



Overview
Fonte: Susannah C. Shissler1, Tonya J. Webb1
1 Dipartimento di Microbiologia e Immunologia, Università del Maryland, Baltimora, MD 21201
L'immunoprecipitazione (IP, noto anche come test "pull-down") è una tecnica ampiamente utilizzata che ha applicazioni in una varietà di campi. Concepito per la prima volta nel 1984, è stato perfezionato nel 1988 (1, 2). L'obiettivo fondamentale dell'IP è la purificazione e l'isolamento di una proteina specifica utilizzando un anticorpo contro quella proteina. La parola "immuno" si riferisce all'uso di un anticorpo mentre la parola "precipitazione" si riferisce all'estrazione di una sostanza specifica da una soluzione. La proteina bersaglio potrebbe essere endogena o ricombinante. La maggior parte delle proteine ricombinanti ha un tag epitopo (cioè myc o bandiera) attaccato a loro per semplificare la successiva purificazione. In genere, è più facile ottimizzare la proteina IP ricombinante perché gli anticorpi contro i tag epitopi ricombinanti sono molto forti ed efficaci. Gli anticorpi contro le proteine endogene hanno un'efficacia estremamente variabile, rendendo molto più difficile ottimizzare queste IP. Un passo necessario dopo l'immunoprecipitazione è la verifica della purificazione. La proteina isolata viene risolta utilizzando SDS-PAGE e successivamente sonde per la purezza da western blots (Figura 1). Un controllo importante è l'uso di un anticorpo diverso durante il Western blot per verificare il pull down della proteina corretta. La combinazione di IP con tecniche successive è un potente strumento di analisi. L'obiettivo dopo la purificazione può essere la caratterizzazione della proteina stessa mediante NMR, spettrometria di massa e saggi in vitro, o l'analisi dei partner interagenti della proteina (cioè proteine, DNA, RNA) (3, 4, 5).
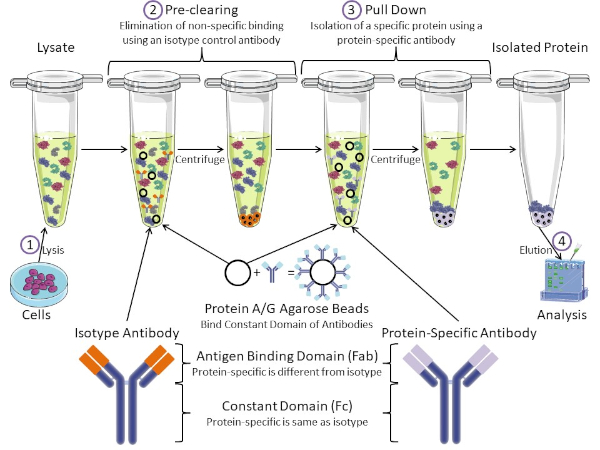
Figura 1: Panoramica della procedura di immunoprecipitazione. L'immunoprecipitazione è l'isolamento di una proteina specifica utilizzando un anticorpo. Dopo la produzione di lisi dalle cellule, ci sono due passaggi principali: pre-pulizia e pull down. Durante la fase di pre-clearing, i lasati cellulari viene pre-eliminata dalle proteine che si legano agli anticorpi in modo non specifico utilizzando un anticorpo di controllo dell'isotipo. Nella fase di pull down, la proteina bersaglio viene tirata verso il basso usando un anticorpo specifico della proteina. La proteina isolata viene quindi analizzata da Western blot. Gli anticorpi isotipia e gli anticorpi specifici della proteina hanno lo stesso dominio costante, ma domini di legame dell'antigene diversi. Un componente chiave di questo protocollo sono le perle di agarose della proteina A / G che legano il dominio costante degli anticorpi, consentendo l'immunoprecipitazione della proteina bersaglio. Fare clic qui per visualizzare una versione più grande di questa figura.
