperlösung hinzu. (Hier wurde Anti-CD107a (LAMP-1) 1:500 verdünnt und Anti-CD1d unverdünnt verwendet (1H6 monoklonaler Antikörper wurde freundlicherweise von Dr. Randy Brutkiewicz zur Verfügung gestellt).)
- Inkubieren Sie die Dias über Nacht bei 4°C.
Hinweis: Wenn Sie nach mehr als einem Ziel suchen, stellen Sie sicher, dass die primären Antikörper unterschiedliche Isotypen sind. Die empfohlenen Antikörperkonzentrationen variieren zwischen den Herstellern und sollten vor der Anwendung titriert werden.
Sekundäre Antikörperinkubation
- Aspirieren Sie die primäre Antikörperlösung aus den Brunnen.
- Waschen Sie die Brunnenkammern 4 mal mit 500 l PBS.
- Fügen Sie dann jedem Brunnen 500 l der verdünnten Sekundärantikörperlösung hinzu. (Hier wurden sowohl die sekundären Antikörper - Anti-Maus-IgG Alexafluor 647 als auch Anti-Ratten-IgG Alexafluor 488 1:2000 im Sperrpuffer) verdünnt.
- Bei Raumtemperatur 1 h im Dunkeln bebrüten.
- Nach der Inkubation die sekundäre Antikörperlösung ansaugen.
- Waschen Sie die Kammern 4 Mal mit 500 L PBS, um alle ungebundenen sekundären Antikörper zu entfernen.
Hinweis: Die empfohlenen Antikörperkonzentrationen variieren je nach Hersteller und sollten vor der Anwendung titriert werden. Bei der Suche nach mehr als einem Ziel müssen sekundäre Antikörper mit verschiedenen Fluorophoren mit einzigartigen Anregungs-/Emissionsspektren konjugiert werden. Beachten Sie auch die Anregung/Emission des nuklearen Gegenflecks (d.h. DAPI) bei der Auswahl der Fluorophore. Die Fluorophorauswahl kann durch die Laserkonfiguration des verwendeten Konfokalmikroskops beeinflusst werden. Die Laserkonfiguration der Maschine bestimmt, welche Fluorophore für das Experiment geeignet sind.
3. Montage von Abdeckungen
- Entfernen Sie zunächst die Kammern vorsichtig von der Rutsche.
- Halten Sie dann die Folie im Winkel über einem empfindlichen Aufgabenwischen und entfernen Sie die Flüssigkeit von den Rändern, ohne die Zellen zu berühren.
- Fügen Sie 1 Tropfen Antifade-Montagemedium, das den kerntechnischen Fleck DAPI enthält, auf jeden Zellabschnitt ein.
- Als nächstes legen Sie einen 20 mm x 60 mm Coverslip über den Schlitten, indem Sie ihn mit den Fingerspitzen über die Kanten halten. (Vermeiden Sie blasenbildung über den Zellen, da sie die Bildgebung stören).
- Wischen Sie jedes zusätzliche Montagemedium an den Seiten mit einem zarten Aufgabenwisch ab und lagern Sie die Dias im Dunkeln bei Raumtemperatur bis zu einer Woche.
4. Konfokale Bildgebung
Bildproben auf einem konfokalen Laserscanmikroskop. Für die in Abbildung 2 dargestellten Daten wurde die Nikon Eclipse Ti A1R mit der Software NIS Elements Advanced Research verwendet. Im folgenden Abschnitt wird das Verfahren zum Erfassen von Bildern mit der oben genannten Software beschrieben.
- Um mit der Bildgebung der Zellen zu beginnen, öffnen Sie die 'NIS Elements Advanced Research Software', indem Sie auf das 'NIS-Softwaresymbol'klicken.
- Als nächstes klicken Sie im Kontrollfenster auf die Registerkarte "TiPad" und wählen Sie das gewünschte Ziel für die Bildgebung aus. (Hier wurde das erste 40x Objektiv verwendet).
- Laden Sie den Dia mit Zellen auf die Bühne und zentrieren Sie ihn unter der Linse.
