



Overview
Fonte: Susannah C. Shissler1, Tonya J. Webb1
1 Departamento de Microbiologia e Imunologia, Universidade de Maryland, Baltimore, MD 21201
Imunoprecipitação (IP, também conhecido como ensaio 'pull-down') é uma técnica amplamente utilizada que tem aplicações em uma variedade de campos. Concebido pela primeira vez em 1984, foi refinado em 1988 (1, 2). O objetivo fundamental do IP é a purificação e isolamento de uma proteína específica usando um anticorpo contra essa proteína. A palavra "imuno" refere-se ao uso de um anticorpo, enquanto a palavra "precipitação" refere-se a retirar uma substância específica de uma solução. A proteína alvo pode ser endógena ou recombinante. A maioria das proteínas recombinantes tem uma tag de epítope (ou seja, myc ou flag) anexada a elas para simplificar a purificação subsequente. Normalmente, é mais fácil otimizar a proteína recombinante IP porque os anticorpos contra tags de epitope recombinantes são muito fortes e eficazes. Anticorpos contra proteínas endógenas têm eficácia extremamente variável - tornando muito mais difícil otimizar esses IPs. Um passo necessário após a imunoprecipitação é a verificação da purificação. A proteína isolada é resolvida usando SDS-PAGE e posteriormente sondada para pureza por manchas ocidentais (Figura 1). Um controle importante é o uso de um anticorpo diferente durante a mancha ocidental para verificar a retirada da proteína correta. A combinação de IP com técnicas subsequentes é uma poderosa ferramenta de análise. O objetivo após a purificação pode ser a caracterização da proteína em si por NMR, espectrometria de massa e ensaios in vitro, ou análise dos parceiros interativos da proteína (ou seja, proteína, DNA, RNA) (3, 4, 5).
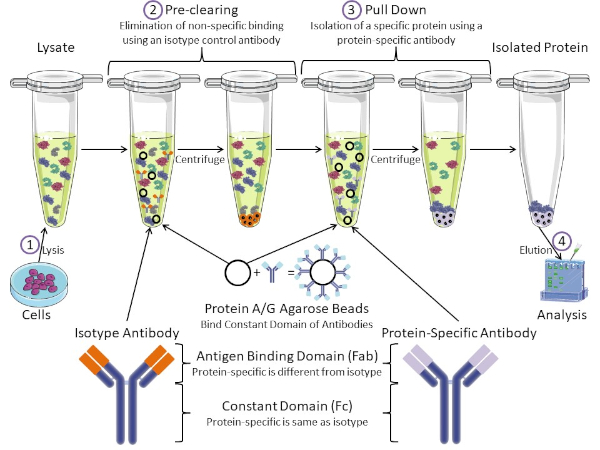
Figura 1: Visão geral do Procedimento de Imunoprecipitação. Imunoprecipitação é o isolamento de uma proteína específica usando um anticorpo. Após a produção de lise das células, há dois passos principais: pré-limpar e puxar para baixo. Durante a etapa de pré-limpeza, os lysatos celulares são pré-eliminados de proteínas que se ligam a anticorpos não especificamente usando um anticorpo de controle de isótipo. Em pull down step, a proteína alvo é puxada para baixo usando um anticorpo específico de proteína. A proteína isolada é então analisada pela mancha ocidental. Anticorpos isótipos e anticorpos específicos de proteína têm o mesmo domínio constante, mas diferentes domínios de ligação de antígeno. Um componente-chave deste protocolo são as contas de agarose da Proteína A/G que ligam o domínio constante dos anticorpos, permitindo a imunoprecipitação da proteína alvo. Clique aqui para ver uma versão maior desta figura.
Os anticorpos são o componente-chave de uma imunoprecipitação que o diferencia de outras formas de purificação de proteínas (ou seja, purificação da coluna de afinidade de níquel). Anticorpos são moléculas feitas por células B que podem reconhecer epítopos proteicos específicos. Os anticorpos possuem dois domínios: ligação constante (Fc) e antígeno (Fab) (Figura 1). O domínio constante identifica o tipo de anticorpo e dita a função in vivo. Normalmente, os domínios constantes dos anticorpos usados para IP são mouse, rat ou rabbit IgG. A porção de ligação de antígeno do anticorpo reconhece um epítope específico de uma proteína específica. Anticorpos podem reconhecer epítopos em proteínas dobradas que podem não existir quando a proteína é desnaturada e vice-versa. Portanto, a disponibilidade do epítope depende da dobra de proteínas - identificando um fator importante a considerar na escolha de anticorpos e condições para IP.