Gli anticorpi sono il componente chiave di un'immunoprecipitazione che la differenziano da altre forme di purificazione delle proteine (cioè la purificazione della colonna di affinità del nichel). Gli anticorpi sono molecole prodotte da cellule B in grado di riconoscere specifici epitopi proteici. Gli anticorpi hanno due domini: costante (Fc) e legame con l'antigene (Fab) (Figura 1). Il dominio costante identifica il tipo di anticorpo e detta la funzione in vivo. Di solito, i domini costanti degli anticorpi utilizzati per l'IP sono IgG di topo, ratto o coniglio. La porzione di legame con l'antigene dell'anticorpo riconosce un epitopo specifico di una proteina specifica. Gli anticorpi possono riconoscere gli epitopi su proteine ripiegate che potrebbero non esistere quando la proteina è denaturata e viceversa. Pertanto, la disponibilità dell'epitopo dipende dal ripiegamento delle proteine, identificando un fattore importante da considerare quando si scelgono anticorpi e condizioni per l'IP.
Sia il sistema procariotico che eucariotico hanno proteine leganti gli anticorpi. Nei sistemi eucariotici, lo scopo è la protezione immunitaria dai batteri mentre nei sistemi procariotici, lo scopo è la protezione dal sistema immunitario. Le proteine leganti gli anticorpi influenzano la metodologia IP in due modi. In primo luogo, c'è una fase di pre-clearing necessaria (Figura 1) per liberare il lisiato dalle proteine che legano gli anticorpi, riducendo così il legame non specifico nel prodotto finale. Questo passaggio utilizza un anticorpo isotipo che ha lo stesso dominio costante ma un dominio di legame anticorpale diverso rispetto all'anticorpo specifico della proteina. Le proteine che legano gli anticorpi batterici sono il secondo componente chiave di questo metodo. Dopo che l'anticorpo proteina-specifica lega la proteina bersaglio, il complesso anticorpo: proteina deve essere tirato verso il basso (Figura 1). Le proteine A, G e L sono proteine batteriche che legano il dominio costante degli anticorpi. Mentre i batteri usano questo per sovvertire il sistema immunitario, i ricercatori hanno cooptato questo sistema per una facile purificazione degli anticorpi, e viene utilizzato sia durante le fasi di pre-clearing che di pull-down. Queste proteine hanno diverse affinità di legame per diverse specie e diversi sottotipi di dominio costante - un altro fattore da considerare quando si scelgono le condizioni per IP. Molte aziende vendono perle di agarose marcate Protein A / G (Figura 1), colonne di spin prefabbricate o resine per realizzare colonne. In generale, le pere e le colonne di spin vengono utilizzate per campioni di dimensioni inferiori, mentre le resine vengono utilizzate per la purificazione alla rinfusa.
In questo esercizio di laboratorio, dimostriamo come purificare la proteina endogena c-myc, dai timociti murini primari, utilizzando la tecnica di immunoprecipitazione di base basata sulle perle di agarose Protein A / G Plus. Il protocollo inizia dalla preparazione del lizzata cellulare e termina con la verifica del successo del pull down proteico utilizzando l'analisi Western blot.
Procedure
1. Immunoprecipitazione con pere di agarose della proteina A/G PLUS
Preparazione del losato cellulare
- Centrifugare 108 timociti in una microcentrifuga a 13.000 giri/min per 3 minuti e rimuovere il surnatante.
Nota: Il numero di cellule varierà a seconda dei livelli di espressione della proteina desiderata e del tipo di cellula scelto. - Sospendere di ricaspendere le cellule nel tampone di lisi da 500 μL RIPA con PMSF.
- Interrompere le cellule usando alcuni impulsi rapidi con un vortice e quindi aspirare il lisirato un paio di volte con un ago da 25 G attaccato a una siringa.
Nota: Evita di creare bolle. Utilizzare un ago più grande come un ago da 21 G per tipi di cellule più grandi. - Incubare il lisiato cellulare sul ghiaccio per 10 minuti.
- Centrifugare il lisidato a 13.000 giri/min per 15 min a 4°C.
- Trasferire il surnatante in un tubo microcentrifuga fresco ed etichettato.
Pre-compensazione
- Aggiungere al lisi 20 μL di proteine A/G PLUS perle di agarose e 1 μg di un anticorpo di controllo dell'isotipo (qui viene utilizzato l'anticorpo di controllo dell'isotipo IgG1 del topo).
