



Overview
Fonte: Jonathan F. Blaize1, Elizabeth Suter1e Christopher P. Corbo1
1 Dipartimento di Scienze Biologiche, Wagner College, 1 Campus Road, Staten Island NY, 10301
La valutazione quantitativa dei procarioti può essere onerosa data la loro abbondanza, la propensione alla proliferazione esponenziale, la diversità delle specie all'interno di una popolazione e le specifiche esigenze fisiologiche. Ad aggravare questa sfida, è la natura a quattro fasi in cui i batteri si replicano (ritardo, log, stazionario e morte). La capacità di stimare con precisione la concentrazione di microrganismi è necessaria per il successo dell'identificazione, dell'isolamento, della coltivazione e della caratterizzazione (6). Pertanto, i microbiologi hanno impiegato la diluizione seriale e varie tecniche di placcatura per oltre un secolo per quantificare in modo affidabile la carica batterica e virale in ambienti di laboratorio clinici, industriali, farmaceutici e accademici (2,4,6). Le descrizioni di questa metodologia apparvero per la prima volta nel 1883 quando lo scienziato e medico tedesco Robert Koch pubblicò il suo lavoro sugli agenti infettivi che causano malattie (2). Spesso indicato come il padre della batteriologia moderna, le tecniche prenominate di Koch sono diventate il gold standard per l'enumerazione di microrganismi, coltivabili o meno, in tutto il mondo.
La diluizione seriale è una riduzione sistematica di un'entità nota o sconosciuta (un soluto, un organismo, ecc.) attraverso la successiva risos sospensione di una soluzione iniziale(soluzione 0) in volumi fissi di un diluente liquido (spazi vuoti). Questi spazi vuoti di solito consistono di 0,45% di soluzione salina, anche se la composizione può essere variata (7). Mentre uno sperimentatore può scegliere qualsiasi volume per ogni diluente, è più spesso un multiplo di 10, facilitando la riduzione logaritmica del campione. Ad esempio, lasoluzione 0 contiene un totale di 100 cellule di E. coli sospese in 10 ml di brodo nutritivo. Se 1 mL disoluzione 0 viene rimosso e aggiunto a 9 mL di soluzione salina (diluente1), la nuovasoluzione (soluzione 1) conterrebbe 1/10 della concentrazione iniziale di E. coli. In questo esempio, la nuova soluzione(soluzione 1) conterrebbe 10 cellule di E. coli. Ripetendo questo processo rimuovendo 1 mL disoluzione 1 e aggiungendolo ad altri 9 mL di soluzione salina (diluente2)si produrrebbela soluzione 2, contenente solo una singola cellula di E. coli. Poiché ogni nuova soluzione (9 mL di diluente + 1 mL di soluzione) contiene un totale di 10mL, possiamo concludere che il fattore di diluizione per questa riduzione è 10 o che si è tratta di una diluizione seriale 10 volte (Figura 1). Poiché abbiamo iniziato solo con 100 cellule in questo esempio e stiamo diluendo di un fattore 10, sono necessari solo due passaggi per raggiungere la concentrazione minima assoluta di 1 cellula.
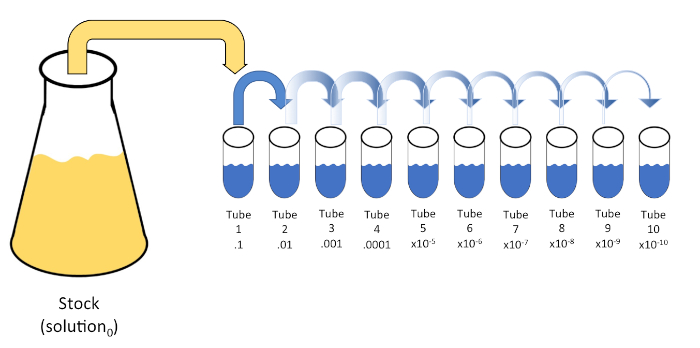
Figura 1: Diluizione seriale di una soluzione stock. Un'aliquota di 1 mL della soluzione stock(soluzione 0) viene aggiunta al tubo 1 che contiene 9 mL di soluzione salina allo 0,45% (dilent1); il prodotto di questa miscela è lasoluzione 1. Ripetere aliquotando 1 mL dellasoluzione appena creata 1 e aggiungendola al tubo 2. L'aliquotazione e la ricaspensione continuano in questo modo fino al raggiungimento del tubo finale, diluendo la concentrazione di stock di un fattore di 10 ciascuno ad ogni passo. Fare clic qui per visualizzare una versione più grande di questa figura.