Ambos os sistemas procarióticos e eucarióticos têm proteínas de ligação de anticorpos. Nos sistemas eucarióticos, o objetivo é a proteção imune contra bactérias enquanto em sistemas procarióticos, o objetivo é a proteção do sistema imunológico. Proteínas de ligação de anticorpos afetam a metodologia IP de duas maneiras. Em primeiro lugar, há uma etapa de pré-compensação necessária (Figura 1) para livrar o lise de proteínas que ligam anticorpos - reduzindo assim a vinculação não específica no produto final. Esta etapa usa um anticorpo isótipo que tem o mesmo domínio constante que, mas um domínio de ligação de anticorpos diferente do seu anticorpo específico para proteínas. Proteínas bacterianas de ligação de anticorpos são o segundo componente-chave deste método. Depois que o anticorpo específico da proteína liga a proteína alvo, o anticorpo: complexo proteico deve ser puxado para baixo (Figura 1). Proteínas A, G e L são proteínas bacterianas que ligam o domínio constante dos anticorpos. Embora as bactérias usem isso para subverter o sistema imunológico, os pesquisadores têm cooptado este sistema para uma fácil purificação de anticorpos, e ele é usado durante os passos pré-clareira e pull-down. Essas proteínas têm diferentes afinidades de ligação para diferentes espécies e diferentes subtipos de domínio constantes - outro fator a considerar ao escolher condições para IP. Muitas empresas vendem contas de agarose rotuladas protein A/G (Figura 1), colunas de spin pré-feitas ou resinas para fazer colunas. Em geral, contas e colunas de spin são usadas para tamanhos amostrais menores, enquanto resinas são usadas para purificação a granel.
Neste exercício de laboratório, demonstramos como purificar a proteína endógena c-myc, dos timócitos murinas primários, utilizando contas de proteína A/G Plus baseadas em imunoprecipitação básica. O protocolo começa a partir da preparação de lisecelulares e termina com a verificação de retirada de proteínas bem sucedidas usando a análise de manchas ocidentais.
Procedure
1. Imunoprecipitação usando contas de agarose de proteína agarose
Preparação de lysato celular
- Centrifugar 108 timócitos em uma microcentrifuuagem a 13.000 rpm por 3 min e remover o supernatante.
Nota: O número celular variará dependendo dos níveis de expressão da proteína desejada e do tipo celular escolhido. - Suspenda-as as células em RIPA tampão de 500 μL com PMSF.
- Interrompa as células usando alguns pulsos rápidos com um vórtice e, em seguida, aspire o lysate algumas vezes com uma agulha de 25 G presa a uma seringa.
Nota: Evite criar bolhas. Use uma agulha maior, como uma agulha 21G para tipos de células maiores. - Incubar a célula no gelo por 10 minutos.
- Centrifugar o lysate a 13.000 rpm por 15 min a 4°C.
- Transfira o supernatante para um tubo de microcentrifuuge fresco e rotulado.
Pré-compensação
- Adicione 20 contas de proteína A/G PLUS de proteína agarose e 1 μg de um anticorpo de controle de isótipo (aqui, o anticorpo de controle de isótipo do mouse IgG1 é usado), ao liseto.
Nota: A escolha do anticorpo isótipo utilizado dependerá do anticorpo específico da proteína usado mais tarde na etapa de puxar para baixo. - Incubar a mistura de lise em uma rotadora centrífuga na sala fria (4°C) por 30 min.
- Centrifugar a amostra a 3200 rpm para 30 s a 4°C.
- Transfira o supernatante pré-limpo para um tubo de microcentrifuge de 1,5 mL fresco. Descarte a pelota.
Determinação da concentração de proteínas
- Determine a concentração proteica do lise celular realizando um Ensaio bradford.
- Aliquot 1000 μL Bradford Reagent em 7 tubos de microcentrífusão.
- Adicione as seguintes quantidades de padrão de proteína BSA (2 mg/mL) em 6 tubos (Tabela 1).
| Número do tubo | Volume BSA (μL) (2 mg/mL) | Concentração de proteínas (μg/μL) |
| 1 | 0 | 0 |
| 2 | 1 | 2 |
| 3 | 2 | 4 |
| 4 | 3 | 6 |
| 5 | 4 | 8 |
| 6 | 5 | 10 |
Tabela 1: Quantidades padrão de proteína BSA
- Notubo 7, adicione 1 μL do lise pré-limpo.