Nota: La scelta dell'anticorpo isotipo utilizzato dipenderà dall'anticorpo proteina-specifica utilizzato più avanti nella fase di pull-down. - Incubare la miscela di lisi su un rotatore centrifugo in cella frigorifera (4°C) per 30 min.
- Centrifugare il campione a 3200 giri/min per 30 s a 4°C.
- Trasferire il surnatante pre-eliminato in un tubo microcentrifuga fresco, etichettato, da 1,5 mL. Scartare il pellet.
Determinazione della concentrazione proteica
- Determinare la concentrazione proteica del lizzata cellulare eseguendo un test di Bradford.
- Aliquota 1000 μL Bradford Reagent in 7 tubi microcentrifuga.
- Aggiungere le seguenti quantità di BSA protein standard (2 mg/mL) in 6 dei tubi (Tabella 1).
| Numero del tubo | Volume BSA (μL) (2 mg/mL) | Concentrazione proteica (μg/μL) |
| 1 | 0 | 0 |
| 2 | 1 | 2 |
| 3 | 2 | 4 |
| 4 | 3 | 6 |
| 5 | 4 | 8 |
| 6 | 5 | 10 |
Tabella 1: Quantità standard di proteine BSA
- Nel 7° tubo, aggiungere 1 μL del lisirato pre-eliminato.
Nota: Per garantire che la concentrazione del campione rientri nell'intervallo di rilevamento del test, preparare e analizzare anche una diluizione del lsato 1:2 o 1:5. - Posizionare 200 μL da ciascuno dei 7 tubi in singoli pozzetti di una piastra a fondo piatto a 96 pozzetti ripetendo ogni campione in triplice copia.
- Lettura della piastra su un lettore di piastre a 595 nm.
- Generare la curva standard in excel e calcolare la concentrazione proteica del lisirato pre-eliminato.
Abbattere
- Etichettare due tubi microcentrifuga freschi da 1,5 ml, uno come "controllo" e l'altro come "test", che in questo esempio è c-myc.
- Inserire 500 μg di lisato pre-eliminato in ciascuno di questi tubi.
Nota: La quantità di proteine utilizzate qui dipenderà dalla quantità di proteina desiderata per essere purificata. - Portare il volume totale per ogni tubo fino a 500 μL utilizzando il tampone di lisi.
- Aggiungere 2 μg di anticorpo anti-c-myc alla provetta del gruppo di prova e 2 μg di anticorpo di controllo dell'isotipo IgG1 del topo al gruppo di controllo.
Nota: la quantità di anticorpi dipenderà dall'efficacia dell'anticorpo e dalla quantità della proteina bersaglio. - Incubare i tubi su un rotatore nella cella frigorifera (4°C) per 2 ore.
- Aggiungere 20 μL di proteine A/G PLUS perle di agarose a ciascun tubo.
Nota: Si consiglia di utilizzare punte per pipette con l'estremità tagliata per evitare danni alle perle. - Incubare su un rotatore in camera fredda (4°C) durante la notte.
Nota: A seconda della proteina bersaglio e dell'efficacia degli anticorpi, questo passaggio può variare da 1 ora a durante la notte. - Centrifugare i tubi a 3200 giri/min per 30 s 4°C per tirare giù le perle.
- Aspirare il surnatante da ciascun tubo.
Nota: La proteina bersaglio è ora legata alle perle. - Lavare le perle due volte utilizzando il PBS di Dulbecco da 500 μL 1X.
- Centrifugare i tubi a 3200 giri/min per 30 s 4°C.
Nota: Per un lavaggio più rigoroso, utilizzare tamponi più rigorosi, come RIPA. - Aspirare il tampone da ciascun tubo. Utilizzando punte per il caricamento del gel, rimuovere qualsiasi tampone residuo dalle pere e mantenere le pere sul ghiaccio per eluire la proteina.
Nota: in questo esempio, la proteina viene eluita nel buffer di esecuzione SDS-PAGE facendo bollire le pere, per l'analisi Western blot. Questo approccio è adatto per verificare i risultati IP o per esaminare le interazioni proteina-proteina. Per altre applicazioni a valle, come la purificazione delle proteine per l'analisi strutturale o enzimatica, vengono utilizzati sistemi più sofisticati, come i tag epitopi (flag-tag o myc-tag) per evitare l'eluizione dell'anticorpo con la proteina di interesse.
