



Overview
Fonte: Ewa Bukowska-Faniband1, Tilde Andersson1, Rolf Lood1
1 Dipartimento di Scienze Cliniche Lund, Divisione di Medicina delle Infezioni, Centro Biomedico, Università di Lund, 221 00 Lund, Svezia
Il pianeta Terra è un habitat per milioni di specie batteriche, ognuna delle quali ha caratteristiche specifiche. L'identificazione delle specie batteriche è ampiamente utilizzata nell'ecologia microbica per determinare la biodiversità dei campioni ambientali e la microbiologia medica per diagnosticare i pazienti infetti. I batteri possono essere classificati utilizzando metodi microbiologici convenzionali, come la microscopia, la crescita su supporti specifici, test biochimici e sierologici e saggi di sensibilità agli antibiotici. Negli ultimi decenni, i metodi di microbiologia molecolare hanno rivoluzionato l'identificazione batterica. Un metodo popolare è il sequenziamento del gene dell'RNA ribosomiale 16S (rRNA). Questo metodo non è solo più veloce e più accurato rispetto ai metodi convenzionali, ma consente anche l'identificazione di ceppi difficili da coltivare in condizioni di laboratorio. Inoltre, la differenziazione dei ceppi a livello molecolare consente la discriminazione tra batteri fenotipicamente identici (1-4).
L'rRNA 16S si unisce a un complesso di 19 proteine per formare una subunità 30S del ribosoma batterico (5). È codificato dal gene rRNA 16S, che è presente e altamente conservato in tutti i batteri grazie alla sua funzione essenziale nell'assemblaggio dei ribosomi; tuttavia, contiene anche regioni variabili che possono fungere da impronte digitali per particolari specie. Queste caratteristiche hanno reso il gene rRNA 16S un frammento genetico ideale da utilizzare nell'identificazione, nel confronto e nella classificazione filogenetica dei batteri (6).
Il sequenziamento del gene rRNA 16S si basa sulla reazione a catena della polimerasi (PCR) (7-8) seguita dal sequenziamento del DNA (9). La PCR è un metodo di biologia molecolare utilizzato per amplificare specifici frammenti di DNA attraverso una serie di cicli che includono:
i) Denaturazione di un modello di DNA a doppio filamento
ii) Ricottura di primer (oligonucleotidi corti) complementari al template
iii) Estensione dei primer da parte dell'enzima DNA polimerasi, che sintetizza un nuovo filamento di DNA
Una panoramica schematica del metodo è illustrata nella Figura 1.
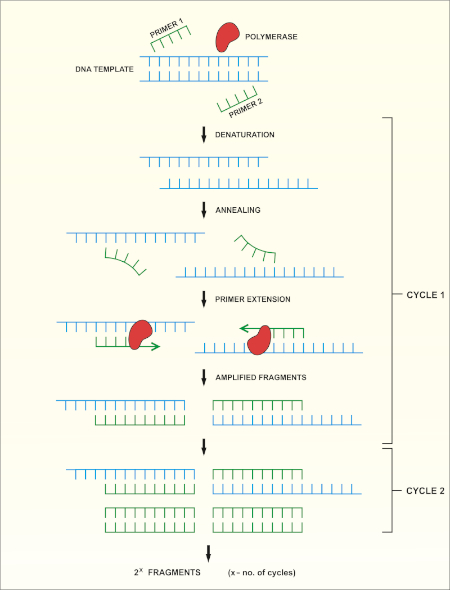
Figura 1: Panoramica schematica della reazione PCR. Fare clic qui per visualizzare una versione più grande di questa figura.
Ci sono diversi fattori che sono importanti per una reazione PCR di successo, uno dei quali è la qualità del modello di DNA. L'isolamento del DNA cromosomico dai batteri può essere eseguito utilizzando protocolli standard o kit commerciali. Particolare attenzione deve essere prestata per ottenere DNA privo di contaminanti che possono inibire la reazione PCR.