La diluizione seriale è la tecnica più semplice per ottenere concentrazioni gestibili di un organismo desiderato ed è completata da striature e spalmature di Petri, solo due delle molte tecniche di placcatura utilizzate dai microbiologi. Questo vantaggio di questo approccio è che lo sperimentatore può raccogliere ceppi puri di una singola specie o ceppi separati da una popolazione mista (7). La striatura si ottiene introducendo un organismo in un mezzo solido (generalmente costituito da agarose) su cui crescerà se sono disponibili i nutrienti appropriati. Spazzare delicatamente un ciclo inoculante sterile attraverso il mezzo (in modo che rimanga una striscia sottile) in un modello sinusoidale rigido distribuirà l'organismo proporzionalmente alla frequenza della forma d'onda dello sperimentatore. Dividere la capsula di Petri in terzi o quarti (striscia di quadrante) e diminuire la frequenza di ogni striscia quando viene inserita una nuova regione del piatto ridurrà gradualmente il numero di microrganismi che possono occupare quella regione, producendo singole colonie invece di un prato batterico non quantificabile. La placcatura diffusa non diluisce ulteriormente i campioni; uno spandi vetro sterile viene utilizzato per distribuire un'aliquota di mezzi di sospensione su un'intera capsula di Petri (Figura 2). Le colonie che crescono sulla piastra diffusa derivano da una singola cellula e ogni colonia sul piatto può essere contata per stimare il numero di unità formanti colonie per millilitro (CFU) in una data sospensione, rappresentata come CFU/mL (6) (Figura 3) L'agar morbido e la replica placcatura sono variazioni delle tecniche di cui sopra e consentono l'isolamento del batteriofago e lo screening mutante, rispettivamente (1,7).
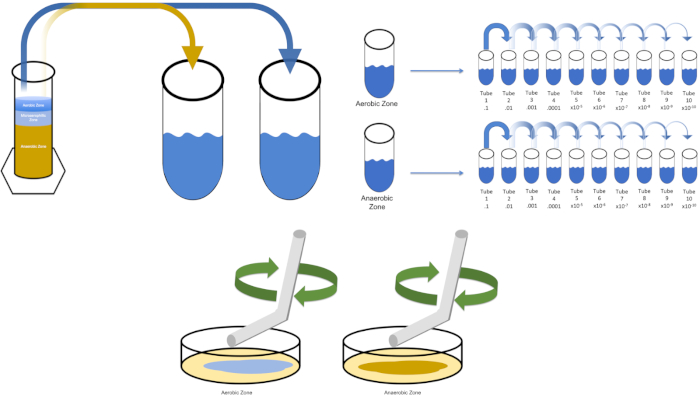
Figura 2: Striature di piastre per l'enumerazione batterica e l'isolamento del ceppo. Etichettare il fondo di una capsula di Petri con informazioni di identificazione (nome dei batteri, data, supporto) e dividere in terzi. Dopo aver selezionato una diluizione appropriata del campione di riserva, prendere un anello di inoculazione sterile (monouso o fiammato) e immergerlo nella provetta (qui, T3). Sollevare leggermente il coperchio della capsula di Petri su un lato in modo che solo l'anello inoculante possa accedere all'agar. Fai scivolare il loop inoculante attraverso la parte superiore del supporto a zig-zag facendo attenzione a non compromettere l'agar. Ruotare la piastra di circa 1/3 (~118°) e ridurre la frequenza del movimento a zig-zag. Ruota un ultimo tempo e riduci ancora una volta la frequenza a zig-zag. Fare clic qui per visualizzare una versione più grande di questa figura.