2. Verifica IP tramite Western Blot Analysis
SDS-PAGE Elettroforesi:
- Sospendere nuovamente le perle in colorante di caricamento SDS-PAGE da 20 μL contenente β-mercapto-ethonolo.
- Far bollire i campioni a 95 °C per 5 minuti.
- Centrifugare le perle a 13.000 giri/min per 10 s a temperatura ambiente.
- Utilizzando punte di caricamento del gel, pipettare con cura i campioni ottenuti dalle perle e caricarli in pozzi di gel SDS-PAGE con gradiente del 4-15%.
- Oltre ai campioni, caricare una corsia con una scala proteica e una corsia con il lisirato pre-eliminato per fungere da controllo del carico.
- Eseguire a 100 V fino a quando la parte anteriore del colorante raggiunge il fondo del gel (~ 1h).
Analisi Western Blot:
- Fai il sandwich Western blot, assicurandoti che la membrana PVDF sia tra gel e catodo rosso.
- Trasferimento per 1 ora a 100 V.
- Posizionare la membrana in un tampone di blocco da 5 ml a temperatura ambiente per 1 ora su un bilanciere a bassa impostazione, per bloccare i siti di legame proteico non specifici.
Nota: potrebbe essere necessario aumentare i volumi del tampone bloccante, dell'anticorpo primario, dell'anticorpo secondario e dei lavaggi per macchie di dimensioni maggiori. - Incubare la macchia con 5 mL di anticorpo anti-c-myc nel tampone bloccante durante la notte a 4 ° C sul bilanciere a un'impostazione bassa.
Nota: l'anticorpo utilizzato qui dovrebbe essere diverso da quello utilizzato nella fase di pull-down. - Lavare la macchia 3-6 volte utilizzando 5 ml di TBST con ogni lavaggio di 5 minuti a temperatura ambiente su un bilanciere a bassa impostazione.
- Incubare la macchia con anticorpo secondario a catena leggera anti-coniglio con etichetta HRP in tampone di blocco, per 1 ora a temperatura ambiente su bilanciere a bassa impostazione.
Nota: la scelta dell'anticorpo secondario dipenderà dall'anticorpo primario utilizzato per il Western blot. Inoltre, nel protocollo viene utilizzato un secondario specifico per catena leggera perché la proteina bersaglio è vicina in peso molecolare alla catena pesante dell'anticorpo. Se la proteina bersaglio è vicina a 50kDa, utilizzare un secondario specifico per la catena leggera. Se la proteina bersaglio è vicina a 25kDa, utilizzare e catena pesante specifica secondaria. - Lavare la macchia 3-6 volte utilizzando 5 ml di TBST con ogni lavaggio di 5 minuti a temperatura ambiente su un bilanciere a bassa impostazione.
- Rimuovere il liquido dalla macchia e tamponare il bordo della macchia sulle salviette da laboratorio per rimuovere il liquido in eccesso.
- Coprire la macchia con 1x reagente di rivelazione chemiluminescente' e incubare per 1 min.
Nota: I seguenti passaggi devono essere eseguiti in rapida successione poiché il reagente di rilevamento è sensibile alla luce e al tempo. - Tamponare il bordo della macchia sulle salviette da laboratorio per rimuovere il reagente di rilevamento in eccesso.
- Posizionare la macchia sulla superficie di imaging del vassoio Imager.
Nota: le macchie chemiluminescenti possono anche essere visualizzate utilizzando la pellicola. - Immagine utilizzando il 'Chemiluminescent Program' per catturare più punti temporali da 10 s a 5 min.
Nota: il momento ottimale può cambiare in base alla quantità di proteine e alla qualità degli anticorpi. - Scegli un'immagine con visibilità ottimale della banda e quindi esporta quell'immagine.
- Prima di spostare la macchia, scatta una foto della macchia usando l'imager, per catturare la posizione della scala. Quindi, esporta anche quell'immagine.