Le regioni conservate del gene rRNA 16S consentono la progettazione di coppie di primer universali (una in avanti e una inversa) che possono legarsi e amplificare la regione bersaglio in qualsiasi specie batterica. L'area di destinazione può variare in termini di dimensioni. Mentre alcune coppie di primer possono amplificare la maggior parte del gene rRNA 16S, altre amplificano solo parti di esso. Esempi di primer di uso comune sono mostrati nella Tabella 1 e i loro siti di legame sono illustrati nella Figura 2.
| Nome del primer | Sequenza (5'→3') | Avanti/indietro | Riferimento |
| 8F b) | AGAGTTTGATCCTGGCTCAG | inoltrare | -1 |
| 27F · | AGAGTTTGATCMTGGCTCAG | inoltrare | -10 |
| 515F · | GTGCCAGCMGCCGCGGTAA | inoltrare | -11 |
| 911R | GCCCCCGTCAATTCMTTTGA | inverso | -12 |
| 1391R | GACGGGCGGTGTGTRCA | inverso | -11 |
| 1492R | GGTTACCTTGTTACGACTT | inverso | -11 |
Tabella 1: Esempi di oligonucleotidi standard utilizzati nell'amplificazione dei geni rRNA 16S a).
a) Le lunghezze previste del prodotto PCR generato utilizzando le diverse combinazioni di primer possono essere stimate calcolando la distanza tra i siti di legame per il primer avanti e il primer inverso (vedi Figura 2), ad esempio la dimensione del prodotto PCR utilizzando la coppia di primer 8F-1492R è ~ 1500 bp e per la coppia di primer 27F-911R ~ 900 bp.
b) noto anche come fD1

Figura 2: Figura rappresentativa della sequenza di rRNA 16S e dei siti di legame del primer. Le regioni conservate sono colorate in grigio e le regioni variabili sono riempite con linee diagonali. Per consentire la massima risoluzione, il primer 8F e 1492R (nome basato sulla posizione sulla sequenza di rRNA) vengono utilizzati per amplificare l'intera sequenza, consentendo il sequenziamento di diverse regioni variabili del gene. Fare clic qui per visualizzare una versione più grande di questa figura.
Le condizioni cicliche per la PCR(cioè la temperatura e il tempo necessari per denaturare il DNA, ricotto con primer e sintetizzato) dipendono dal tipo di polimerasi utilizzata e dalle proprietà dei primer. Si consiglia di seguire le linee guida del produttore per una particolare polimerasi.
Dopo aver completato il programma PCR, i prodotti vengono analizzati mediante elettroforesi su gel di agarose. Una PCR di successo produce una singola banda di dimensioni previste. Il prodotto deve essere purificato prima del sequenziamento per rimuovere i primer residui, i desossiribonucleotidi, la polimerasi e il tampone che erano presenti nella reazione PCR. I frammenti di DNA purificati vengono solitamente inviati per il sequenziamento ai servizi di sequenziamento commerciali; tuttavia, alcune istituzioni eseguono il sequenziamento del DNA presso le proprie strutture principali.
La sequenza di DNA viene generata automaticamente da un cromatogramma del DNA da un computer e deve essere attentamente controllata per verificare la qualità, poiché a volte è necessaria la modifica manuale. Seguendo questo passaggio, la sequenza genica viene confrontata con le sequenze depositate nel database dell'rRNA 16S. Vengono identificate le regioni di somiglianza e vengono consegnate le sequenze più simili.
Procedure
1. Configurazione
- Durante la manipolazione dei microrganismi, è necessario seguire una buona pratica microbiologica. Tutti i microrganismi, in particolare i campioni sconosciuti, devono essere trattati come potenziali agenti patogeni. Seguire la tecnica asettica per evitare di contaminare i campioni, i ricercatori o il laboratorio. Lavarsi le mani prima e dopo aver maneggiato i batteri, usare guanti e indossare indumenti protettivi.
- Effettuare una valutazione del rischio per il protocollo sperimentale per l'isolamento del DNA genomico e la purificazione del prodotto PCR. Alcuni reagenti possono essere dannosi!