Nota: Para garantir que a concentração da amostra esteja dentro da faixa de detecção de ensaios, prepare e analise uma diluição de 1:2 ou 1:5lysate também. - Coloque 200 μL de cada um dos 7 tubos em poços individuais de uma placa de fundo plano, 96-bem repetindo cada amostra em triplicado.
- Leia placa em um leitor de placas a 595 nm.
- Gere a curva padrão no excel e calcule a concentração proteica do lysate pré-limpo.
Demolir
- Rotule dois tubos frescos de microcentrifuuge de 1,5 mL, um como "controle" e outro como "teste", que neste exemplo é c-myc.
- Coloque 500 μg de lise pré-limpa em cada um desses tubos.
Nota: A quantidade de proteína utilizada aqui dependerá da quantidade da proteína desejada para ser purificada. - Levante o volume total de cada tubo até 500 μL usando tampão de lise.
- Adicione 2 μg de anticorpo anti-c-myc ao tubo de grupo de ensaio e 2 μg mouse IgG1 anticorpo de controle de isótipo ao grupo controle.
Nota: A quantidade de anticorpos dependerá da eficáciado anticorpo e da quantidade da proteína alvo. - Incubar os tubos em uma rotadora na sala fria (4°C) por 2h.
- Adicione 20 contas de proteína A/G PLUS agarose a 20 μL agarose a cada tubo.
Nota: É aconselhável usar pontas de pipeta com a extremidade cortada para evitar danos às contas. - Incubar em uma rotadora em sala fria (4°C) durante a noite.
Nota: Dependendo da proteína alvo e da eficácia do anticorpo, esta etapa pode variar de 1h a durante a noite. - Centrifugar os tubos a 3200 rpm para 30 s 4°C para puxar para baixo as contas.
- Aspire o supernatante de cada tubo.
Nota: A proteína alvo está agora ligada às contas. - Lave as contas duas vezes usando o PBS de 500 μL 1X Dulbecco.
- Centrifugar os tubos a 3200 rpm para 30 s 4°C.
Nota: Para uma lavagem mais rigorosa, use buffers mais rigorosos, como o RIPA. - Aspire o tampão de cada tubo. Usando pontas de carregamento de gel, remova qualquer tampão que sobrou das contas e mantenha as contas no gelo para estotar a proteína.
Nota: Neste exemplo, a proteína é eluida em buffer de execução SDS-PAGE fervendo as contas, para análise de manchas ocidentais. Esta abordagem é adequada para verificar resultados ip ou para examinar interações proteína-proteína. Para outras aplicações a jusante, como a purificação de proteínas para análise estrutural ou enzimática, sistemas mais sofisticados, como tags de epítope (sinalização ou etiqueta de mamão) são usados para evitar a elução do anticorpo com a proteína de interesse.
2. Verificação ip através da análise de manchas ocidentais
Eletroforese SDS-PAGE:
- Suspender de nova resolução em 20 μL de corante de carregamento SDS-PAGE contendo β-mercapto-ethonol.
- Ferva as amostras a 95 °C por 5 minutos.
- Centrifugar as contas a 13.000 rpm para 10 s em temperatura ambiente.
- Utilizando pontas de carregamento de gel, encobante cuidadosamente as amostras obtidas das contas e carregue-as em poços de gel SDS-PAGE de 4-15% gradiente.
- Além das amostras, carregue uma pista com uma escada de proteína, bem como uma pista com o lysate pré-limpo para servir como controle de carga.
- Corra a 100 V até que a frente de corante atinja a parte inferior do gel (~1h).
Análise de manchas ocidentais:
- Faça sanduíche de mancha ocidental, garantindo que a membrana PVDF esteja entre gel e cátodo vermelho.
- Transferência para 1 h a 100 V.
- Coloque a membrana em tampão de bloqueio de 5 mL à temperatura ambiente por 1h em um roqueiro em um ajuste baixo, para bloquear os locais de ligação proteica não específico.
Nota: Os volumes de tampão de bloqueio, anticorpo primário, anticorpos secundários e lavagens podem precisar ser aumentados para manchas de tamanho maior. - Incubar a mancha com anticorpo anti-c-myc de 5 mL no tampão de bloqueio durante a noite a 4°C no roqueiro em um ajuste baixo.