- Utilizzando un software di preparazione delle diapositive (ad esempio PowerPoint), allineare le bande e le immagini ladder per formare una singola immagine.
L'immunoprecipitazione, o IP, è una tecnica ampiamente utilizzata per isolare una proteina di interesse da un lisato cellulare o tissutale o da un fluido corporeo per la caratterizzazione proteica o per studiare le interazioni proteina-proteina.
Il processo inizia con un anticorpo, che ha un'alta affinità e specificità per la proteina bersaglio. Questo anticorpo viene miscelato con il campione, consentendo la formazione di complessi anticorpo-bersaglio. Qualsiasi proteina legata alla proteina bersaglio viene anche indirettamente attaccata all'anticorpo nel processo. Successivamente, la soluzione viene incubata con pere di agarosio, coniugate a una proteina batterica, che ha una forte affinità per la regione costante degli anticorpi. La proteina batterica si lega all'anticorpo e collega i complessi anticorpo-bersaglio alle perle. Quindi, la soluzione viene centrifugata per far precipitare le perle, estraendo così l'intero complesso contenente l'anticorpo legante, la proteina bersaglio e qualsiasi proteina interagente. Infine, le proteine legate vengono estratte dalle perle e rilasciate l'una dall'altra e vengono utilizzate per ulteriori analisi con tecniche come il Western blotting.
Diverse varianti di diverse parti di questa tecnica sono comunemente usate, come la pre-compensazione, l'uso di tag peptidici o perle magnetiche o l'analisi di altri partner non leganti proteine. L'IP può essere preceduto da una fase di pre-clearing, per rimuovere le proteine che legano anticorpi non specifiche nel campione e ridurre al minimo lo sfondo. Ciò comporta prima l'incubazione del campione con anticorpi di controllo dell'isotipo, consentendo loro di legarsi a queste proteine, e quindi l'uso di perle di agarose per precipitare i complessi. L'esempio è quindi pronto per procedere all'IP effettivo.
I tag peptidici sono utili se un anticorpo specifico non è disponibile per IP. Qui, la proteina bersaglio può essere geneticamente modificata per contenere un tag epitopo peptidico e un anticorpo contro il tag è in grado di estrarre la proteina di interesse. Le pere magnetiche sono spesso usate al posto dell'agarose per far precipitare il bersaglio. Dopo il legame al complesso anticorpo-bersaglio, la provetta campione viene posta in un forte campo magnetico, che estrae le pere dalla soluzione. Ciò elimina la necessità di centrifugazione e migliora la velocità e la praticità.
L'immunoprecipitazione viene anche utilizzata per studiare le proteine leganti il DNA o l'RNA e sono note rispettivamente come immunoprecipitazione della cromatina e immunoprecipitazione dell'RNA. Queste variazioni sono utili per la risoluzione dei problemi e l'adattamento del metodo per diverse applicazioni sperimentali. In questo video, osserverai come pre-cancellare un lasato cellulare ed eseguire l'immunoprecipitazione per estrarre una proteina di interesse, seguita dall'analisi Western blot per convalidare l'esperimento.
Per iniziare, posizionare le cellule pre-raccolte in una microcentrifuga e ruotare a 13 mila giri / min per tre minuti. Dopo la rotazione, rimuovere il surnatante e quindi risospepare le cellule in 500 microlitri di tampone di lisi RIPA con PMSF. Ora, interrompere le cellule usando alcuni impulsi rapidi con un vortice e quindi aspirare il lisirato un paio di volte con un ago calibro 25 attaccato a una siringa, avendo cura di evitare di creare bolle. Mettere le cellule sul ghiaccio per 15 minuti. Dopo aver incubato i campioni su ghiaccio, centrifugare il lisirato per 15 minuti a quattro gradi Celsius.