- La coltura pura è essenziale per il sequenziamento dell'rRNA 16S. Prima di procedere all'isolamento del DNA genomico, assicurarsi che il materiale di partenza sia completamente puro. Questo può essere fatto con la placcatura a strisce per isolare le singole colonie. Questi possono essere ulteriormente coltivati striati su piatti singolarmente o in brodo, se necessario.
- Attrezzature di laboratorio necessarie:
- Termocicclatore per PCR. La funzione del termocicatore è quella di alzare e abbassare la temperatura secondo un programma prestabilito. Durante la creazione del programma ti verrà chiesto di inserire i valori di temperatura e tempo per ogni fase della PCR e il numero totale di cicli.
- Sistema di elettroforesi su gel di acarosio. Viene utilizzato per separare i frammenti di DNA in base alle loro dimensioni e carica. In questo protocollo, l'elettroforesi su gel di acarosio verrà utilizzata per visualizzare la qualità del DNA genomico isolato e dei prodotti PCR.
2. Protocollo
Nota: Il protocollo dimostrato si applica al sequenziamento del gene rRNA 16S da una coltura pura di batteri. Non si applica agli studi metagenomici.
-
Coltura di batteri per l'isolamento del DNA genomico (gDNA).
- Fai crescere il tuo microrganismo su un terreno adatto. Sia i mezzi liquidi che solidi possono essere utilizzati in questa fase. Scegli le condizioni che producono la migliore crescita. Durante la pianificazione dell'esperimento, tieni presente che i batteri a crescita lenta potrebbero richiedere diversi giorni per raggiungere la fase di crescita tardiva / stazionaria. In questo protocollo, Bacillus subtilis 168 è stato coltivato in brodo di lipogenia (LB) durante la notte in un incubatore vibrante impostato a 200 giri / min, 37 ° C.
-
Isolamento del gDNA.
- Se i batteri sono stati coltivati su un terreno solido, raschiare alcune cellule usando un anello sterile e risuscimarle in 1 mL di acqua distillata
- Se i batteri sono stati coltivati in mezzo liquido, utilizzare circa 1,5 ml di una coltura notturna.
- Pellet le cellule mediante centrifugazione (1 minuto, 12.000 - 16.000 × g), rimuovere il surnatante e utilizzare le cellule per l'isolamento del gDNA utilizzando un kit commerciale o protocolli standard[ad esempio preparazione totale del DNA CTAB (13) o estrazione fenolo-cloroformio (14)]. Qui, un kit commerciale è stato utilizzato per isolare gDNA da 1,5 mL di B. subtilis 168 coltura notturna, OD600 = 1,5.
Nota 1: Per alcuni batteri Gram-negativi questo passaggio può essere omesso e sostituito da un semplice rilascio di DNA dalle cellule mediante ebollizione. Sospendare il pellet batterico in acqua distillata e incubare in un blocco riscaldante impostato a 100 °C per 10 minuti.
Nota 2: Le cellule batteriche Gram-positive sono difficili da distruggere. Si raccomanda quindi di scegliere un metodo o un kit di isolamento del gDNA dedicato all'isolamento da questo gruppo di batteri.
-
Controllo qualità gDNA.
- Controllare la qualità del gDNA isolato mediante elettroforesi su gel di agarosio. In primo luogo, mescolare 5 μL del gDNA isolato con 1 μL del colorante di carico (6x) e caricare il campione su un gel di agarosio allo 0,8% che contiene un reagente di colorazione del DNA.
- Caricare uno standard di massa molecolare ed eseguire l'elettroforesi fino a quando il fronte del colorante raggiunge il fondo del gel.
- Una volta completata l'elettroforesi, visualizzare il gel su un transilluminatore adatto (luce UV o blu). Il gDNA appare come una banda molecolare alta spessa (superiore a 10 kb). Un esempio del controllo di qualità del gDNA è mostrato nella Figura 3.
- Se il gDNA supera il controllo di qualità(cioèla banda molecolare alta è presente e c'è poco o nessuna sbavatura del gDNA), diluire il gDNA in serie etichettando prima 3 tubi microcentrifuga come segue: "10x", "100x" e "1000x".
- Pipettare 90 μL di acqua distillata sterile in ciascuno dei 3 tubi.
