



Overview
Fuente: Susannah C. Shissler1, Tonya J. Webb1
1 Departamento de Microbiología e Inmunología, Universidad de Maryland, Baltimore, MD 21201
La inmunoprecipitación (IP, también conocida como ensayo "pull-down") es una técnica ampliamente utilizada que tiene aplicaciones en una variedad de campos. Concebido por primera vez en 1984, fue refinado en 1988 (1, 2). El objetivo fundamental de la P.I. es la purificación y el aislamiento de una proteína específica utilizando un anticuerpo contra esa proteína. La palabra "inmuno" se refiere al uso de un anticuerpo, mientras que la palabra "precipitación" se refiere a extraer una sustancia específica de una solución. La proteína diana puede ser endógena o recombinante. La mayoría de las proteínas recombinantes tienen una etiqueta de epítopo (es decir, mico o bandera) unida a ellas para simplificar la purificación posterior. Típicamente, es más fácil optimizar la proteína recombinante IP porque los anticuerpos contra las etiquetas de epítopos recombinantes son muy fuertes y eficaces. Los anticuerpos contra las proteínas endógenas tienen una eficacia extremadamente variable, lo que hace mucho más difícil optimizar estas IP. Un paso necesario después de la inmunoprecipitación es la verificación de la purificación. La proteína aislada se resuelve utilizando SDS-PAGE y posteriormente sondeada para la pureza por manchas occidentales (Figura 1). Un control importante es el uso de un anticuerpo diferente durante la mancha occidental para verificar el tirón de la proteína correcta. La combinación de IP con técnicas posteriores es una poderosa herramienta de análisis. El objetivo después de la purificación puede ser la caracterización de la proteína en sí por RMN, espectrometría de masas y ensayos in vitro, o el análisis de los socios que interactúan con la proteína (es decir, proteína, ADN, ARN) (3, 4, 5).
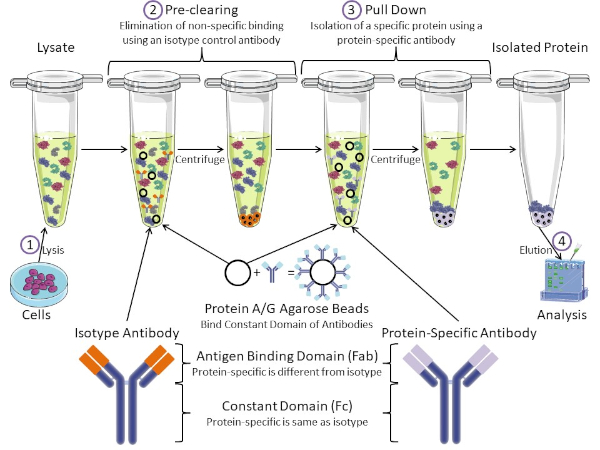
Figura 1: Visión general del procedimiento de inmunoprecipitación. La inmunoprecipitación es el aislamiento de una proteína específica utilizando un anticuerpo. Después de la producción de lisado a partir de células, hay dos pasos principales: pre-clearing y pull down. Durante el paso de pre-limpieza, los lisados celulares se eliminan previamente de proteínas que se unen a los anticuerpos no específicamente utilizando un anticuerpo de control de isotipo. En el paso de tracción hacia abajo, la proteína diana se tira hacia abajo usando un anticuerpo específico de la proteína. La proteína aislada es entonces analizada por western blot. Los anticuerpos isotipo y los anticuerpos específicos de proteínas tienen el mismo dominio constante, pero diferentes dominios de unión de antígenos. Un componente clave de este protocolo son las perlas de agarosa de proteína A/G que unen el dominio constante de los anticuerpos, lo que permite la inmunoprecipitación de la proteína diana. Haga clic aquí para ver una versión más grande de esta figura.