Etichettare un nuovo tubo microcentrifuga da 1,5 millilitro. Dopo la rotazione, trasferire il surnatante nel tubo appena etichettato ed eliminare il pellet. Successivamente, pre-cancellare il lisato di contaminanti che si legano in modo non specifico alle perle di agarose o all'anticorpo primario aggiungendo 20 microlitri della proteina A / G PLUS-agarose perle e un microgrammo di un anticorpo di controllo dell'isotipo al lisato, che in questo esempio è un controllo dell'isotipo IgG1 del topo. Incubare il tubo su un rotatore in una cella frigorifera per 30 minuti. Dopo aver ruotato il lisito nella cella frigorifera per 30 minuti, centrifugare il campione a 3200 giri / min per 30 secondi a quattro gradi Celsius. Rimuovere il tubo dalla centrifuga e trasferire il surnatante pre-eliminato in un tubo microcentrifuga da 1,5 millilitro appena etichettato. Scartare il pellet.
Ora, determinare la concentrazione proteica del lizzata cellulare eseguendo un test di Bradford. Etichetta sette 1. Tubi di microcentrifuga da 5 millilitri da uno a sei e campione e aliquota 1000 microlitri del reagente bradford in ciascun tubo. Sei dei tubi saranno utilizzati per creare una curva standard aggiungendo varie quantità di quantità note di BSA a ciascun tubo. Gli importi da aggiungere sono elencati in questa tabella. Nella settima provetta del campione, aggiungere un microlitro del lisirato pre-eliminato. Posizionare 200 microlitri da ciascuno dei sette tubi in singoli pozzetti di una piastra a fondo piatto da 96 pozzetti, ripetendo ogni campione in triplice copia in modo che ci siano tre colonne di sette campioni. Leggere la piastra su un lettore di piastre, utilizzando una lunghezza d'onda di 595 nanometri. Dopo aver creato una curva standard in Excel, calcolare la concentrazione proteica del lisirato pre-eliminato.
Quindi, etichettare due tubi microcentrifuga da 1,5 millilitro, uno come controllo e l'altro come test, che in questo esempio sarà l'anticorpo c-myc. Posizionare 500 microgrammi di lisato pre-eliminato in ciascuno di questi tubi e quindi portare il volume totale per ciascun tubo fino a 500 microlitri utilizzando il tampone di lisi. Quindi, aggiungere due microgrammi dell'anticorpo anti-c-myc alla provetta del gruppo di prova. Per il controllo, aggiungere due microgrammi dell'anticorpo di controllo dell'isotipo IgG1 del topo. Una volta aggiunti gli anticorpi ai tubi, posizionare i campioni su un rotatore in una cella frigorifera e incubare per due ore. Ora, aggiungi le pere di agarose. Per fare ciò, si consiglia di tagliare l'estremità di una punta della pipetta e quindi, utilizzando questa punta modificata, aggiungere 200 microlitri delle pere di agarose protein A / G PLUS a ciascun tubo. Incubare i tubi su un rotatore nella cella frigorifera durante la notte.
Dopo l'incubazione, rimuovere i tubi dal rotatore e ruotare i lasati nella microcentrifuga per tirare giù le perle. Una volta completata la rotazione, rimuovere i tubi dalla centrifuga e aspirare il surnatante da ciascun tubo. Quindi, lavare le perle utilizzando 500 microlitri del PBS di 1X Dulbecco. Posizionare i tubi in una microcentrifuga e ruotare verso il basso per 30 secondi a quattro gradi Celsius. Successivamente, rimuovere il surnatante. Ripetere ancora una volta i passaggi di lavaggio e centrifuga per un totale di due volte. Rimuovere i tubi dalla microcentrifuga e aspirare il tampone da ciascun tubo. Utilizzando le punte di caricamento del gel, rimuovere qualsiasi tampone residuo dalle perle, mantenendo le perle sul ghiaccio per eluire la proteina legata.
In questo esempio, la proteina viene eluita nel buffer di esecuzione SDS-PAGE mediante ebollizione per l'analisi Western blot. Per fare questo, riconseminare le perle in 20 microlitri di colorante di caricamento SDS-PAGE contenente beta-mercaptoetanolo o BME. Far bollire i campioni a 95 gradi Celsius per cinque minuti per dissociare gli immunocomplessi dalle perle. Quindi, centrifugare le perle alla massima velocità per 10 secondi a temperatura ambiente. Rimuovere i tubi dalla microcentrifuga e tenerli in un rack a temperatura ambiente. Utilizzando le punte di caricamento del gel, pipettare accuratamente i campioni dalle perle e caricarli in pozzeggi di un gel SDS-PAGE con gradiente dal 4 al 15%. Oltre ai campioni, caricare una corsia con una scala proteica e una corsia con il lisirato pre-eliminato per fungere da controllo del carico. Una volta caricato il gel, far funzionare il gel a 100 volt.