- Prendere 10 μL della soluzione di gDNA e aggiungerla al tubo contrassegnato "10x".
- Pipettare accuratamente l'intero volume(cioè 100 μL) su e giù per garantire che la soluzione sia miscelata uniformemente. Quindi, prendere 10 μL della soluzione da questo tubo e trasferirlo sul tubo contrassegnato "100x".
- Miscelare come descritto in precedenza e ripetere la stessa procedura trasferendo 10 μL della soluzione dal tubo "100x" al tubo "1000x". Queste diluizioni saranno utilizzate come modello nella reazione PCR.

Figura 3: Elettroforesi su gel di agarosio di gDNA isolato da Bacillus subtilis. Corsia 1: M - marcatore di massa molecolare (dall'alto verso il basso: 10000 bp, 8000 bp, 6000 bp, 5000 bp, 4000 bp, 3500 bp, 3000 bp, 2500 bp, 2000 bp, 1500 bp, 1000 bp). Corsia 2: gDNA - DNA genomico isolato da Bacillus subtilis. Fare clic qui per visualizzare una versione più grande di questa figura.
-
Amplificazione del gene rRNA 16S mediante PCR.
Nota: Il protocollo PCR riportato di seguito è ottimizzato per una particolare coppia di DNA polimerasi e primer 8F - 1492R (vedere Tabella 1). L'ottimizzazione del protocollo è necessaria per ogni coppia di polimerasi e primer.- Scongelare tutti i reagenti sul ghiaccio.
- Preparare la miscela master PCR come mostrato nella Tabella 2. Poiché la DNA polimerasi è attiva a temperatura ambiente, la configurazione della reazione deve essere eseguita su ghiaccio, cioè i tubi PCR e i componenti della reazione devono essere mantenuti sul ghiaccio tutto il tempo. Preparare una reazione per ogni campione di gDNA e una reazione per il controllo negativo. Il controllo negativo è una miscela PCR senza il modello gDNA e viene utilizzato per garantire che gli altri componenti della reazione non siano contaminati.
Nota: In caso di più campioni, viene comunemente preparato un mix master. Master mix è una soluzione contenente tutti i componenti di reazione ad eccezione del modello. Aiuta a omettere il pipettaggio ripetitivo, evitare errori di pipettaggio e garantisce un'elevata coerenza tra i campioni. Per preparare il master mix, moltiplicare il volume di ciascun componente (ad eccezione del modello di DNA) per il numero di campioni testati. Mescolare tutti i componenti in tubo microcentrifuga e pipettare l'intero volume su e giù più volte. - Aliquota 49 μL della miscela master nei singoli tubi PCR.
- Aggiungere un modello da 1 μL nei tubi con miscela master. Per il controllo negativo aggiungere 1 μL di acqua sterile. Per garantire che i componenti siano ben miscelati, pipettare delicatamente la miscela su e giù ~ 10 volte con una pipetta impostata su 30-50μL.
- Impostare la macchina PCR con il programma mostrato nella Tabella 3.
- Inserire i tubi nella macchina PCR e avviare il programma.
- Una volta completato il programma, esaminare la qualità del prodotto PCR mediante elettroforesi su gel di agarose.
- Una reazione PCR di successo utilizzando la coppia di primer 8F-1492R produce una singola banda di circa 1,5 kb (Figura 4). Se sono presenti altre bande (cioè prodotti non specifici), ottimizzare il programma PCR regolando la temperatura di ricottura. Se è presente una singola banda di dimensioni previste, procedere al passaggio successivo. Qui, la reazione PCR con modello di gDNA diluito 100x ha prodotto il miglior prodotto in quanto aveva una banda netta di dimensioni previste e mancava di prodotti non specifici. Quindi è stato scelto per essere purificato e inviato per il sequenziamento.