Los anticuerpos son el componente clave de una inmunoprecipitación que la diferencia de otras formas de purificación de proteínas (es decir, purificación de columnas de afinidad de níquel). Los anticuerpos son moléculas producidas por células B que pueden reconocer epítopos proteicos específicos. Los anticuerpos tienen dos dominios: constante (Fc) y enlace de antígeno (Fab) (Figura 1). El dominio constante identifica el tipo de anticuerpo y dicta la función in vivo. Por lo general, los dominios constantes de los anticuerpos utilizados para IP son IgG de ratón, rata o conejo. La porción de unión al antígeno del anticuerpo reconoce un epítopo específico de una proteína específica. Los anticuerpos pueden reconocer epítopos en proteínas plegadas que pueden no existir cuando la proteína está desnaturalizada y viceversa. Por lo tanto, la disponibilidad del epítopo depende del plegado de proteínas, identificando un factor importante a tener en cuenta a la hora de elegir anticuerpos y condiciones para la P.I.
Tanto los sistemas procaario como el eucariota tienen proteínas de unión a anticuerpos. En los sistemas eucariotas, el propósito es la protección inmune contra las bacterias mientras que en los sistemas procariotas, el propósito es la protección del sistema inmunológico. Las proteínas de unión a anticuerpos afectan a la metodología IP de dos maneras. En primer lugar, hay un paso necesario de pre-limpieza (Figura 1) para eliminar el lisado de proteínas que unen anticuerpos - reduciendo así la unión no específica en el producto final. Este paso utiliza un anticuerpo isotipo que tiene el mismo dominio constante que un dominio de unión de anticuerpos diferente al de su anticuerpo específico de proteína. Las proteínas bacterianas de unión a anticuerpos son el segundo componente clave de este método. Después de que el anticuerpo específico de la proteína une la proteína diana, el anticuerpo: complejo proteico debe ser arrastrado hacia abajo (Figura 1). Las proteínas A, G y L son proteínas bacterianas que unen el dominio constante de los anticuerpos. Mientras que las bacterias utilizan esto para subvertir el sistema inmunológico, los investigadores han cooptado este sistema para una fácil purificación de anticuerpos, y se utiliza durante los pasos previos a la limpieza y el tirón. Estas proteínas tienen diferentes afinidades de unión para diferentes especies y diferentes subtipos de dominio constante - otro factor a tener en cuenta a la hora de elegir condiciones para la P.I. Muchas empresas venden cuentas de agarosa etiquetadas con proteína A/G (Figura 1), columnas de espín prefabricadas o resinas para hacer columnas. En general, las perlas y las columnas de espín se utilizan para tamaños de muestra más pequeños, mientras que las resinas se utilizan para la purificación a granel.
En este ejercicio de laboratorio, demostramos cómo purificar la proteína endógena c-myc, a partir de timocitos murinoprimarios primarios, utilizando la técnica básica de inmunoprecipitación basada en la proteína A/G Plus. El protocolo comienza a partir de la preparación del lisado celular y termina con la verificación de la extracción exitosa de proteína hacia abajo usando el análisis de la mancha occidental.
Procedure
1. Inmunoprecipitación con abalorios de agarosa de proteína A/G PLUS
Preparación de lisato celular
- Centrifugar 108 timiocitos en una microcentrífuga a 13.000 rpm durante 3 min y retirar el sobrenadante.
Nota: El número de células variará dependiendo de los niveles de expresión de la proteína deseada y el tipo de célula elegido. - Vuelva a suspender las celdas en el búfer de lisis de 500 l RIPA con PMSF.
- Interrumpe las células usando unos pulsos rápidos con un vórtice y luego aspira el lisado unas cuantas veces con una aguja de 25 G unida a una jeringa.
Nota: Evite crear burbujas. Utilice una aguja más grande, como una aguja 21G para tipos de células más grandes. - Incubar el lisado celular sobre hielo durante 10 minutos.
- Centrifugar el lisado a 13.000 rpm durante 15 min a 4oC.
- Transfiera el sobrenadante a un tubo de microcentrífuga fresco y etiquetado.
Pre-clearing
- Añadir cuentas de agarosa de 20 l de proteína A/G PLUS y 1 g de un anticuerpo de control de isotipo (aquí, se utiliza el anticuerpo de control de isotipo IgG1 de ratón), al lisado.