Dopo che il fronte del colorante ha raggiunto il fondo del gel, che dovrebbe richiedere circa un'ora, fermare il gel e fare un sandwich Western blot, assicurandosi che la membrana PVDF sia tra il gel e il catodo. Posizionare il sandwich Western blot nell'apparato di trasferimento e trasferire le proteine sul gel alla membrana per un'ora a 100 volt. Al termine del trasferimento, posizionare la membrana in cinque millilitri di blocco per evitare che gli anticorpi si lesino in modo non specifico alla membrana. Roccia a un ambiente basso per un'ora a temperatura ambiente. Quando il timer suona, rimuovere il buffer di blocco. Aggiungere cinque millilitri del tampone bloccante con l'anticorpo di rilevamento alla membrana. Qui viene utilizzato un anticorpo anti-c-myc, diverso da quello utilizzato per il pull down.
Incubare la macchia durante la notte, a quattro gradi Celsius su un bilanciere a un'impostazione bassa. Dopo l'incubazione, rimuovere l'anticorpo e il tampone di blocco. Lavare la macchia, usando cinque millilitri di TBST per cinque minuti a temperatura ambiente, su un bilanciere a bassa impostazione. Questa fase di lavaggio deve essere ripetuta da due a cinque volte per un totale di tre o sei lavaggi, utilizzando TBST fresco per ogni lavaggio. Aggiungere cinque millilitri da uno a 1000 anticorpi secondari e tampone bloccante alla macchia. In questo caso, l'anticorpo secondario è la catena leggera anti-coniglio marcata con HRP. Incubare la macchia su un bilanciere a un'impostazione bassa per uno a temperatura ambiente. Quindi, rimuovere il tampone e lavare la macchia con cinque millilitri di TBST. Incubare questo lavaggio su un bilanciere a bassa impostazione per cinque minuti a temperatura ambiente. Ripeti questo lavaggio per un totale di sei-12 lavaggi, ciascuno con cinque millilitri freschi di TBST. Rimuovere il lavaggio finale versando prima il liquido dalla macchia. Quindi, usando una pinzetta, tamponare il bordo della macchia su una salvietta da laboratorio per rimuovere il liquido in eccesso e quindi posizionare la macchia in un contenitore fresco. Quindi, coprire la macchia con 1X Chemiluminescent Detection Reagent e incubare per un minuto.
Lavorando rapidamente, tamponare il bordo della macchia su una salvietta da laboratorio per rimuovere qualsiasi reagente di rilevamento in eccesso e quindi posizionare la macchia sulla superficie di imaging del vassoio Imager. Immagine utilizzando il programma Chemiluminescent per catturare più punti temporali da 10 a 30 secondi. Dopo aver ripreso l'immagine, scegli un'immagine con visibilità ottimale della banda e quindi esporta quell'immagine. Prima di spostare la macchia, utilizzare l'Imager per scattare una foto della macchia per acquisire la posizione della scala. Quindi, esporta anche quell'immagine. Infine, utilizzando un software di preparazione delle diapositive, come PowerPoint, allineare le bande e le immagini ladder per formare un'unica immagine.
Questa immagine mostra il risultato Western blot per l'immunoprecipitazione della proteina c-myc dalle cellule timocitarie. Da sinistra a destra, le corsie rappresentano il controllo dell'isotipo, l'IP c-myc e l'ingresso del lisi pre-cancellato. La corsia all'estrema destra è un'immagine unita della scala del peso molecolare. La banda forte, a circa 25 kilodaltons proviene dalla catena leggera e quella a 50 kilodaltons proviene dalla catena pesante dell'anticorpo legante e non è specifica per l'IP o i campioni. C-myc corre intorno a 67 kilodalton su western blots ed è solitamente visibile appena sotto la banda della scala di 75 kilodalton. In questa macchia, la banda c-myc è visibile nella seconda corsia ma assente nella prima corsia, indicando che l'anticorpo IP ha tirato giù con successo c-myc. Non esiste una banda visibile nella corsia del lisi pre-eliminato, suggerendo che questa proteina ha bassi livelli di espressione endogena.