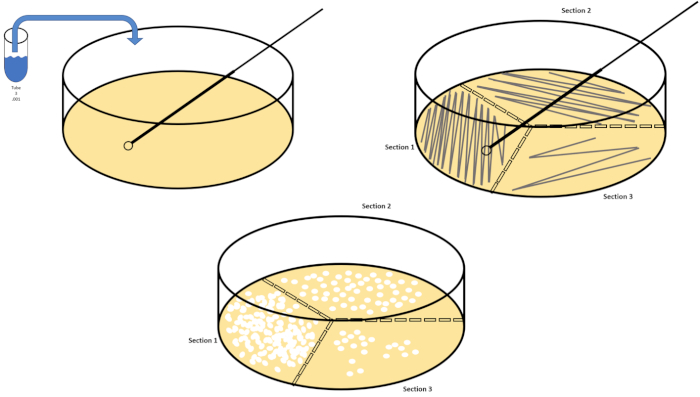
Figura 3: Placcatura diffusa. 1 g della zona aerobica è stato sospenso in T1 e poi diluito in serie. Un'asta sterile di spargimento monouso in vetro o plastica viene utilizzata per distribuire l'inoculo in ogni piatto. Questo è stato ripetuto con 1 g della zona anaerobica. Fare clic qui per visualizzare una versione più grande di questa figura.
Come per le diluizioni seriali, viene utilizzata una scala logaritmica per esprimere la concentrazione dell'organismo. Il numero di colonie coltivate in piastre di Petri standard di 100 mm x15 mm può essere enumerato manualmente (o automatizzato con l'aiuto dell'elaborazione computazionale) identificando cluster isolati di crescita. I conteggi che ammontano a meno di 30 o superiore a 300 devono essere definiti rispettivamente come troppo pochi da contare (TFTC) o troppo numerosi da contare (TNTC). Nel caso di quest'ultimo, deve essere eseguita una diluizione seriale per ridurre la concentrazione prima di riaffermire una nuova capsula di Petri. Facendo la media del numero di colonie autonome identificate da tre piastre di Petri separate e moltiplicando la media per il fattore di diluizione si otterrà CFU/mL; tracciare il log10 di CFU/mL contro il tempo rivelerà il tempo medio di generazione dell'organismo (7).
Procedure
1. Configurazione
- Un diagramma di flusso che elenca tutti i materiali, il protocollo sperimentale graduale e il metodo per scartare le forniture dovrebbe essere scritto in un quaderno di laboratorio e tenuto vicino allo spazio di lavoro sperimentale.
- Gli spazi di lavoro devono essere sterilizzati con un antisettico appropriato (70% di etanolo) e lo sperimentatore dovrebbe mitigare il rischio di contaminazione indossando indumenti da laboratorio puliti che li proteggano anche dalle anomalie di esposizione. Gli indumenti adatti includono, ma non sono limitati a un cappotto da laboratorio, guanti in lattice o nitrile, google, respiratori e scarpe chiuse. È fondamentale mantenere la tecnica asettica in ogni momento.
- Preparare 90 ml di soluzione salina al 0,45%. Utilizzando un cilindro graduato pulito, misurare 90 ml di acqua sterile e trasferirlo in un matraccio Erlenmeyer pulito etichettato 0,45% soluzione salina. Pesare .405 g di cloruro di sodio (Sigma-Aldrich NaCl S9888) e aggiungerlo al matraccio etichettato 0,45% salino. Ruota ripetutamente fino a quando nessun soluto rimane visibile.
- Al termine, lo sperimentatore dovrebbe ri-sterilizzare tutte le superfici e scartare eventuali organismi indesiderati, scorte di diluenti, piastre di Petri o anelli di inoculazione usa e getta secondo le linee guida OSHA. Gli indumenti da laboratorio possono essere rimossi prima di lavarsi le mani.
2. Preparazione dei supporti
- Selezionare i mezzi appropriati per la coltivazione di un organismo desiderato. Nella maggior parte degli scenari, un brodo consentirebbe una crescita batterica sufficiente. Poiché qui si desiderano organismi di un protocollo Winogradsky, una colonna composta da carbonato di calcio, zolfo, cellulosa e fango è stata assemblata e lasciata indisturbata per 7 giorni. La colonna con nome è separata in sezioni aerobiche, microaerofile e anaerobiche.
- Scegli un mezzo appropriato per placcare l'organismo di interesse. L'integrazione di mezzi liquidi con agar di grado microbiologico è tipicamente impiegata come agente solidificante. LB medium/agar è sufficiente quando si raccolgono campioni dalle regioni aerobica, microaerofila e anaerobica della colonna prenaminata. Nota: i campioni della regione microaerofila non sono stati raccolti per questa procedura. Tuttavia, questi organismi dovrebbero essere coltivati in barattoli di candele. L'introduzione di una candela in questa camera di coltivazione prima della sigillatura crea un ambiente a basso contenuto di ossigeno adatto alla proliferazione microaerofila.