Nota: La elección del anticuerpo isotipo utilizado dependerá del anticuerpo específico de proteína utilizado más adelante en el paso desplegable. - Incubar la mezcla de lisado en un rotador centrífugo en la cámara fría (4oC) durante 30 min.
- Centrifugar la muestra a 3200 rpm durante 30 s a 4oC.
- Transfiera el sobrenadante pre-claro a un tubo de microcentrífuga fresco, etiquetado, de 1,5 ml. Deseche el pellet.
Determinación de la concentración de proteínas
- Determinar la concentración de proteína del lisado celular mediante la realización de un ensayo de Bradford.
- Reactivo Bradford Aliquot 1000 l en 7 tubos de microcentrífuga.
- Añadir las siguientes cantidades de proteína BSA estándar (2 mg/ml) en 6 de los tubos (Tabla 1).
| Número de tubo | Volumen de la AB (L) (2 mg/ml) | Concentración de proteínas (g/L) |
| 1 | 0 | 0 |
| 2 | 1 | 2 |
| 3 | 2 | 4 |
| 4 | 3 | 6 |
| 5 | 4 | 8 |
| 6 | 5 | 10 |
Tabla 1: Cantidades estándar de proteína BSA
- En eltubo 7, añadir 1 l del lisado pre-claro.
Nota: Para asegurarse de que la concentración de la muestra se encuentra dentro del rango de detección de ensayo, prepare y analice también una dilución de lisado 1:2 o 1:5. - Coloque 200 ml de cada uno de los 7 tubos en pozos individuales de una placa de fondo plano de 96 pocillos repitiendo cada muestra en triplicado.
- Lea la placa en un lector de placas a 595 nm.
- Genere la curva estándar en Excel y calcule la concentración de proteínas del lisado pre-borrado.
Tire hacia abajo
- Etiquete dos tubos de microcentrífuga nuevos de 1,5 ml, uno como "control" y otro como "prueba", que en este ejemplo es c-myc.
- Coloque 500 g de lisado pre-claro en cada uno de estos tubos.
Nota: La cantidad de proteína utilizada aquí dependerá de la cantidad de la proteína deseada para ser purificada. - Traiga el volumen total para cada tubo hasta 500 l utilizando el tampón de lisis.
- Añadir 2 g de anticuerpo anti-c-mico al tubo del grupo de ensayo y 2 g de anticuerpo de control de isotipo IgG1 del ratón al grupo de control.
Nota: La cantidad de anticuerpos dependeráde la eficacia de los anticuerpos y de la cantidad de la proteína diana. - Incubar los tubos de un rotador en la cámara frigorífica (4oC) durante 2 h.
- Añadir 20 éL de proteína A/G PLUS perlas de agarosa a cada tubo.
Nota: Es aconsejable utilizar puntas de pipeta con el extremo cortado para evitar daños en las perlas. - Incubar sobre un rotador en cámara frigorífica (4oC) durante la noche.
Nota: Dependiendo de la proteína diana y la eficacia de los anticuerpos, este paso puede variar de 1 h a la noche. - Centrifugar los tubos a 3200 rpm a 30 s 4oC para tirar hacia abajo de las perlas.
- Aspirar el sobrenadante de cada tubo.
Nota: La proteína diana ahora está unida a las cuentas. - Lave las perlas dos veces con PBS de 500 L 1X Dulbecco.
- Centrifugar los tubos a 3200 rpm durante 30 s 4oC.
Nota: Para un lavado más estricto, utilice búferes más estrictos, como RIPA. - Aspirar el tampón de cada tubo. Usando puntas de carga de gel, elimina cualquier tampón sobrante de las cuentas y mantén las cuentas en hielo para eluir la proteína.