- Jetzt, auf der Registerkarte 'A1plus Compact GUI', richten Sie die Laser passend für die verwendeten Fluorophore ein. Klicken Sie auf das Zahnradsymbol, um das Farb- und Spektraleinstellungsmenü zu öffnen und die benötigten Kanäle auszuwählen und den Laser für jeden Kanal einzustellen.
- Wählen Sie dann die entsprechenden Emissionen im Dropdown-Menü unter dem ersten dichroitischen Spiegel aus.
- Als nächstes, unter 'A1plus Compact GUI' Fenster, klicken Sie auf 'Ch. Series', um die Line-Channel-Serie einzurichten, die festlegt, ob die verwendeten Laser gleichzeitig oder sequenziell auf die Probe feuern. (Hier wurden sequentielle Pässe gewählt, beginnend mit Kanal 1, gefolgt von Kanal 2, dann 4).
- Danach beginnen Sie mit dem Scannen, indem Sie oben auf das Symbol "Pfeilspitze"klicken. An diesem Punkt, während die Bildgebung live ist, klicken Sie unter 'A1plus Compact GUI' Fenster auf die Schiebeskala und ändern Sie die Lochgröße, um sicherzustellen, dass das Fokuslicht nicht mehr angezeigt wird. (Hier wurde die niedrigste verfügbare Einstellung (0,5) verwendet.
- Passen Sie als Nächstes die Einstellungen "Hochspannung"und"Offset"unter jedem Laser auf entsprechende Werte an, indem Sie die Gleitwaagen verwenden, um die Erkennung der spezifischen Färbung zu ermöglichen und gleichzeitig mögliche Hintergrundfärbungen zu begrenzen. Wenn eine positiv befleckende Probe verfügbar ist, beginnen Sie mit der Abbildung dieser Probe für jeden Kanal, um sicherzustellen, dass die Lasereinstellungen optimale Signal-Rausch-Verhältnisse liefern.
Achtung: Eine hohe Laserintensität über einen längeren Zeitraum kann zu Photobleichungen führen. - Nachdem Sie die optimalen HV- und Offsetwerte für jeden Laser eingestellt haben, klicken Sie auf die Registerkarte "ND-Erfassung"und wählen Sie dann das 'Z'-Symbol aus, um die Parameter für die Z-Serie einzurichten. Als nächstes, beim Erfassen eines Live-Bildes der Probe, setzen Sie zuerst den unteren Rand des Bildes, indem Sie den unteren Rand des Bildes finden und auf die Schaltfläche"unten"klicken, dann finden Sie die obere Position des Beispiels und klicken Sie auf die Schaltfläche "oben". Legen Sie die Schrittgröße fest, indem Sie für jeden Schritt die bevorzugte Schrittgröße in die Größe von m eingeben oder angeben, wie viele Schritte insgesamt erforderlich sind.
- Nachdem die Parameter der Z-Serie eingestellt wurden, wählen Sie die gewünschte Größe/Pixelauflösung des Bildes aus. Klicken Sie dazu auf das Fenster 'A1plus Compact GUI' und wählen Sie unter dem Symbol 'Größe'die gewünschte Auflösung aus. Um das Rauschen des Bildes zu verringern, kann man das Dropdown-Menü neben dem Symbol'' wählen, um die ausgewählte Anzahl von Bildern zu durchschnittlichzumittelieren.
- Klicken Sie nun auf die Registerkarte 'Jetzt ausführen' im' ND-Erfassungsmenü', um mit der Bildgebung des Beispiels zu beginnen.
- Nachdem die Bildgebung abgeschlossen ist, speichern Sie das Bild, indem Sie auf 'Datei' und dann 'Speichern unter' klicken, wodurch die Bilddatei mit der Erweiterung'.nd2'exportiert wird. Wiederholen Sie schließlich den Vorgang für jede der anderen Stichproben.