- Poiché desideriamo preparare 250 ml, utilizzare fiaschi Erlenmeyer da 500 ml (o più grandi) per evitare l'ebollizione durante l'autoclave. Etichetta uno "Brodo" e l'altro "Agar".
- Determinare la quantità di supporti necessari per creare ogni soluzione seguendo le raccomandazioni di concentrazione del produttore. LB Agar, usato qui, viene preparato combinando 25g / L con acqua ultrapura. Il nostro volume di 250 mL richiede una soluzione di 6,25 LB Agar/250 mL di acqua. Allo stesso modo, LB Broth viene preparato combinando lo stesso rapporto di LB Broth e acqua. Poiché non è integrato con un agente solidificante, non si indurirà quando raffreddato.
- Pesare il supporto e mescolarlo con acqua in proporzioni coerenti con le raccomandazioni del produttore. Aggiungere 6,25 g di LB Agar a un pallone etichettato "Agar" e 6,25 g di LB Broth a un pallone etichettato "Brodo". Aggiungere 250 ml di acqua ultrapura a ciascun pallone.
- Avvolgere un foglio di alluminio su ogni pallone e utilizzare un'autoclave per sterilizzare i mezzi per un minimo di 15 minuti a 121 ° C, 15 psi.
- Utilizzando un guanto o un cuscinetto resistente al calore, rimuovere i palloni dall'autoclave al termine del ciclo e metterli in un bagno d'acqua a 40-50 ° C.
- Una volta raggiunta la temperatura appropriata, versare il contenuto del matraccio etichettato "Brodo" in un Erlenmeyer da 250 ml o in un matraccio a fondo tondo. Etichettare il matraccio da 250 ml"soluzione 0".
- Ottenere piastre di Petri sterili da 10, 100 mm x 15 mm ed etichettarle con la data, il nome, il tipo di mezzo utilizzato e la zona della colonna Winogradsky da cui verranno raccolti gli organismi.
- Rimuovere il pallone etichettato "Agar" dal bagno d'acqua e iniziare a versare in ciascuna delle 10 piastre di Petri. Non più di 15 ml dovrebbero essere aggiunti a ciascun piatto. Questo può anche essere eseguito con un pipettor e una pipetta sierologica da 25 ml per migliorare la precisione. Utilizzare una punta di pipetta sterile per rimuovere eventuali bolle presenti, quindi coprire con i coperchi della piastra e lasciare solidificare durante la notte.
3. Preparazione del diluente
- Preparare dieci provette in grado di conservare 20 ml o più in un rack ed etichettarle T1-T10. Ogni numero di tubo è coerente con il fattore di diluizione a cui corrisponde (cioè T4 = 1x10-4 o 0,0001 o 1/10.000 di concentrazione di stock).
- Pipettare 9 mL di soluzione salina allo 0,45% in ciascuna delle 10 provette.
- Gli spazi vuoti salini sono ora pronti per essere sterilizzati in autoclave. Utilizzare un foglio di alluminio per coprire ciascuna delle 10 provette e quindi trasferirle su un rack per provette compatibile con l'autoclave. Sterilizzare per un minimo di 15 minuti a 121°C, 15 psi.
- Rimuovere gli spazi vuoti utilizzando guanti resistenti al calore e lasciare raffreddare. Coprire e conservare a 4°C fino a quando i tubi non hanno raggiunto la temperatura ambiente o quando sono freddi al tatto.
4. Coltivazione dell'organismo bersaglio
- Inoculare"soluzione 0"con una singola colonia da una piastra precedentemente striata o 50 μL di un brodo congelato. Lasciare all'organismo bersaglio il tempo di replicarsi posizionando la"soluzione 0"inoculata in un incubatore a 37°C durante la notte con agitazione (se necessario). (Nota: il pallone deve essere coperto per evitare contaminazioni. Se l'organismo bersaglio è aerobico, utilizzare una garza sterile e tappi di cotone per prevenire la contaminazione. Se si valutano le regioni della colonna Winogradsky, è sufficiente rimuovere 1 grammo da ciascuna zona desiderata (aerobica e anaerobica ai fini di questo studio) e risospentare in T1 prima di procedere al passaggio 5.3.
5. Diluizione seriale
- Ottenere il pallone etichettato "Brodo nutriente" dall'incubatrice e agitare vigorosamente.