Nota: En este ejemplo, la proteína se eluye en el búfer de ejecución SDS-PAGE hirviendo las perlas, para el análisis de manchas occidentales. Este enfoque es adecuado para verificar los resultados de la P.I. o para examinar las interacciones proteínas y proteínas. Para otras aplicaciones posteriores, como la purificación de proteínas para análisis estructurales o enzimáticos, se utilizan sistemas más sofisticados, como etiquetas de epítopos (etiqueta de bandera o etiqueta micálica) para evitar la elución del anticuerpo con la proteína de interés.
2. Verificación de la P.I. a través del análisis de Western Blot
Electroforesis SDS-PAGE:
- Vuelva a suspender las perlas en un tinte de carga SDS-PAGE de 20 l que contenga el mercapto-etonol.
- Hervir las muestras a 95 oC durante 5 min.
- Centrifugar las perlas a 13.000 rpm durante 10 s a temperatura ambiente.
- Usando puntas de carga de gel, entube cuidadosamente las muestras obtenidas de las cuentas y cárguelas en pozos de 4-15% de gradiente SDS-PAGE gel.
- Además de las muestras, cargue un carril con una escalera de proteínas, así como un carril con el islapreo pre-despejado para servir como control de carga.
- Corre a 100 V hasta que el frente del tinte alcance la parte inferior del gel (1h).
Análisis de Western Blot:
- Haga un sándwich western blot, asegurándose de que la membrana PVDF esté entre el gel y el cátodo rojo.
- Traslado por 1 h a 100 V.
- Coloque la membrana en tampón de bloqueo de 5 ml a temperatura ambiente durante 1 h en un balancín en un ajuste bajo, para bloquear los sitios de unión a proteínas no específicos.
Nota: Es posible que sea necesario aumentar los volúmenes de tampón de bloqueo, anticuerpoprimario, anticuerpo secundario y lavados para manchas de mayor tamaño. - Incubar la mancha con un anticuerpo anti-c-mico de 5 ml en el tampón de bloqueo durante la noche a 4oC en balancín en un ajuste bajo.
Nota: El anticuerpo utilizado aquí debe ser diferente del utilizado en el paso desplegable. - Lave la mancha 3-6 veces usando 5 ml de TBST con cada lavado siendo 5 min a temperatura ambiente en un balancín en un ajuste bajo.
- Incubar la mancha con anticuerpo secundario de cadena ligera anticonejo con etiqueta HRP en tampón de bloqueo, durante 1 h a temperatura ambiente en balancín a un ajuste bajo.
Nota: La elección del anticuerpo secundario dependerá del anticuerpo primario utilizado para la mancha occidental. Además, una cadena ligera específica secundaria se utiliza en el protocolo porque la proteína diana está cerca en peso molecular de la cadena pesada del anticuerpo. Si la proteína diana está cerca de 50kDa, utilice una cadena ligera específica secundaria. Si la proteína diana está cerca de 25kDa, utilice y una cadena pesada específica secundaria. - Lave la mancha 3-6 veces usando 5 ml de TBST con cada lavado siendo 5 min a temperatura ambiente en un balancín en un ajuste bajo.
- Retire el líquido de la mancha y el borde de la mancha en las toallitas de laboratorio para eliminar el exceso de líquido.
- Cubra la mancha con 1 x reactivo de detección quimioluminiscente' e incubar durante 1 min.
Nota: Los siguientes pasos deben realizarse en rápida sucesión, ya que el reactivo de detección es sensible a la luz y al tiempo. - Dab borde de mancha en toallitas de laboratorio para eliminar el exceso de reactivo de detección.
- Coloque la mancha en la superficie de imagen de la bandeja Imager.
Nota: Las manchas quimioluminiscentes también se pueden visualizar utilizando película. - Imagen usando el 'Programa Chemiluminescent' para capturar múltiples puntos de tiempo de 10 s a 5 min.
Nota: El tiempo óptimo puede cambiar en función de la cantidad de proteínas y la calidad de los anticuerpos. - Elija una imagen con una visibilidad óptima de la banda y, a continuación, exporte esa imagen.
- Antes de mover la mancha, tome una foto de la mancha usando el imager, para capturar la ubicación de la escalera. Luego, exporta esa imagen también.