Die konfokale Fluoreszenzmikroskopie ist eine spezialisierte bildgebende Technik zur Lokalisierung eines Proteins oder Antigens von Interesse in einer Zell- oder Gewebeprobe, indem das Antigen mit einem antikörperkonjugierten Fluoreszenzfarbstoff gekennzeichnet und das fluoreszierende Signal erkannt wird. Es bietet eine höhere räumliche Auflösung als weiträumige Fluoreszenzmikroskopie, mit Hilfe von zwei Lochlöchern, die an den Brennebenen der Objektivlinse platziert werden, was ihm den Namen konfokale gibt. Es ermöglicht Benutzern, die Färbung auf subzellulärer Ebene zu visualisieren, wie die Unterscheidung zwischen Oberflächenmembranfärbung von intrazellulärer Färbung.
Ein konfokales Mikroskop folgt einem ähnlichen Grundprinzip wie ein klassisches Fluoreszenzmikroskop. Der Strahl einer Lichtquelle, in der Regel ein Laser für konfokale, wird von einem dichroitischen Spiegel reflektiert und durch eine Objektivlinse auf die Probe fokussiert. Dieses Licht regt die Fluorophore zu einer anderen Wellenlänge an, die durch die Objektivlinse und den dichroitischen Spiegel zu einer Kamera oder einem Okular zurückfließt.
Die verbesserte Auflösung eines konfokalen Mikroskops ist hauptsächlich auf das Vorhandensein von zwei Lochlöchern zurückzuführen, bei denen es sich um sehr kleine Löcher handelt, die Licht auf den Anregungs- und Emissionslichtpfaden passieren kann. Die Lochlöcher werden strategisch auf der Brennebene der Objektivlinse platziert. Wechseln wir nun zu einem Seitenansichtsschema der Mikroskopanordnung, um den Lichtpfad zu überprüfen. Nach dem Durchlaufen des Anregungslochs hat der Anregungslichtstrahl den Effekt, dass er von einem Brennpunkt ausgeht, was es der Objektivlinse ermöglicht, das Licht dann auch auf einen Punkt auf die Probe zu fokussieren. Der Emissionsstrahl von diesem Brennpunkt konvergiert am Emissionsloch, wodurch er passieren kann. Während der Anregung sind die Fluorophore innerhalb des Lichtweges oberhalb und unterhalb des Brennpunkts auch leicht aufgeregt. Während das vom Brennpunkt ausgehende Emissionslicht durch das Loch geht, konvergieren die Emissionen der out-of-fokus-Punkte vor oder nach dem Emissionsloch und werden daher blockiert, was zu einer reduzierten Hintergrundfluoreszenz führt.
Der Anregungs-Emissions-Erkennungszyklus muss für jeden Bildpunkt im Interessenbereich wiederholt werden, was auf verschiedene Weise erfolgen kann. Beispielsweise verwendet das Laserscanning confocal galvanometer-Scanning-Spiegel, die das Anregungslicht in verschiedenen Winkeln ablenken. Daher den Lichtstrahl über die Probe in der XY-Ebene zu fegen. Spinning Disc konfokale verwendet eine Scheibe mit einer Reihe von Lochlöchern, die sich dreht, um die Anordnung der Pinholes zu verschieben. Auf diese Weise können Benutzer jedes Mal mehrere kleine Bildstellen in der Probe beleuchten und nach und nach den gesamten Bereich abdecken, während sich die Scheibe dreht. Durch die Lochlöcher stellt das XY-Bild am Detektor eine schmale Z-Ebene dar. Daher können Bilder aus einer Reihe aufeinander folgender Z-Ebenen gesammelt werden, die oft als Z-Stack bezeichnet werden. Aus diesen Bildern kann eine entsprechende Software eine 3D-Darstellung des Fluoreszenzsignalmusters in der Probe erzeugen.
In diesem Protokoll beobachten Sie die Immunostainierung von Maus-Fibroblasten, gefolgt von einer Bildgebung auf einem konfokalen Mikroskop, um ein Zelloberflächenprotein und ein lysosomales Protein differenziell zu visualisieren.