- Pipettare 1 mL di"soluzione 0"nella provetta etichettata T1. Vortice T1. Se si valutano gli espianti di Winogradsky, pesare 1 grammo della zona desiderata e aggiungerlo a T1 prima del vortice. (Nota: qui viene utilizzato 1 mL per semplicità- possono essere utilizzati anche volumi più piccoli o più grandi di diluente).
- Rimuovere 1 mL dalla provetta T1 e aggiungerlo alla provetta T2. Vortice T2.
- Rimuovere 1 mL dalla provetta T2 e aggiungerlo alla provetta T3. Vortice T3.
- Rimuovere 1 mL dalla provetta T3 e aggiungerlo alla provetta T4. Vortice T4.
- Rimuovere 1 mL dalla provetta T4 e aggiungerlo alla provetta T5. Vortice T5.
- Rimuovere 1 mL dalla provetta T5 e aggiungerlo alla provetta T6. Vortice T6.
- Rimuovere 1 mL dalla provetta T6 e aggiungerlo alla provetta T7. Vortice T7.
- Rimuovere 1 mL dalla provetta T7 e aggiungerlo alla provetta T8. Vortice T8.
- Rimuovere 1 mL dalla provetta T8 e aggiungerlo alla provetta T9. Vortice T9.
- Rimuovere 1 mL dalla provetta T9 e aggiungerlo alla provetta T10.
6. Placcatura diffusa
- Pipetta 100 μL di un campione diluito da T1 direttamente su una capsula di Petri. Questo passaggio può essere, ma non è necessario che sia, ripetuto per ogni tubo.
- Ottenere un'asta di diffusione sterile e monouso o una fiamma sterilizzare un'asta di diffusione del vetro. In senso orario/antiorario, far scivolare la parte orizzontale dell'asta di diffusione per distribuire equamente il campione all'interno della capsula di Petri.
- Ripetere l'operazione per ogni zona della colonna Winogradsky da valutare.
- Incubare le piastre in un incubatore a 37°C per 24 ore. Per gli organismi anaerobici, utilizzare una camera anaerobica.
7. Striature
- Selezionare una diluizione appropriata dell'organismo bersaglio. Ad esempio,la soluzione 4 produrrà una diluizione di 1/10.000 della concentrazione iniziale. Tipicamente, le diluizioni di 1/1.000th (T3/Solution), 1/1.000.000th (T6/Solution6) e 1/1.000.000.000th (T9/Solution9) vengono valutate per enumerare i microbi.
- Utilizzando un anello di inoculazione monouso sterile in plastica o un anello di inoculazione in metallo riutilizzabile che è stato sotto tiro per non meno di 10 secondi, immergersi nella soluzione desiderata dal passaggio 5. I circuiti di inoculazione calibrati devono trasferire 0,01 ml. (Attenzione: non lasciare che il loop fiammato contatti i batteri immediatamente dopo la rimozione dal calore)
- Sollevare leggermente il coperchio della capsula di Petri su un lato in modo che solo l'anello inoculante possa accedere all'agar. Fai scivolare il loop inoculante attraverso la parte superiore del supporto a zig-zag facendo attenzione a non compromettere l'agar. Abbassare il coperchio della capsula di Petri.
- Utilizzare un nuovo ciclo di inoculazione monouso o risterizzare il ciclo riutilizzabile.
- Ruotare la piastra di circa 1/3 (~118°) e ridurre la frequenza del movimento a zig-zag.
- Ancora una volta, utilizzare un nuovo loop usa e getta o ri-sterilizzare un loop metallico prima di ruotare un tempo finale e ridurre ancora una volta la frequenza a zig-zag. Abbassare il coperchio della capsula di Petri.
- Ripetere i passaggi 7.2 - 7.6 fino a quando almeno tre piastre di Petri sono state striate per tre diverse diluizioni, utilizzando un nuovo loop usa e getta o riaccheggiando un loop riutilizzabile (Figura 2).
- Mettere le piastre di Petri striate in un'incubatrice a 37 ° C durante la notte. Per gli organismi anaerobici, utilizzare una camera anerobica.
8. Analisi dei dati e risultati
- Le colture sono state raccolte dalle zone ossiche e anossiche di una colonna Winogradsky di 7 giorni. Queste zone sono adatte rispettivamente per anaerobi eterotrofici e ferro-ossidanti.
- Gli espianti a colonna sono stati diluiti in serie prima di striature o diffusione su piastre LB Agar.