- Con un software de preparación de diapositivas (como PowerPoint), alinee las bandas y las imágenes de escalera para formar una sola imagen.
La inmunoprecipitación, o IP, es una técnica ampliamente utilizada para aislar una proteína de interés de un lisado celular o tisular o un fluido corporal para la caracterización de proteínas o para investigar interacciones proteína-proteína.
El proceso comienza con un anticuerpo, que tiene una alta afinidad y especificidad para la proteína diana. Este anticuerpo se mezcla con la muestra, lo que permite la formación de complejos de anticuerpos objetivo. Cualquier proteína unida a la proteína diana también se une indirectamente al anticuerpo en el proceso. A continuación, la solución se incuba con cuentas de agarosa, conjugada con una proteína bacteriana, que tiene una fuerte afinidad por la región constante de los anticuerpos. La proteína bacteriana se une al anticuerpo y conecta los complejos objetivo de anticuerpos con las cuentas. A continuación, la solución se centrifuga para precipitar las perlas, extrayendo así todo el complejo que contiene el anticuerpo de unión, la proteína diana y cualquier proteína que interactúe. Finalmente, las proteínas unidas se extraen de las cuentas y se liberan entre sí y se utilizan para el análisis posterior por técnicas como la hincha occidental.
Varias variaciones de diferentes partes de esta técnica se utilizan comúnmente, como pre-clearing, usando etiquetas de péptidos o cuentas magnéticas, o el análisis de otros socios de unión no proteica. La IP puede ser preceed por un paso de pre-limpieza, para eliminar proteínas no específicas de unión a anticuerpos en la muestra y minimizar el fondo. Esto implica primero incubar la muestra con anticuerpos de control de isotipo, permitiéndoles unirse a estas proteínas, y luego usar cuentas de agarosa para precipitar los complejos. El ejemplo entonces está listo para proceder a la IP real.
Las etiquetas de péptidos son útiles si un anticuerpo específico no está disponible para IP. Aquí, la proteína diana puede ser modificada genéticamente para contener una etiqueta de epítopeno péptido y un anticuerpo contra la etiqueta es capaz de extraer la proteína de interés. Las cuentas magnéticas se utilizan a menudo en lugar de agarosa para precipitar el objetivo. Después de unirse al complejo de anticuerpos, el tubo de muestra se coloca en un campo magnético fuerte, que extrae las perlas de la solución. Esto elimina la necesidad de centrifugación y mejora la velocidad y la comodidad.
La inmunoprecipitación también se utiliza para estudiar proteínas de unión al ADN o al ARN y se conocen como inmunoprecipitación de cromatina e inmunoprecipitación de ARN, respectivamente. Estas variaciones son útiles para solucionar problemas y adaptar el método para diferentes aplicaciones experimentales. En este video, observará cómo pre-borrar un lisado celular y realizar inmunoprecipitación para extraer una proteína de interés, seguido de análisis de manchas occidentales para validar el experimento.
Para empezar, coloque las células pre-recogidas en una microcentrífuga y gire a 13 mil rpm durante tres minutos. Después del giro, retire el sobrenadante y luego resuspenda las células en 500 microlitros de lisis amortiguan RIPA con PMSF. Ahora, interrumpa las células usando unos pulsos rápidos con un vórtice y luego aspirar el lisado unas cuantas veces con una aguja de calibre 25 unida a una jeringa, teniendo cuidado de evitar la creación de burbujas. Coloque las células en hielo durante 15 minutos. Después de incubar las muestras en hielo, centrifugar el lisado durante 15 minutos a cuatro grados centígrados.