Um mit sterilen Techniken zu beginnen, die Zellen von Interesse in 500 Mikroliter Wachstumsmedien pro Brunnen wieder auszusetzen, und sie dann in die Brunnen einer Vier-Brunnen-Kammerrutsche säen. Hier verwenden wir Maus-Fibroblasten, die transfiziert wurden, um das antigenpräsentierende Molekül CD1d auszudrücken. Damit die Zellen am Glas haften können, schieben Sie die Kammer in einen 5% Kohlendioxid-Inkubator bei 37 Grad Celsius und inkubieren Sie sie über Nacht. Am Morgen, aspirieren Sie die Medien aus jedem Brunnen, und dann waschen Sie die Zellen einmal mit 500 Mikroliter PBS für ein paar Sekunden.
Um die Zellen zu fixieren, fügen Sie 500 Mikroliter 1% Paraformaldehydlösung in jeden Brunnen ein und inkubieren Sie 15 Minuten bei Raumtemperatur. Nach der Inkubation das Paraformaldehyd in einen geeigneten Behälter für gefährliche Flüssigkeiten sammeln und dann alle Reste des Fixativs entfernen, indem Sie die Zellen dreimal mit PBS für ein paar Sekunden waschen.
Um das Eindringen von Antikörpern in die Zellen zu ermöglichen, fügen Sie jedem Bohrkörper 500 Mikroliter Permeabilisationspuffer hinzu und brüten sie 15 Minuten bei Raumtemperatur auf der Bank. Nach der Permeabilisation die Zellen mit 500 Mikroliter PBS kurz waschen. Als nächstes fügen Sie 500 Mikroliter Blockierpuffer zu jedem Brunnen hinzu und brüten für eine Stunde bei vier Grad Celsius, um eine unspezifische Antikörperbindung zu verhindern.
Bereiten Sie die primären Antikörper, Anti-CD1d und Anti-LAMP-1, in entsprechenden Arbeitskonzentrationen vor. Dann den Puffer aus den Brunnen ansaugen und die Zellen in jedem Brunnen mit 500 Mikrolitern verdünnter Primärantikörperlösung bedecken und dann den Schlitten über Nacht bei vier Grad Celsius auf einer ebenen Oberfläche bebrüten. Am nächsten Morgen verdünnen Sie die sekundären Antikörper, in diesem Fall einen Anti-Maus- und Rattenanti-Antikörper mit deutlichen Fluoreszenz-Tags, im Blockierpuffer zu entsprechenden Arbeitskonzentrationen. Als nächstes die primäre Antikörperlösung aus den Brunnen aspirieren und dann die Zellen viermal mit 500 Mikroliter PBS waschen. Dann 500 Mikroliter der verdünnten Sekundärantikörperlösung zu jedem Brunnen hinzufügen und bei Raumtemperatur für eine Stunde im Dunkeln inkubieren. Nach der Inkubation die sekundäre Antikörperlösung ansaugen und die Brunnen viermal mit 500 MikroliterPBS waschen, um alle ungebundenen Sekundärantikörper zu entfernen.
Um die Proben nach der letzten Wäsche zu montieren, lösen Sie die Kammern vorsichtig und entfernen Sie sie von der Rutsche. Um die verbleibende PBS zu entfernen, halten Sie die Folie in einem Winkel über einem empfindlichen Aufgabenwisch, und entfernen Sie die Flüssigkeit von den Rändern, ohne die Zellen zu berühren. Sobald die überschüssige PBS entfernt wurde, fügen Sie einen Tropfen Antifade-Montagemedium, das den kerntechnischen Fleck DAPI enthält, auf jeden Zellabschnitt ein. Nehmen Sie als Nächstes einen 20 mal 60-Millimeter-Abdeckungsslip, und mit nur Fingerspitzen beginnen Sie, den Coverslip langsam auf beiden Kanten zu senken, wobei darauf zu achten ist, dass Blasenbildung über den Zellen vermieden wird. Wischen Sie jedes zusätzliche Montagemedium auf den Dias mit einem zarten Aufgabenwisch ab und lagern Sie die Dias bei Raumtemperatur bis zu einer Woche im Dunkeln.