- Streaking ha rivelato una popolazione mista da ciascuna delle zone Winogradsky valutate. Le piastre diffuse hanno prodotto risultati simili.
- Per calcolare CFU/mL o CFU/g, mediare il numero di colonie contate da tre piastre. Moltiplicare il numero medio di colonie per il fattore di diluizione e dividere per la quantità aliquota. Ad esempio, se una media di 65 colonie fosse contate su piastre inoculate con 0,1 mL disoluzione 6 (T6) la formula descritta in precedenza sarebbe pari a 650.000.000 CFU/mL.
- Le colonie isolate possono ora essere scelte da ciascuna piastra per l'uso in saggi di arricchimento per determinare l'identità delle specie.
A volte, per identificare e studiare i batteri, dobbiamo prima isolarli e arricchirli da un campione. Ad esempio, i campioni ottenuti da una colonna Winogradsky sono misti, il che significa che contengono più specie o ceppi di batteri, quindi studiare un singolo batterio o enumerare i diversi tipi presenti può essere difficile. A tal fine, le tecniche di diluizione seriale e placcatura sono tipicamente impiegate per quantificare in modo affidabile la carica batterica e isolare le singole colonie.
La diluizione seriale è un processo attraverso il quale la concentrazione di un organismo, batteri in questo esempio, viene sistematicamente ridotta attraverso una successiva risospensione in volumi fissi di diluente liquido. Di solito il volume del diluente è un multiplo di 10 per facilitare la riduzione logaritmica dell'organismo campione. Ad esempio, un grammo di sedimento viene prima rimosso dalla zona di interesse di Winogradsky e aggiunto a 10 millilitri di un mezzo liquido appropriato. Quindi, un millilitro di questa prima diluizione viene aggiunto a un altro tubo contenente nove millilitri di mezzo. Il processo può essere ripetuto fino a quando non sono state preparate diverse concentrazioni di batteri. La diluizione seriale è la chiave per l'enumerazione dei batteri in questo esempio, poiché i campioni misti di una colonna Winogradsky contengono un numero sconosciuto, spesso elevato, di batteri.
Successivamente, la placcatura a striature e la placcatura diffusa consentono rispettivamente l'isolamento e l'enumerazione dei batteri all'interno di un campione. La striatura si ottiene introducendo un campione diluito in una sezione del mezzo solido integrata con nutrienti, che è diviso in terzi. Questo inoculo viene quindi distribuito su ogni terzo della piastra in uno schema a zig-zag. Poiché diverse sezioni della piastra sono striate, incrociando dal campione precedente solo una volta, il campione viene distribuito più sottilmente. Ciò significa che potrebbe essere necessario eseguire solo una striscia da una diluizione per ottenere singole colonie nelle sezioni successive. Dopo l'incubazione, le placche striate consentono osservazioni della morfologia della colonia, informazioni che possono aiutare a distinguere tra diverse specie batteriche.
In alternativa, se l'obiettivo principale è l'enumerazione dei batteri nella placcatura di diffusione del campione può essere utilizzata. Nella placcatura diffusa, un'aliquota di un singolo campione viene distribuita uniformemente su tutta la superficie del mezzo solido. In genere, poiché non conosciamo i numeri batterici nel campione misto, viene realizzata una piastra diffusa per ciascuna delle diluizioni o un campione rappresentativo di esse. Dopo l'incubazione, l'enumerazione può essere eseguita utilizzando queste piastre di diffusione. Qualsiasi placca con numero di colonie inferiore a 30 dovrebbe essere scartata, poiché i piccoli conteggi sono soggetti a un errore maggiore. Allo stesso modo, qualsiasi conteggio superiore a 300 dovrebbe essere scartato perché l'affollamento e la sovrapposizione delle colonie possono portare a sottovalutare il numero di colonie. Se il conteggio delle colonie di ciascuno di questi piatti rimanenti viene registrato e moltiplicato per il fattore di diluizione, e quindi diviso per il volume placcato, questo produce le unità formanti la colonia, o CFU, per millilitro di sospensione. In questo video, imparerai come valutare qualitativamente e quantitativamente un campione contenente un batterio noto e le comunità microbiche contenute in varie regioni di una colonna Winogradsky tramite diluizione seriale, placcatura diffusa e placcatura a strisce.