Etiquete un nuevo tubo de microcentrífuga de 1,5 mililitros. Después del giro, transfiera el sobrenadante al tubo recién etiquetado y deseche el pellet. A continuación, pre-borrar el lisado de contaminantes que se unen no específicamente a las perlas de agarosa o el anticuerpo primario mediante la adición de 20 microlitros de las cuentas de proteína A/G PLUS-agarose y un microgramo de un anticuerpo de control de isotipos al lisado, que en este ejemplo es un control de isotipo IgG1 de ratón. Incubar el tubo en un rotador en una sala fría durante 30 minutos. Después de girar el lysato en la cámara frigorífica durante 30 minutos, centrifugar la muestra a 3200 rpm durante 30 segundos a cuatro grados centígrados. Retire el tubo de la centrífuga y transfiera el sobrenadante pre-claro a un tubo de microcentrífuga de 1,5 mililitros con etiqueta fresca. Deseche el pellet.
Ahora, determinar la concentración de proteína del lisado celular mediante la realización de un ensayo de Bradford. Etiqueta siete 1. Tubos de microcentrífuga de 5 mililitros de uno a seis y muestra y alícuota 1000 microlitros del reactivo Bradford en cada tubo. Seis de los tubos se utilizarán para hacer una curva estándar mediante la adición de varias cantidades de cantidades conocidas de BSA a cada tubo. Los importes que se van a agregar se enumeran en esta tabla. En el séptimo tubo de muestra, agregue un microlitro del islato pre-claro. Coloque 200 microlitros de cada uno de los siete tubos en pozos individuales de una placa de fondo plano de 96 pocillos, repitiendo cada muestra en triplicado para que haya tres columnas de siete muestras. Lea la placa en un lector de placas, utilizando una longitud de onda de 595 nanómetros. Después de crear una curva estándar en Excel, calcule la concentración de proteínas del lisado preborrado.
A continuación, etiquetar dos tubos de microcentrífuga de 1,5 mililitros, uno como control y el otro como prueba, que en este ejemplo, será el anticuerpo c-myc. Coloque 500 microgramos del lisado pre-claro en cada uno de estos tubos y luego traiga el volumen total para cada tubo hasta 500 microlitros usando tampón de lisis. A continuación, agregue dos microgramos del anticuerpo anti-c-mico al tubo del grupo de ensayo. Para el control, agregue dos microgramos del anticuerpo de control de isotipo IgG1 del ratón. Una vez que los anticuerpos se añaden a los tubos, coloque las muestras en un rotador en una cámara fría e incubar durante dos horas. Ahora, agrega las cuentas de agarose. Para ello, se recomienda cortar el extremo de una punta de pipeta y luego, usando esta punta modificada, añadir 200 microlitros de las cuentas de proteína A/G PLUS-agarose a cada tubo. Incubar los tubos de un rotador en la cámara fría durante la noche.
Después de la incubación, retire los tubos del rotador y gire los lysates en el microcentrífugo para tirar hacia abajo de las perlas. Una vez completado el giro, retire los tubos de la centrífuga y aspire el sobrenadante de cada tubo. A continuación, lave las perlas con 500 microlitros de PBS de 1X Dulbecco. Coloque los tubos en una microcentrífuga y gire hacia abajo durante 30 segundos a cuatro grados centígrados. Después de esto, retire el sobrenadante. Repita los pasos de lavado y centrífuga una vez más para un total de dos veces. Retire los tubos de la microcentrífuga y aspire el tampón de cada tubo. Usando puntas de carga de gel, elimina cualquier tampón sobrante de las cuentas, manteniendo las cuentas en hielo para eluir la proteína enlazada.
En este ejemplo, la proteína se eluye en el búfer de ejecución SDS-PAGE hirviendo para el análisis de manchas occidentales. Para ello, resuspenda las perlas en 20 microlitros de tinte de carga SDS-PAGE que contenga beta-mercaptoetanol o BME. Hervir las muestras a 95 grados centígrados durante cinco minutos para disociar los inmunocomplejos de las cuentas. A continuación, centrifugar las perlas a la velocidad máxima durante 10 segundos a temperatura ambiente. Retire los tubos de la microcentrífuga y sosténgalos en un bastidor a temperatura ambiente. Usando puntas de carga de gel, pipetee cuidadosamente las muestras de las perlas y cárguelas en pozos de un gel SDS-PAGE de 4 a 15% de gradiente. Además de las muestras, cargue un carril con una escalera de proteínas, así como un carril con el islapreo pre-despejado para servir como control de carga. Una vez cargado el gel, ejecute el gel a 100 voltios.