Um mit der Abbildung der Zellen zu beginnen, klicken Sie zuerst auf das NIS-Softwaresymbol auf dem Desktop. Sobald Sie im Kontrollfenster sind, klicken Sie oben auf die Registerkarte TiPad, und wählen Sie das gewünschte Ziel für die Bildgebung aus. Laden Sie dann den Dia mit Zellen auf die Bühne und zentrieren Sie ihn unter der Linse. Als nächstes richten Sie auf der Registerkarte A1plus Compact GUI neben dem TiPad-Tab die Fürdier verwendeten Laser ein. Klicken Sie auf das Zahnradsymbol, um das Menü Farb- und Spektraleinstellungen zu öffnen. Sobald das Menü farb- und spektrale Einstellungen geöffnet ist, wählen Sie die benötigten Kanäle aus und stellen Sie den Laser für jeden Kanal ein. Wählen Sie dann die entsprechenden Emissionen im Dropdown-Menü unter dem ersten dichroitischen Spiegel aus. Klicken Sie anschließend unter dem A1plus Compact GUI-Fenster auf Ch.Series, um die Linienkanalserie einzurichten, die festlegt, ob die verwendeten Laser gleichzeitig oder sequenziell auf die Probe feuern.
Danach beginnen Sie mit dem Scannen, indem Sie oben auf das Pfeilspitzensymbol klicken. An dieser Stelle, während die Bildgebung live ist, klicken Sie unter dem A1plus Compact GUI-Fenster auf die Schiebeskala, und ändern Sie die Lochgröße, um die Begrenzung von Nichtfokuslicht zu gewährleisten. Passen Sie als Nächstes die Hochspannungs- und Offseteinstellungen unter jedem Laser auf entsprechende Ebenen an, indem Sie die Gleitwaagen verwenden, um die Erkennung der spezifischen Färbung zu ermöglichen und gleichzeitig mögliche Hintergrundfärbungen einzuschränken. Wenn eine positive Färbeprobe verfügbar ist, beginnen Sie mit der Abbildung dieser Probe für jeden Kanal, um sicherzustellen, dass die Lasereinstellungen optimale Signal-Rausch-Verhältnisse ergeben. Nachdem Sie die optimalen HV- und Offsetwerte für jeden Laser eingestellt haben, klicken Sie auf die Registerkarte ND-Erfassung, und wählen Sie dann das Z-Symbol aus, um die Parameter für die Z-Serie einzurichten.
Wenn Sie als Nächstes ein Livebild des Beispiels abrufen, legen Sie zuerst den unteren Bereich des Bildes fest, indem Sie den unteren Rand des Bildes finden und auf die untere Schaltfläche klicken. Suchen Sie dann die obere Position des Beispiels und klicken Sie auf die obere Schaltfläche. Legen Sie die Schrittgröße fest, indem Sie die bevorzugte Schrittgröße für jeden Schritt in Mikrometer eingeben oder angeben, wie viele Schritte insgesamt erforderlich sind. Um die gewünschte Größe/Pixelauflösung des Bildes auszuwählen, klicken Sie auf das Aiplus Compact GUI-Fenster, und wählen Sie unter dem Größensymbol die gewünschte Auflösung aus.
Um das Rauschen des Bildes zu verringern, können Sie das Dropdown-Menü neben dem Theta-Symbol auswählen, um die ausgewählte Anzahl von Bildern zu durchschnittlich zu haben. Klicken Sie anschließend im Menü ND-Erfassung auf die Registerkarte Jetzt ausführen, um mit der Bildgebung des Beispiels zu beginnen. Nachdem die Bildgebung abgeschlossen ist, speichern Sie das Bild, indem Sie auf Datei klicken, und speichern Sie dann unter, wodurch die Bilddatei mit der Erweiterung dot-nd2 exportiert wird. Wiederholen Sie schließlich den Vorgang für jede der anderen Stichproben.
In diesem Experiment wurden Maus-Fibroblasten, die das Oberflächenglykoprotein-Gen CD1d exzessierten, fixiert, immungefärbt und auf einem konfokalen Mikroskop abgebildet. Dieses Bild zeigt einen einzelnen Abschnitt eines Z-Stacks bei 40-facher Vergrößerung, wobei CD1d rot gefärbt ist. Die Probe wurde mit LAMP-1, einem lysosomalen Marker, in Grün gecostainiert. Kernfleck DAPI wurde verwendet, um die Kerne der Zellen zu zeigen.