- Prima del sequenziamento, il prodotto deve essere ripulito da primer residui, desossiribonucleotidi, polimerasi e tampone che erano presenti nella reazione PCR. I prodotti PCR possono essere isolati utilizzando un kit di purificazione PCR commerciale. La reazione PCR viene caricata su una colonna che contiene una matrice legante il DNA. Il prodotto PCR si associa alla colonna, mentre altri componenti scorrono attraverso la colonna. La colonna viene quindi lavata con tampone di lavaggio e, infine, il DNA viene eluito nel tampone scelto. Verificare che il buffer di eluizione integrato con il kit sia compatibile con il sequenziamento.
- Inviare il prodotto PCR purificato per il sequenziamento del DNA. Seguire le linee guida per la presentazione dei campioni di sequenziamento presso l'impianto di sequenziamento scelto. Per la migliore copertura della sequenza, utilizzare i primer di amplificazione PCR (gli stessi utilizzati nella sezione 2.4.1) come primer di sequenziamento. Qui, i primer 8F e 1492R sono stati utilizzati per sequenziare il prodotto PCR.
| Componente | Concentrazione finale | Volume per reazione | Volume per x reazioni (master mix) |
| 5x tampone di reazione | 1x | 10 μL | 10 μL × x |
| 10 mM dNTPs | 200 μM | 1 μL | 1 μL × x |
| 10 μM Primer 8F | 0,5 μM | 2,5 μL | 2,5 μL × x |
| 10 μM Primer 1492R | 0,5 μM | 2,5 μL | 2,5 μL × x |
| Phusion polimerasi | 1 unità | 0,5 μL | 0,5 μL × x |
| Modello DNA * | - | 1 μL | - |
| ddH2O | - | 32,5 μL | 32,5 μL × x |
| Volume totale | 50 μL | 49 μL × x |
Tabella 2: Componenti di reazione PCR. * utilizzare il gDNA diluito 10x, 100x o 1000x dal passaggio 2.3.
| Passo | Temperatura | Ore | Cicli |
| Denaturazione iniziale | 98°C | 30 secondi | |
| Denaturazione | 98°C | 10 secondi | 25-30 |
| Ricottura | 60°C | 30 secondi | |
| Estensione | 72°C | 45 secondi | |
| Estensione finale | 72°C | 7 minuti | |
| Tenere | 4°C | ∞ |
Tabella 3: Programma PCR per l'amplificazione del gene rRNA 16S.

Figura 4: Elettroforesi su gel di agarosio di prodotti PCR amplificata utilizzando primer 8F e 1492R e gDNA come modello. Il campione di gDNA di B. subtilis (vedere Figura 3) è stato diluito 10, 100 e 1000 volte al fine di testare il miglior risultato. Corsia 1: M - marcatore di massa molecolare (dall'alto verso il basso: 10000 bp, 8000 bp, 6000 bp, 5000 bp, 4000 bp, 3500 bp, 3000 bp, 2500 bp, 2000 bp, 1500 bp, 1000bp, 750 bp, 500 bp, 250 bp). Corsia 2: Reazione PCR con modello diluito 10x. Corsia 3: Reazione PCR con modello diluito 100x. Corsia 4: Reazione PCR con modello diluito 1000x. Corsia 5: (C-) - controllo negativo (reazione senza il modello di DNA). Fare clic qui per visualizzare una versione più grande di questa figura.
3. Analisi dei dati e risultati
Nota: Il prodotto PCR viene sequenziato utilizzando i primer avanti (qui 8F) e inverso (qui 1492R). Pertanto, vengono generati due set di sequenza di dati, uno per il primer in avanti e uno per il primer inverso. Per ogni sequenza vengono generati almeno due tipi di file: i) un file di testo contenente la sequenza del DNA e ii) un cromatogramma del DNA, che mostra la qualità del sequenziamento.
- Per il primer in avanti, aprire il cromatogramma ed esaminare attentamente la sequenza. Un cromatogramma ideale per una sequenza di qualità dovrebbe avere picchi uniformemente distanziati e segnali di fondo scarsi o nulli (Figura 5A).
- Se il cromatogramma non è di alta qualità, la sequenza deve essere scartata o il file di testo della sequenza deve essere rivisto in base a quanto segue:
- La presenza di doppi picchi in tutto il cromatogramma indica la presenza di più modelli di DNA. Questo può essere il caso se la coltura batterica non era pura. Tale sequenza dovrebbe essere scartata (Figura 5B).