Nota: O anticorpo aqui utilizado deve ser diferente do usado na etapa de puxar para baixo. - Lave a mancha 3-6 vezes usando 5 mL TBST com cada lavagem sendo 5 min à temperatura ambiente em um roqueiro em um ajuste baixo.
- Incubar a mancha com anticorpo secundário de corrente anti-coelho com marca HRP no buffer de bloqueio, por 1 h à temperatura ambiente no roqueiro em um ajuste baixo.
Nota: A escolha do anticorpo secundário dependerá do anticorpo primário usado para a mancha ocidental. Além disso, uma cadeia leve específica secundária é usada no protocolo porque a proteína alvo está próxima em peso molecular à cadeia pesada do anticorpo. Se a proteína alvo estiver perto de 50kDa, use uma cadeia de luz específica secundária. Se a proteína alvo estiver próxima de 25kDa, use e cadeia pesada específica secundária. - Lave a mancha 3-6 vezes usando 5 mL TBST com cada lavagem sendo 5 min à temperatura ambiente em um roqueiro em um ajuste baixo.
- Remova o líquido da mancha e dab borda da mancha em lenços de laboratório para remover o excesso de líquido.
- Borra de cobertura com 1x reagente de detecção de chemiluminescente' e incubar por 1 min.
Nota: As etapas a seguir devem ser feitas em rápida sucessão, pois o reagente de detecção é sensível à luz e ao tempo. - Borda dab de mancha em lenços de laboratório para remover o reagente de detecção em excesso.
- Coloque a mancha na superfície de imagem da bandeja Imager.
Nota: Manchas de chemiluminescentes também podem ser visualizadas usando filme. - Imagem usando o 'Programa Chemiluminescent' para capturar vários pontos de tempo de 10 s a 5 min.
Nota: O tempo ideal pode mudar com base na quantidade de proteína e na qualidade do anticorpo. - Escolhai uma imagem com visibilidade de banda ideal e, em seguida, exporte essa imagem.
- Antes de mover a mancha, tire uma foto da mancha usando o imager, para capturar a localização da escada. Então, exporte essa imagem também.
- Usando um software de preparação de slides (como o PowerPoint), alinhe as bandas e as imagens da escada para formar uma única imagem.
A imunoprecipitação, ou IP, é uma técnica amplamente utilizada para isolar uma proteína de interesse de uma célula ou tecido ou um fluido corporal para caracterização de proteínas ou para investigar interações proteína-proteína.
O processo começa com um anticorpo, que tem alta afinidade e especificidade para a proteína alvo. Este anticorpo é misturado com a amostra, permitindo que complexos de alvo de anticorpos se formem. Qualquer proteína ligada à proteína alvo também fica indiretamente ligada ao anticorpo no processo. Em seguida, a solução é incubada com contas de agarose, conjugadas a uma proteína bacteriana, que tem forte afinidade com a região constante dos anticorpos. A proteína bacteriana se liga ao anticorpo e conecta os complexos de alvo de anticorpos às contas. Em seguida, a solução é centrifugada para precipitar as contas, extraindo assim todo o complexo contendo o anticorpo de ligação, a proteína alvo e quaisquer proteínas interativas. Finalmente, as proteínas vinculadas são extraídas das contas e liberadas umas das outras e são usadas para análises posteriores por técnicas como a mancha ocidental.
Várias variações de diferentes partes desta técnica são comumente usadas, como pré-limpeza, uso de etiquetas de peptídeos ou contas magnéticas, ou análise de outros parceiros de ligação não-proteica. O IP pode ser preceed por uma etapa de pré-compensação, para remover proteínas não específicas de ligação de anticorpos na amostra e minimizar o fundo. Isso envolve primeiro incubar a amostra com anticorpos de controle de isótipo, permitindo que eles se liguem a essas proteínas e, em seguida, usando contas de agarose para precipitar os complexos. A amostra está então pronta para prosseguir para o IP real.
As tags peptídeos são úteis se um anticorpo específico não estiver disponível para IP. Aqui, a proteína alvo pode ser geneticamente modificada para conter uma tag de epítope de peptídeo e um anticorpo contra a etiqueta é capaz de extrair a proteína de interesse. Contas magnéticas são frequentemente usadas em vez de agarose para precipitar o alvo. Após a ligação ao complexo de alvo de anticorpos, o tubo de amostra é colocado em um campo magnético forte, que extrai as contas da solução. Isso elimina a necessidade de centrifugação e melhora a velocidade e a conveniência.