Después de que el frente del tinte ha llegado a la parte inferior del gel, que debe tomar aproximadamente una hora, detener el gel y hacer un sándwich de mancha occidental, asegurando que la membrana PVDF está entre el gel y el cátodo. Coloque el sándwich Western blot en el aparato de transferencia y transfiera las proteínas del gel a la membrana durante una hora a 100 voltios. Una vez completada la transferencia, coloque la membrana en cinco mililitros de bloque para evitar que los anticuerpos se adhieran no específicamente a la membrana. Roca a un ajuste bajo durante una hora a temperatura ambiente. Cuando suene el temporizador, quite el búfer de bloqueo. Añadir cinco mililitros del tampón de bloqueo con el anticuerpo de detección a la membrana. Aquí, se utiliza un anticuerpo anti-c-mico, que es diferente al utilizado para el pull down.
Incubar la mancha durante la noche, a cuatro grados centígrados en un balancín en un ajuste bajo. Después de la incubación, retire el anticuerpo y el tampón de bloqueo. Lave la mancha, usando cinco mililitros de TBST durante cinco minutos a temperatura ambiente, en un balancín en un ajuste bajo. Este paso de lavado debe repetirse de dos a cinco veces para un total de tres a seis lavados, utilizando TBST fresco para cada lavado. Añadir cinco mililitros de uno a 1000 anticuerpos secundarios y tampón de bloqueo a la mancha. En este caso, el anticuerpo secundario es la cadena ligera anti-conejo con etiqueta HRP. Incubar la mancha en un balancín en un ajuste bajo para uno nuestro a temperatura ambiente. A continuación, retire el tampón y lave la mancha con cinco mililitros de TBST. Incubar este lavado en un balancín a un ajuste bajo durante cinco minutos a temperatura ambiente. Repita este lavado para un total de seis a 12 lavados, cada uno con cinco mililitros frescos de TBST. Retire el lavado final vertiendo primero el líquido de la mancha. Luego, usando pinzas, coloque el borde de la mancha en una toallita de laboratorio para eliminar cualquier exceso de líquido y luego coloque la mancha en un recipiente nuevo. A continuación, cubra la mancha con 1X Reactivo de Detección Quimioluminiscente e incubar durante un minuto.
Trabajando rápidamente, coloque el borde de la mancha en una toallita de laboratorio para eliminar cualquier exceso de reactivo de detección y luego coloque la mancha en la superficie de imagen de la bandeja imager. Imagen utilizando el programa Chemiluminescent e chemiluminescente para capturar múltiples puntos de tiempo de 10 a 30 segundos. Después de crear una imagen de la mancha, elija una imagen con una visibilidad óptima de la banda y, a continuación, exporte esa imagen. Antes de mover la mancha, utilice el Imager para tomar una foto de la mancha para capturar la ubicación de la escalera. Luego, exporte esa imagen también. Por último, utilizando un software de preparación de diapositivas, como PowerPoint, alinee las bandas y las imágenes de escalera para formar una imagen.
Esta imagen muestra el resultado de la mancha occidental para la inmunoprecipitación de la proteína c-mico de las células de timiocitos. De izquierda a derecha, los carriles representan el control de isotipo, la IP c-myc y la entrada de lisado pre-borrada. El carril en el extremo derecho es una imagen fusionada de la escalera de peso molecular. La banda fuerte, alrededor de 25 kilodaltones es de la cadena ligera y la de 50 kilodaltones es de la cadena pesada del anticuerpo de unión y no son específicos de la IP o las muestras. C-myc corre alrededor de 67 kilodaltones en manchas occidentales y por lo general es visible justo debajo de la banda de escalera de 75 kilodaltones. En esta mancha, la banda c-myc es visible en el segundo carril pero ausente en el primer carril, lo que indica que el anticuerpo IP tiró con éxito por c-myc. No hay banda visible en el carril de lisado pre-claro, lo que sugiere que esta proteína tiene bajos niveles de expresión endógena.