- Un cromatogramma ambiguo potrebbe derivare dalla presenza di picchi colorati diversi nella stessa posizione. Uno degli errori più comuni è la presenza di due picchi colorati diversi nella stessa posizione e l'assegnazione impropria delle basi da parte del software di sequenziamento (Figura 5C). Correggere manualmente eventuali nucleotidi assegnati in modo errato e modificarli nel file di testo.
- I cromatogrammi a bassa risoluzione possono provocare "picchi ampi" che spesso causano un conteggio errate dei nucleotidi in queste regioni (Figura 5D). Questo errore è difficile da correggere e pertanto eventuali discrepanze nell'ulteriore fase di allineamento non devono essere considerate affidabili.
- La scarsa qualità di lettura del cromatogramma e la presenza di picchi multipli sono comunemente osservate alle estremità 5' e 3' della sequenza. Alcuni software sequencer rimuovono automaticamente questi frammenti di bassa qualità (Figura 5E) e i nucleotidi non sono inclusi nel file di testo. Se la sequenza non è stata troncata automaticamente, determinare i frammenti di bassa qualità(ad esempio segnale debole, picchi sovrapposti, perdita di risoluzione) alle estremità e rimuovere le rispettive basi dal file di testo.

Figura 5: Esempi di risoluzione dei problemi relativi al sequenziamento del DNA. A) Un esempio di sequenza cromatografica di qualità (picchi uniformemente distanziati e non ambigui). B) Sequenza di scarsa qualità che di solito si verifica all'inizio del cromatogramma. L'area della zona grigia è considerata di bassa qualità e rimossa automaticamente dal software di sequenziamento. Più basi possono essere tagliate manualmente. C) Presenza di picchi doppi (indicati da frecce). Un nucleotide indicato dalla freccia rossa è stato letto dal sequencer come "T" (picco rosso), ma il picco blu è più forte e può anche essere interpretato come "C". D) I picchi sovrapposti indicano la contaminazione da DNA(cioè più di un modello). E) Perdita di risoluzione e i cosiddetti "picchi larghi" (contrassegnati da rettangolo) che impediscono una chiamata di base affidabile. Fare clic qui per visualizzare una versione più grande di questa figura.
- Ripetere 3.1 e 3.2 per il primer inverso.
- Infine, assemblate le sequenze avanti e indietro in un'unica sequenza contigua. Una buona sequenza produce una sequenza fino a 1100 bp. Considerando che il prodotto PCR è lungo ~ 1500 bp, le sequenze ottenute utilizzando primer avanti e indietro dovrebbero parzialmente sovrapporsi.
- Unisci le due sequenze usando il programma di assemblaggio della sequenza del DNA, ad esempio uno strumento gratuito come CAP3 (http://doua.prabi.fr/software/cap3) (15).
- Inserire le due sequenze in formato FASTA nella casella indicata. Fai clic sul pulsante "Invia" e attendi che i risultati restituiscano.
- Per visualizzare la sequenza assemblata premere "Contigs" nella scheda dei risultati. Per visualizzare i dettagli dell'allineamento premere "Dettagli assemblaggio".
Nota 1: Se il software CAP3 viene utilizzato per l'assemblaggio contig, non è necessario convertire la sequenza di primer inversa in reverse-complementary; tuttavia, questo passaggio potrebbe essere necessario se viene utilizzato un altro programma.
Nota 2: Il formato FASTA è un formato basato su testo per rappresentare la sequenza nucleotidica. La prima riga (la riga di descrizione) in un file FASTA inizia con un simbolo ">" seguito dal nome o da un identificatore univoco della sequenza. Segue la linea di descrizione la sequenza nucleotidica. Incollate le sequenze nel seguente formato:
>sequence_frw_primer
Incolla qui la sequenza dal file di testo
>sequence_rvs_primer
Incolla qui la sequenza dal file di testo
- Effettuare una ricerca nel database visitando il sito Web per lo strumento di ricerca di allineamento locale di base (BLAST; https://blast.ncbi.nlm.nih.gov/Blast.cgi).