In einem zusammengesetzten Bild, in dem die drei verschiedenen Kanäle zusammengeführt werden, ergibt sich das Auftreten von Gelb aus der Überlappung der roten und grünen Kanäle und zeigt einen Bereich an, in dem CD1d und LAMP-1 in den Lysosomen kolokalisiert sind. Bereiche, in denen nur eine Farbe vorhanden ist, zeigen das Vorhandensein von CD1d oder LAMP-1 ohne Kolokalisierung an. Dieses Bild zeigt eine 3D-Rendering der Zellen aus Bildern im Z-Stack aufgenommen erstellt und diese Methode ermöglichte die Konstruktion einer Seitenansicht dieser Gruppe von Zellen. Die folgende Abbildung zeigt ein Segment aus dem Z-Stack bei 100-facher Vergrößerung, das die Expressionsmuster dieser beiden Proteine genauer demonstriert. Das rosa umrissene Feld auf der rechten Seite des Bildes zeigt den Querschnitt der x-Koordinate an, die durch die rosa Linie im Bild gekennzeichnet ist, die die Seitenansicht an der rosa Linie darstellt. In ähnlicher Weise zeigt das blau umrissene Feld am unteren Rand des Bildes den Querschnitt der y-Koordinate, der durch die blaue Linie im Bild bezeichnet wird, die die Vorderansicht der blauen Linie darstellt. Das 3D-Rendering des Z-Stack-Bildes ermöglicht es Benutzern, das Bild in 3D anzuzeigen und alle x-, y- und z-Ebenen zu visualisieren. Dies kann verwendet werden, um die Kolokalisierung der verschiedenen Flecken an verschiedenen Regionen innerhalb der Zelle zu untersuchen.
Subscription Required. Please recommend JoVE to your librarian.
Results
In diesem Experiment wurden Maus-Fibroblasten, die das Oberflächenglykoprotein-Gen CD1d exzessierten, fixiert, immungefärbt und auf einem konfokalen Mikroskop abgebildet. Abbildung 4 zeigt ein repräsentatives Bild, das mit dem obigen Protokoll erstellt wurde. Im oberen Bereich von A werden einkanalige Bilder dargestellt, die das Färbemuster jedes einzelnen Ziels zeigen. Diese Bilder bestehen aus einem einzelnen Abschnitt (Slice) des z-Stacks, der erfasst wurde. Das rechte Panel zeigt die DAPI-Färbung von Kernen der Zellen. Die mittelzentralen Paneele zeigen CD1d in rot und LAMP-1, ein lysosomaler Marker, grün gebeizt. Das linke Bedienfeld ist ein zusammengesetztes Bild, in dem die drei verschiedenen Kanäle zusammengeführt werden. Das Auftreten von Gelb ergibt sich aus überlappenden roten und grünen Kanälen und zeigt einen Bereich an, in dem CD1d und LAMP-1 gemeinsam lokalisiert sind. Die Ergebnisse der Färbung bestätigen, dass CD1d in den LAMP-1+ endosomalen Fächern lokalisiert ist. Es gibt auch Bereiche, in denen nur eine Farbe vorhanden ist, was auf das Vorhandensein von CD1d oder LAMP-1 ohne Kolokalisierung hinweist. Die untere Seite von A zeigt eine 3D-Rendering der Zellen aus Bildern im Z-Stack aufgenommen konstruiert.
Panel B zeigt eine Scheibe aus dem Z-Stack bei 100-facher Vergrößerung, die die Expressionsmuster dieser beiden Proteine genauer demonstriert. Das rosa umrissene Feld auf der rechten Seite des Bildes zeigt den Querschnitt der x-Koordinate an, der durch die rosa Linie im Bild gekennzeichnet ist, die die Seitenansicht an der rosa Linie darstellt. In ähnlicher Weise zeigt das blau umrissene Feld am unteren Rand des Bildes den Querschnitt der y-Koordinate, der durch die blaue Linie im Bild bezeichnet wird, die die Vorderansicht der blauen Linie darstellt. Das 3D-Rendering des Z-Stack-Bildes ermöglicht es Benutzern, das Bild in 3D anzuzeigen und alle x-, y- und z-Ebenen zu visualisieren.