In primo luogo, indossare qualsiasi dispositivo di protezione individuale appropriato tra cui un cappotto da laboratorio, guanti e occhiali. Quindi, sterilizzare lo spazio di lavoro con etanolo al 70% e pulire la superficie. Quindi, raccogliere due fiaschi Erlenmeyer da 500 millilitro ed etichettare un brodo e l'altro agar. Per preparare la soluzione di agar LB, mescolare circa 6,25 grammi di agar LB, tre grammi di agar tecnico e 250 millilitri di acqua distillata nel pallone etichettato agar.
Quindi, preparare il brodo LB combinando 2. 5 grammi di LB media e 100 millilitri di acqua distillata nel brodo etichettato del pallone. Dopo l'autoclave dei palloni, utilizzare un guanto resistente al calore per rimuovere i palloni dall'autoclave e metterli in un bagno d'acqua da 40 a 50 gradi Celsius. Una volta che i palloni sono a 50 gradi Celsius, preparare con cura tre aliquote da 100 millilitro della soluzione di brodo ed etichettare ogni soluzione aliquota zero. Quindi, raccogli 10 piastre di Petri sterili ed etichettale con la data, il nome, il tipo di supporto utilizzato e la zona della colonna Winogradsky da cui verranno raccolti gli organismi. Pipettare 15 millilitri di agar dal pallone di agar in ogni capsula di Petri. Quindi, utilizzare la punta della pipetta per rimuovere eventuali bolle, sostituire i coperchi delle piastre e consentire loro di solidificarsi sul piano di lavoro durante la notte.
Il giorno dopo, pulire il piano di lavoro con il 70% di etanolo. Quindi, etichettare 10 provette da 20 millilitro da T1 a T10 e metterle in un rack. Pipettare nove millilitri di soluzione salina dello 0,45% in ciascun tubo. Ora, coprire ciascuna delle 10 provette liberamente con i loro tappi e trasferirle in un rack per provette compatibile con l'autoclave. Al termine del ciclo, rimuovere gli spazi vuoti salini utilizzando guanti resistenti al calore e lasciarli raffreddare. Conservare i tubi a temperatura ambiente fino a quando non hanno raggiunto circa 22 gradi Celsius.
Per coltivare un organismo bersaglio noto, E. coli in questo esempio, inoculare 100 millilitri di soluzione zero con una singola colonia da una piastra precedentemente striata. Quindi, coprire il tubo e incubarlo durante la notte a 37 gradi Celsius. Per valutare le regioni di una colonna di Winogradsky, aggiungere circa un grammo di materiale dalla zona aerobica a T1 e risospendare mediante vortice. Quindi, ripetere questo processo con un grammo di materiale dalla zona anaerobica.
Rimuovere il tubo contenente la soluzione zero inoculata con E. coli dall'incubatrice e agitarlo. Quindi, pipettare un millilitro della soluzione in una provetta T1 e vortice per mescolare. Rimuovere un millilitro di soluzione da T1 e trasferirlo in T2, vorticosamente per mescolare. Ripetere questo processo attraverso il tubo T10. Per valutare le zone aerobiche e anaerobiche della colonna Winogradsky, rimuovere un millilitro di soluzione da ciascuno dei tubi T1 precedentemente preparati e trasferirlo ai tubi T2 appropriati. Quindi, continuare le diluizioni seriali attraverso i tubi T10 come precedentemente dimostrato.
Per distribuire la piastra, pipettare 100 microlitri del campione diluito da ciascun tubo T3 alla capsula di Petri corrispondente. Quindi, utilizzare un'asta di diffusione sterile per distribuire delicatamente il campione sulla capsula di Petri e sostituire il coperchio della piastra. Ripetere questo processo per le diluizioni T6 e T9, come dimostrato in precedenza. Incubare le piastre contenenti organismi aerobici in un incubatore a 37 gradi Celsius per 24 ore. Incubare le piastre contenenti organismi anaerobici in una camera anaerobica impostata a 37 gradi Celsius per 24 ore. Il giorno successivo rimuovere le piastre di diluizione T3, T6 e T9 dall'incubatore e dalla camera anaerobica e trasferirle sul piano di lavoro. Lavorando con una piastra alla volta, scivola un anello di inoculazione sterile attraverso la parte superiore del supporto in uno schema a zig-zag. Quindi, sostituire il coperchio della capsula di Petri. Quindi, ruotare la piastra di 1/3 e sterilizzare il loop per ridurre la frequenza del modello a zig-zag precedentemente realizzato. Ancora una volta, dopo aver sterilizzato il loop, ruotare la piastra di 1/3, ridurre la frequenza del modello a zig-zag un'ultima volta e sostituire il coperchio. Ripetere questo metodo di striature per le piastre rimanenti, come mostrato in precedenza. Quindi, posizionare le piastre striate contenenti organismi aerobici in un incubatore a 37 gradi Celsius durante la notte e le piastre striate contenenti organismi anaerobici in una camera anaerobica impostata a 37 gradi Celsius durante la notte.