- Selezionare lo strumento "Nucleotide BLAST" per confrontare la sequenza con il database.
- Inserisci la tua sequenza (il contig assemblato in 3.5) nella casella di testo "Sequenza di query", quindi seleziona il database "Sequenze di rRNA 16S (batteri e Archea)" nel menu a scorrimento verso il basso.
- Premere il pulsante "BLAST" nella parte inferiore della pagina. Verranno restituite le sequenze più simili. Un risultato BLAST di esempio è mostrato nella Figura 6. Nell'esperimento presentato il primo colpo è il ceppo B. subtilis 168, che mostra l'identità al 100% con la sequenza disponibile nel database BLAST.
- Se il primo colpo non mostra l'identità al 100%, vai all'allineamento e controlla le mancate corrispondenze. Facendo clic sul colpo in alto, verrai indirizzato ai dettagli dell'allineamento. I nucleotidi allineati saranno uniti da brevi linee verticali mentre i nucleotidi non corrispondenti hanno uno spazio tra di loro. Tornate al cromatogramma che avete ricevuto dalla società di sequenziamento e rivedete la sequenza ancora una volta con l'attenzione sulla regione non corrispondente. Correggere la sequenza se vengono trovati altri errori. Eseguire nuovamente BLAST utilizzando la sequenza corretta.

Figura 6: Esempio del risultato del nucleotide BLAST. La sequenza del gene rRNA 16S da coltura pura di B. subtilis 168 è stata utilizzata come sequenza di query. Il top hit mostra l'identità al 100% (sottolineata) al ceppo B. subtilis 168, come previsto. Fare clic qui per visualizzare una versione più grande di questa figura.
La Terra ospita milioni di specie batteriche, ognuna con caratteristiche uniche. L'identificazione di queste specie è fondamentale nella valutazione dei campioni ambientali. I medici devono anche distinguere diverse specie batteriche per diagnosticare i pazienti infetti.
Per identificare i batteri, è possibile utilizzare una varietà di tecniche, tra cui l'osservazione microscopica della morfologia o della crescita su un mezzo specifico per osservare la morfologia della colonia. L'analisi genetica, un'altra tecnica per identificare i batteri, è cresciuta in popolarità negli ultimi anni, in parte a causa del sequenziamento del gene dell'RNA ribosomiale 16S.
Il ribosoma batterico è un complesso di RNA proteico costituito da due subunità. La subunità 30S, la più piccola di queste due subunità, contiene rRNA 16S, che è codificato dal gene rRNA 16S contenuto nel DNA genomico. Regioni specifiche di rRNA 16S sono altamente conservate, grazie alla loro funzione essenziale nell'assemblaggio dei ribosomi. Mentre altre regioni, meno critiche per funzionare, possono variare tra le specie batteriche. Le regioni variabili nell'rRNA 16S possono servire come impronte digitali molecolari uniche per le specie batteriche, permettendoci di distinguere ceppi fenotipicamente identici.
Dopo aver ottenuto un campione di qualità di gDNA, può iniziare la PCR del gene codificante l'rRNA 16S. La PCR è un metodo di biologia molecolare comunemente usato, costituito da cicli di denaturazione del modello di DNA a doppio filamento, ricottura di coppie di primer universali, che amplificano regioni altamente conservate del gene e l'estensione dei primer da parte della DNA polimerasi. Mentre alcuni primer amplificano la maggior parte del gene codificante l'rRNA 16S, altri ne amplificano solo frammenti. Dopo la PCR, i prodotti possono essere analizzati tramite elettroforesi su gel di agarose. Se l'amplificazione ha avuto successo, il gel dovrebbe contenere una singola banda di dimensioni previste, a seconda della coppia di primer utilizzata, fino a 1500bp, la lunghezza approssimativa del gene rRNA 16S.