A imunoprecipitação também é usada para estudar proteínas de ligação de DNA ou RNA e são conhecidas como imunoprecipitação de cromatina e imunoprecipitação de RNA, respectivamente. Essas variações são úteis para a solução de problemas e adaptação do método para diferentes aplicações experimentais. Neste vídeo, você observará como pré-limpar uma célula lysate e realizar imunoprecipitação para extrair uma proteína de interesse, seguido pela análise de manchas ocidentais para validar o experimento.
Para começar, coloque as células pré-coletadas em uma microcentrifuagem e gire a 13 mil rpm por três minutos. Após o giro, remova o supernascer e, em seguida, resuspenja as células em 500 microliters de tampão de lise RIPA com PMSF. Agora, interrompa as células usando alguns pulsos rápidos com um vórtice e, em seguida, aspire o lysate algumas vezes com uma agulha calibre 25 presa a uma seringa, tomando cuidado para evitar criar bolhas. Coloque as células no gelo por 15 minutos. Depois de incubar as amostras no gelo, centrifugar o lysate por 15 minutos a quatro graus celsius.
Rotule um novo tubo de microcentrífuga de 1,5 mililitro. Após o giro, transfira o sobrenante para o tubo recém-rotulado e descarte a pelota. Em seguida, pré-limpe o lysate de contaminantes que se ligam não especificamente às contas de agarose ou ao anticorpo primário adicionando 20 microliters das contas de proteína A/G PLUS-agarose e um micrograma de um anticorpo de controle de isótipo ao lysato, que neste exemplo é um controle isótipo do mouse IgG1. Incubar o tubo em uma rotadora em uma sala fria por 30 minutos. Depois de rodar o lysate na sala fria por 30 minutos, centrifugar a amostra a 3200 rpm por 30 segundos a quatro graus celsius. Remova o tubo da centrífuga e transfira o supernanato pré-limpo para um tubo de microcentrífuga de 1,5 mililitro fresco. Descarte a pelota.
Agora, determine a concentração de proteína do lysate celular realizando um ensaio de Bradford. Rotule sete 1. Tubos de microcentrífuga de 5 mililitros de um a seis e amostra e alíquota de 1000 microliters do reagente bradford em cada tubo. Seis dos tubos serão usados para fazer uma curva padrão adicionando várias quantidades de quantidades conhecidas de BSA a cada tubo. Os valores a adicionar estão listados nesta tabela. No sétimo tubo de amostra, adicione um microliter do lysate pré-limpo. Coloque 200 microliters de cada um dos sete tubos em poços individuais de uma placa de fundo plano de 96 poços, repetindo cada amostra em triplicado para que haja três colunas de sete amostras. Leia a placa em um leitor de placas, usando um comprimento de onda de 595 nanômetros. Depois de criar uma curva padrão no Excel, calcule a concentração proteica do lysate pré-limpo.
Em seguida, rotule dois tubos de microcentrífuga de 1,5 mililitro, um como controle e outro como teste, que neste exemplo, será o anticorpo c-myc. Coloque 500 microgramas do lysate pré-limpo em cada um desses tubos e, em seguida, traga o volume total para cada tubo até 500 microliters usando tampão de lise. Em seguida, adicione dois microgramas do anticorpo anti-c-myc ao tubo de grupo de ensaio. Para o controle, adicione dois microgramas do anticorpo de controle isótipo IgG1 do mouse. Uma vez que os anticorpos são adicionados aos tubos, coloque as amostras em uma rotadora em uma sala fria e incubar por duas horas. Agora, adicione as contas de agarose. Para isso, recomenda-se cortar a ponta de uma ponta de pipeta e, em seguida, usando esta ponta modificada, adicionar 200 microliters das contas protein A/G PLUS-agarose em cada tubo. Incubar os tubos em uma rotadora na sala fria durante a noite.
Após a incubação, remova os tubos da rotadora e gire os lises no microcentrifuuge para puxar as contas. Após a rotação ser completa, remova os tubos da centrífuga e aspire o sobrenatante de cada tubo. Em seguida, lave as contas usando 500 microliters do PBS 1X Dulbecco. Coloque os tubos em uma microcentrifuagem e gire por 30 segundos a quatro graus celsius. Depois disso, remova o supernatante. Repita os passos de lavagem e centrífuga mais uma vez para um total de duas vezes. Remova os tubos do microcentrifuuge e aspire o tampão de cada tubo. Usando pontas de carregamento de gel, remova qualquer tampão que sobrou das contas, mantendo as contas no gelo para elutar a proteína ligada.