Subscription Required. Please recommend JoVE to your librarian.
Results
I risultati della procedura descritta sopra sono mostrati nella Figura 2. Da sinistra a destra, le corsie contengono il gruppo di controllo (isotipo), il gruppo di prova (c-myc), il lisirato pre-eliminato (lisi) e la scala di peso molecolare (scala). Le bande della scala da 25 e 75 kDa sono contrassegnate. Le due bande prominenti a ~ 25 kDa e 50 kDa sono rispettivamente la catena leggera e pesante dell'anticorpo legante e non sono specifiche per l'IP o i campioni. Proteina c-myc che corre intorno a 67kDa su Western blots ed è solitamente visibile appena sotto la banda della scala 75 kDa. In questa macchia, la banda c-myc è visibile nella seconda corsia, ma assente nella prima corsia, indicando che l'anticorpo IP ha tirato giù con successo c-myc. Non esiste una banda visibile nella corsia del lisi pre-eliminato, suggerendo che questa proteina ha bassi livelli di espressione endogena.

Figura 2: Risultati di un'analisi Western Blot, utilizzata per valutare la purificazione di c-myc mediante immunoprecipitazione. Una banda a 67 kDa, corrispondente a c-myc, è visibile nella corsia anti-c-myc, ma non nella corsia di controllo dell'isotipo. Si noti che i livelli di c-myc non erano abbastanza alti da essere visualizzati nella corsia di lisi. Fare clic qui per visualizzare una versione più grande di questa figura.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
In breve, l'immunoprecipitazione è l'isolamento di una proteina specifica utilizzando un anticorpo. In questo esempio, i risultati dell'immunoprecipitazione sono stati analizzati da Western blot per valutarne la purezza. La proteina isolata potrebbe essere utilizzata in una serie di applicazioni in seguito, tra cui: NMR per la struttura proteica, Spettrometria di massa per la sequenza di amminoacidi o saggi in vitro per la caratterizzazione enzimatica. Gli IP possono anche caratterizzare i partner interagenti delle proteine. Ad esempio, dopo l'isolamento, il DNA o l'RNA potrebbero essere isolati per il sequenziamento. Le co-immunoprecipitazioni valutano le interazioni proteina-proteina. Quando la proteina bersaglio viene tirata giù durante un IP, le proteine interagenti possono anche essere abbattute. Questi partner interagenti possono essere valutati mediante spettrometria di massa e Western blot. L'immunoprecipitazione è una tecnica potente per studiare la biologia delle proteine.
Subscription Required. Please recommend JoVE to your librarian.
References
- Olliver, C. L. and Boyd, C. D. (1984). Immunoprecipitation of In Vitro Translation Products with Protein A Bound to Sepharose. In J. M. Walker (eds), Nucleic Acids. Methods in Molecular Biology (pp. 157-160). New Jersey: Humana Press.
- Thurston, C. F. and Henley, L. F. (1988). Direct Immunoprecipitation of Protein. In J. M. Walker (eds), New Protein Techniques. Methods in Molecular Biology (pp. 149-158). New Jersey: Humana Press.
- Anderson, N. G. (1998). Co-immunoprecipitation: Identification of Interacting Proteins. In R. A. Clegg (eds), Protein Targeting Protocols.Methods in Molecular Biology (pp. 35-45). New Jersey: Humana Press.
- Jackson, D. I. and Dickson, C. (1999). Protein Techniques: Immunoprecipitation, In Vitro Kinase Assays, and Western Blotting. In P.T. Sharpe and I. Mason (eds), Molecular Embryology. Methods in Molecular Biology (pp. 699-708). New Jersey: Humana Press.
- Trieu, E. P. and Targoff, I. N. (2015). Immunoprecipitation: Western Blot for Proteins of Low Abundance. In B. Kurien and R. Scofield (eds), Western Blotting. Methods in Molecular Biology (pp. 327-342). New York, NY: Humana Press.