Le colture sono state raccolte dalle zone aerobiche e anaerobiche di una colonna Winogradsky di sette giorni. Quindi, le colture sono state diluite in serie prima di striature e diffusione su piastre di agar LB. Lo streaking ha rivelato una popolazione mista da ciascuna delle zone Winogradsky valutate e le piastre diffuse hanno prodotto risultati simili. Un piatto striato da una popolazione mista si tradurrà in colonie batteriche di diverse forme, dimensioni, trame e colori. Al contrario, le placche striate e diffuse contenenti l'organismo noto, E. coli, hanno dimostrato una popolazione omologa. In generale, è meglio calcolare le CFU per millilitro utilizzando il conteggio medio delle colonie di tre piastre distribuite con lo stesso campione e fattore di diluizione. Moltiplicare il numero medio di colonie per il fattore di diluizione e dividere per la quantità aliquota. Infine, le colonie isolate scelte da ciascuna piastra possono essere utilizzate in ulteriori saggi di arricchimento per determinare l'identità delle specie.
Applications and Summary
L'enumerazione batterica e l'isolamento dei ceppi mediante placcatura richiedono concentrazioni gestibili di organismi bersaglio. Il successo della placcatura è quindi subordinato alla diluizione seriale. In quanto tali, le suddette tecniche rimangono la pietra angolare dell'esame e della sperimentazione microbiologica. Sebbene semplici per progettazione, i fattori di diluizione e la tecnica di placcatura possono essere modificati dallo sperimentatore per rafforzare i risultati senza compromettere l'integrità di ciascun metodo. Tracciare le quattro fasi della crescita batterica può essere utile quando si caratterizzano i microbi desiderati. Queste fasi, ritardo, log, stazionarie e morte, sono caratterizzate da cambiamenti nella replicazione batterica. La fase di ritardo presenta una crescita lenta dovuta all'adattamento fisiologico, la fase logarittiva è il periodo di massima proliferazione con un aumento esponenziale delle cellule vitali, la fase stazionaria viene poi raggiunta a causa di limitazioni ambientali e accumuli di tossine, prima della fase di morte in cui la conta cellulare inizia a diminuire. Ciò può essere ottenuto diluendo in serie (o diluendo in 1 fase per evitare confusione)Soluzione 0 ogni ora per un totale di 8 ore, a partire dal tempo0 (la soluzione 0 deve essere restituita a un incubatore vibrante dopo ogni diluizione). Calcolare il log10 di CFU/ml per un singolo diluente di Tempo0 e tracciare sull'asse Y. Ripetere questo calcolo per il tempocampione 1 (assicurarsi di calcolare CFU/mL utilizzando lo stesso fattore di diluizione del tempo0). Ripetete fino a quando ogni volta (Tempo1-Tempo8) vengono tracciati sull'asse X.
References
- Allen, M.E., Gyure, R.A. (2013) An Undergraduate Laboratory Activity Demonstrating Bacteriophage Specificity. Journal of Microbial Biological Education 14: 84-92.
- Ben-David, A., Davidson, C.E. (2014) Estimation Method for Serial Dilution Experiments. Journal of Microbiological Methods 107:214-221.
- Goldman, E., Green, L.H. (2008) Practical Handbook of Microbiology.
- Koch, R. (1883) New Research Methods for Detection of Microcosms in Soil, Air and Water.
- Lederberg, J., Lederberg, E.M. (1952) Replica Plating and Indirect Selection of Bacterial Mutants. Journal of Bacteriology 63:399-406
- Pepper, I., Gerba, C., Ikner, L. (2019) Bacterial Growth Curve Analysis and its Environmental Changes. JoVE Science Education Database. Environmental Microbiology.
- Sanders., E.R. (2012) Aseptic Laboratory Technique: Plating Methods. JoVE 63:e3063.