Neste exemplo, a proteína é eluida em buffer de execução SDS-PAGE, fervendo para análise de manchas ocidentais. Para isso, resuspenque as contas em 20 microliters de corante de carregamento SDS-PAGE contendo beta-mercaptoetanol, ou BME. Ferva as amostras a 95 graus celsius por cinco minutos para dissociar os imunocomplexos das contas. Em seguida, centrifugar as contas em velocidade máxima por 10 segundos à temperatura ambiente. Remova os tubos do microcentrifuuge e segure-os em um rack à temperatura ambiente. Usando pontas de carregamento de gel, pipeta cuidadosamente as amostras das contas e carregue-as em poços de um gel SDS-PAGE de 4 a 15% gradiente. Além das amostras, carregue uma pista com uma escada de proteína, bem como uma pista com o lysate pré-limpo para servir como controle de carga. Uma vez que o gel é carregado, execute o gel a 100 volts.
Depois que a frente de corante atingir o fundo do gel, que deve levar aproximadamente uma hora, pare o gel e faça um sanduíche de mancha ocidental, garantindo que a membrana PVDF esteja entre o gel e o cátodo. Coloque o sanduíche de mancha ocidental no aparelho de transferência e transfira as proteínas no gel para a membrana por uma hora a 100 volts. Após a transferência concluída, coloque a membrana em cinco mililitros de bloco para evitar que os anticorpos se liguem não especificamente à membrana. Abale em um ajuste baixo por uma hora em temperatura ambiente. Quando o temporizador soar, remova o buffer de bloqueio. Adicione cinco mililitros do tampão de bloqueio com o anticorpo de detecção à membrana. Aqui, um anticorpo anti-c-myc, que é diferente do usado para puxar para baixo, é usado.
Incubar a mancha durante a noite, a quatro graus celsius em um roqueiro em um cenário baixo. Após a incubação, remova o anticorpo e bloqueie o buffer. Lave a mancha, usando cinco mililitros de TBST por cinco minutos em temperatura ambiente, em um roqueiro em um ajuste baixo. Esta etapa de lavagem deve ser repetida duas a cinco vezes para um total de três a seis lavagens, utilizando TBST fresco para cada lavagem. Adicione cinco mililitros de um a 1000 anticorpos secundários e bloqueando o buffer à mancha. Neste caso, o anticorpo secundário é a cadeia de luz anti-coelho marcada pelo HRP. Incubar a mancha em um roqueiro em um cenário baixo para um nosso à temperatura ambiente. Em seguida, remova o tampão e lave a mancha com cinco mililitros de TBST. Incubar esta lavagem em um roqueiro em um ajuste baixo por cinco minutos em temperatura ambiente. Repita esta lavagem para um total de seis a 12 lavagens, cada uma com um fresco cinco mililitros de TBST. Remova a lavagem final primeiro despejando o líquido da mancha. Em seguida, usando pinças, coloque a borda da mancha em uma limpeza de laboratório para remover qualquer excesso de líquido e, em seguida, coloque a mancha em um recipiente fresco. Em seguida, cubra a mancha com 1X Reagente de Detecção Chemiluminescente e incubar por um minuto.
Trabalhando rapidamente, dab a borda da mancha em um limpador de laboratório para remover qualquer reagente de detecção em excesso e, em seguida, colocar a mancha na superfície de imagem da bandeja Imager. Imagem usando o programa Chemiluminescent para capturar vários pontos de tempo de 10 a 30 segundos. Depois que a mancha for imagem, escolha uma imagem com visibilidade ideal da banda e, em seguida, exporte essa imagem. Antes de mover a mancha, use o Imager para tirar uma foto da mancha para capturar a localização da escada. Então, exporte essa imagem também. Finalmente, usando um software de preparação de slides, como o PowerPoint, alinhe as bandas e as imagens da escada para formar uma imagem.