Abbildung 4: Färbung von CD1d und LAMP1. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
A, oberes Panel: LCD1-Zellen wurden fixiert, permeabilisiert und mit Antikörpern gegen CD1d (rot) und LAMP-1 (grün, ein Marker des lysosomalen Fachs) gefärbt. DAPI (blau, wurde verwendet, um den Kern zu visualisieren). Das Merge (linkes Bedienfeld) zeigt an, dass CD1d im LAMP-1-positiven späten endosomalen/lysosomalen Fach (gelb) lokalisiert ist.
A, unterer Bereich: 3D-Rendering der gleichen Zellen im oberen Bereich. Die Bilder wurden mit einem 40-fachen Öl-Immersion-Objektiv auf der Nikon Eclipse Timit der Software NIS Elements Advanced Research aufgenommen.
B: 100x Bild von LCD1d-Zellen, die wie in A gebeizt sind, mit Stapelinformationen für eine bestimmte y-Koordinate (bezeichnet durch die blaue Linie) am unteren Rand des Bildes (blaue Box). Die Stapelinformationen für eine bestimmte x-Koordinate (bezeichnet durch die rosa Linie) werden auf der rechten Seite des Bildes angezeigt (rosa Box).
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
Konfokale Fluoreszenzfärbung ist ein relativ einfaches Verfahren, das zu extrem hochwertigen Bildern von Proben führt, die ähnlich wie bei der herkömmlichen Fluoreszenzmikroskopie hergestellt werden. Kurz gesagt, Proben werden fixiert, permeabilisiert und dann blockiert. Primäre Antikörper gegen ein Protein oder Proteine von Interesse dürfen binden, dann werden fluorophorkonjugierte sekundäre Antikörper verwendet, um die Färbung zu visualisieren. Die konfokale Fluoreszenzmikroskopie hat Anwendungen in vielen Forschungsbereichen. Zum Beispiel kann durch Die Färbung von Markern subzellulärer Organellen zusammen mit einem Protein von Interesse die konfokale Mikroskopie verwendet werden, um die subzellulären Standorte verschiedener Proteine zu bestimmen. Im Vergleich zur herkömmlichen Fluoreszenzmikroskopie kann die konfokale Bildgebung effektiver zwischen Zelloberfläche und intrazellulärer Position eines Proteins unterscheiden. Darüber hinaus kann die konfokale Bildgebung auch verwendet werden, um zu bestimmen, ob zwei Proteine innerhalb der Zelle kolokalisieren. Obwohl in diesem Protokoll nicht beschrieben, kann die konfokale Fluoreszenzmikroskopie auch an lebenden Zellen durchgeführt werden, um dynamische Veränderungen zu erkennen.

Video 1: Video, das in der NIS Elements Advanced Research-Software erstellt wurde und die Möglichkeit hervorhebt, sich durch das 3D-Rendering der Bilder zu bewegen. Bitte klicken Sie hier, um dieses Video anzusehen (Rechtsklick zum Download).
Subscription Required. Please recommend JoVE to your librarian.
References
- Claxton, N. S., Fellers, T. J. and Davidson, M. W. Laser scanning confocal microscopy. Department of Optical Microscopy and Digital Imaging, National High Magnetic Field Laboratory, Florida State University, 37 p., Unpublished (2010). Available at- http://www.vertilon.com/pdf/PP6207.pdf.
- Ojcius, D. M., Niedergang, F., Subtil, A., Hellio, R. and Dautry-Varsat, A. Immunology and the confocal microscope. Research in Immunology, 147 (3),175-88 (1996).
- Paddock, S. W. and Eliceiri K. W. Laser scanning confocal microscopy: history, applications, and related optical sectioning techniques. Methods in Molecular Biology, 1075, 9-47 (2014).
- Hoff. F. How to prepare your specimen for immunofluorescence microscopy. Philipps University Marburg, Institute of Cytobiology and Cytopathology, Germany. (2015) Available at- http://www.leica-microsystems.com.