Subscription Required. Please recommend JoVE to your librarian.
Results
Los resultados del procedimiento detallado anteriormente se muestran en la Figura 2. De izquierda a derecha, los carriles contienen el grupo de control (isotipo), el grupo de prueba (c-mic), el islapredado pre-claro (lisado) y la escalera de peso molecular (escalera). Las bandas de escalera de 25 y 75 kDa están marcadas. Las dos bandas prominentes a 25 kDa y 50 kDa son la cadena ligera y pesada del anticuerpo de unión, respectivamente, y no son específicas de la IP o de las muestras. proteína c-myc que dura alrededor de 67kDa en manchas occidentales y generalmente es visible justo debajo de la banda de escalera de 75 kDa. En esta mancha, la banda c-myc es visible en el segundo carril, pero ausente en el primer carril, lo que indica que el anticuerpo IP tiró con éxito por c-myc. No hay banda visible en el carril de lisado pre-claro, lo que sugiere que esta proteína tiene bajos niveles de expresión endógena.

Figura 2: Resultados de un análisis de Western Blot, utilizado para evaluar la purificación de c-myc por inmunoprecipitación. Una banda de 67 kDa, correspondiente a c-myc, es visible en el carril anti-c-mico, pero no en el carril de control de isotipo. Tenga en cuenta que los niveles de c-myc no eran lo suficientemente altos como para ser visualizados en el carril de lisado. Haga clic aquí para ver una versión más grande de esta figura.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
En resumen, la inmunoprecipitación es el aislamiento de una proteína específica utilizando un anticuerpo. En este ejemplo, los resultados de la inmunoprecipitación fueron analizados por Western blot para evaluar la pureza. La proteína aislada podría utilizarse en una serie de aplicaciones posteriores, incluyendo: RMN para la estructura proteica, Espectrometría de masas para secuencia de aminoácidos, o ensayos in vitro para la caracterización enzimática. Las IPs también pueden caracterizar a los socios que interactúan con las proteínas. Por ejemplo, después del aislamiento, el ADN o el ARN podrían aislarse para la secuenciación. Las inmunoprecipitaciones evalúan las interacciones proteína-proteína. Cuando la proteína diana se tira hacia abajo durante una IP, las proteínas que interactúan también se pueden tirar hacia abajo. Estos socios que interactúan pueden ser evaluados por espectrometría de masas y mancha occidental. La inmunoprecipitación es una técnica poderosa para estudiar la biología proteica.
Subscription Required. Please recommend JoVE to your librarian.
References
- Olliver, C. L. and Boyd, C. D. (1984). Immunoprecipitation of In Vitro Translation Products with Protein A Bound to Sepharose. In J. M. Walker (eds), Nucleic Acids. Methods in Molecular Biology (pp. 157-160). New Jersey: Humana Press.
- Thurston, C. F. and Henley, L. F. (1988). Direct Immunoprecipitation of Protein. In J. M. Walker (eds), New Protein Techniques. Methods in Molecular Biology (pp. 149-158). New Jersey: Humana Press.
- Anderson, N. G. (1998). Co-immunoprecipitation: Identification of Interacting Proteins. In R. A. Clegg (eds), Protein Targeting Protocols.Methods in Molecular Biology (pp. 35-45). New Jersey: Humana Press.
- Jackson, D. I. and Dickson, C. (1999). Protein Techniques: Immunoprecipitation, In Vitro Kinase Assays, and Western Blotting. In P.T. Sharpe and I. Mason (eds), Molecular Embryology. Methods in Molecular Biology (pp. 699-708). New Jersey: Humana Press.
- Trieu, E. P. and Targoff, I. N. (2015). Immunoprecipitation: Western Blot for Proteins of Low Abundance. In B. Kurien and R. Scofield (eds), Western Blotting. Methods in Molecular Biology (pp. 327-342). New York, NY: Humana Press.