Esta imagem mostra o resultado da mancha ocidental para a imunoprecipitação da proteína c-myc das células de timócito. Da esquerda para a direita, as pistas representam o controle do isótipo, o IP c-myc e a entrada de lise pré-limpa. A pista da extrema direita é uma imagem mesclada da escada de peso molecular. A banda forte, em torno de 25 kilodaltons é da cadeia leve e a de 50 kilodaltons é da cadeia pesada do anticorpo de ligação e não são específicas para o IP ou as amostras. C-myc corre em torno de 67 kilodaltons em manchas ocidentais e geralmente é visível logo abaixo da banda de escada kilodalton de 75 kilodalton. Nesta mancha, a banda c-myc é visível na segunda pista, mas ausente na primeira pista, indicando que o anticorpo IP puxou com sucesso c-myc. Não há faixa visível na pista de lise pré-limpa, sugerindo que essa proteína tem baixos níveis de expressão endógena.
Subscription Required. Please recommend JoVE to your librarian.
Results
Os resultados do procedimento detalhado acima são mostrados na Figura 2. Da esquerda para a direita, as pistas contêm o grupo controle (isótipo), o grupo de teste (c-myc), o liseto pré-limpo (lysate) e a escada de peso molecular (escada). As bandas de escada de 25 e 75 kDa estão marcadas. As duas bandas proeminentes a ~25 kDa e 50 kDa são a cadeia leve e pesada do anticorpo de ligação, respectivamente e não são específicas para o IP ou as amostras. proteína c-myc que corre em torno de 67kDa em manchas ocidentais e geralmente é visível logo abaixo da faixa de escada de 75 kDa. Nesta mancha, a banda c-myc é visível na segunda pista, mas ausente na primeira pista, indicando que o anticorpo IP puxou com sucesso c-myc. Não há faixa visível na pista de lise pré-limpa, sugerindo que essa proteína tem baixos níveis de expressão endógena.

Figura 2: Resultados de uma Análise de Manchas Ocidentais, utilizada para avaliar a purificação de c-myc por imunoprecipitação. Uma banda a 67 kDa, correspondente ao c-myc, é visível na pista anti-c-myc, mas não na faixa de controle do isótipo. Note que os níveis de c-myc não eram altos o suficiente para serem visualizados na pista de lise. Clique aqui para ver uma versão maior desta figura.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
Em suma, a imunoprecipitação é o isolamento de uma proteína específica usando um anticorpo. Neste exemplo, os resultados da imunoprecipitação foram analisados pela mancha ocidental para avaliar a pureza. A proteína isolada poderia ser usada em uma série de aplicações posteriores, incluindo: NMR para estrutura proteica, Espectrometria de Massa para sequência de aminoácidos, ou ensaios in vitro para caracterização enzimática. Os IPs também podem caracterizar os parceiros interajantes das proteínas. Por exemplo, após o isolamento, DNA ou RNA podem ser isolados para sequenciamento. Co-imunoprecipitações avaliam interações proteína-proteína. Quando a proteína alvo é puxada para baixo durante um IP, proteínas interativas também podem ser puxadas para baixo. Esses parceiros de interação podem ser avaliados por espectrometria de massa e mancha ocidental. A imunoprecipitação é uma técnica poderosa para estudar biologia proteica.
Subscription Required. Please recommend JoVE to your librarian.
References
- Olliver, C. L. and Boyd, C. D. (1984). Immunoprecipitation of In Vitro Translation Products with Protein A Bound to Sepharose. In J. M. Walker (eds), Nucleic Acids. Methods in Molecular Biology (pp. 157-160). New Jersey: Humana Press.
- Thurston, C. F. and Henley, L. F. (1988). Direct Immunoprecipitation of Protein. In J. M. Walker (eds), New Protein Techniques. Methods in Molecular Biology (pp. 149-158). New Jersey: Humana Press.
- Anderson, N. G. (1998). Co-immunoprecipitation: Identification of Interacting Proteins. In R. A. Clegg (eds), Protein Targeting Protocols.Methods in Molecular Biology (pp. 35-45). New Jersey: Humana Press.
- Jackson, D. I. and Dickson, C. (1999). Protein Techniques: Immunoprecipitation, In Vitro Kinase Assays, and Western Blotting. In P.T. Sharpe and I. Mason (eds), Molecular Embryology. Methods in Molecular Biology (pp. 699-708). New Jersey: Humana Press.
- Trieu, E. P. and Targoff, I. N. (2015). Immunoprecipitation: Western Blot for Proteins of Low Abundance. In B. Kurien and R. Scofield (eds), Western Blotting. Methods in Molecular Biology (pp. 327-342). New York, NY: Humana Press.