Dopo la purificazione e il sequenziamento, le sequenze ottenute possono quindi essere inserite nel database BLAST, dove possono essere confrontate con le sequenze di rRNA 16S di riferimento. Poiché questo database restituisce corrispondenze basate sulla più alta somiglianza, ciò consente di confermare l'identità dei batteri di interesse. In questo video, osserverai il sequenziamento del gene rRNA 16S, tra cui PCR, analisi e modifica della sequenza del DNA, assemblaggio della sequenza e ricerca nel database.
Quando si maneggiano microrganismi, è essenziale seguire una buona pratica microbiologica, compreso l'uso di tecniche asettiche e l'uso di adeguati dispositivi di protezione individuale. Dopo aver eseguito un'appropriata valutazione del rischio per il microrganismo o il campione ambientale di interesse, ottenere una coltura di prova. In questo esempio, viene utilizzata una cultura pura di Bacillus subtilis.
Per iniziare, fai crescere il tuo microrganismo su un terreno adatto nelle condizioni appropriate. In questo esempio, Bacillus subtilis 168 viene coltivato in brodo LB durante la notte in un incubatore vibrante impostato a 200 giri / min a 37 gradi Celsius. Successivamente, utilizzare un kit disponibile in commercio per isolare il DNA genomico o il gDNA da 1,5 millilitri della coltura notturna di B. subtilis.
Per verificare la qualità del DNA isolato, mescolare prima cinque microlitri del gDNA isolato con un microlitro di colorante a carica di gel di DNA. Quindi, caricare il campione su un gel di agarose allo 0,8%, contenente reagente per la colorazione del DNA, come SYBR safe o EtBr. Successivamente, caricare uno standard di massa molecolare di un kilobase sul gel ed eseguire l'elettroforesi fino a quando il colorante anteriore è a circa 0,5 centimetri dal fondo del gel. Una volta completata l'elettroforesi su gel, visualizzare il gel su un transilluminatore a luce blu. Il gDNA dovrebbe apparire come una banda spessa, di dimensioni superiori a 10 kilobase e avere sbavature minime.
Successivamente, per creare diluizioni seriali del gDNA, etichettare tre tubi microcentrifuga come 10X, 100X e 1000X. Quindi, utilizzare una pipetta per erogare 90 microlitri di acqua distillata sterile in ciascuno dei tubi. Quindi, aggiungere 10 microlitri della soluzione gDNA al tubo 10X. Pipettare l'intero volume su e giù per garantire che la soluzione sia miscelata accuratamente. Quindi, rimuovere 10 microlitri della soluzione dal tubo 10X e trasferirlo al tubo 100X. Mescolare la soluzione come descritto in precedenza. Infine, trasferire 10 microlitri della soluzione nel tubo 100X, al tubo 1000X.
Per iniziare il protocollo PCR, scongelare i reagenti necessari sul ghiaccio. Quindi, preparare il mix master PCR. Poiché la DNA polimerasi è attiva a temperatura ambiente, la reazione impostata deve avvenire sul ghiaccio. Aliquot 49 microlitri della miscela master in ciascuno dei tubi PCR. Quindi, aggiungere un microlitro di modello a ciascuno dei tubi sperimentali e un microlitro di acqua sterile al tubo di controllo negativo, pipettando su e giù per mescolare. Successivamente, impostare la macchina PCR in base al programma descritto nella tabella. Posizionare i tubi nel termociclatore e avviare il programma.
Una volta completato il programma, esaminare la qualità del prodotto tramite elettroforesi su gel di acarosio, come dimostrato in precedenza. Una reazione di successo utilizzando il protocollo descritto dovrebbe produrre una singola banda di circa 1,5 kilobase. In questo esempio, il campione contenente gDNA diluito 100X ha prodotto il prodotto di altissima qualità. Successivamente, purificare il miglior prodotto PCR, in questo caso, il gDNA 100X, con un kit disponibile in commercio. Ora il prodotto PCR può essere inviato per il sequenziamento.
In questo esempio, il prodotto PCR viene sequenziato utilizzando primer avanti e indietro. Pertanto, vengono generati due set di dati, ciascuno contenente una sequenza di DNA e un cromatogramma di DNA: uno per il primer in avanti e l'altro per il primer inverso. In primo luogo, esaminare i cromatogrammi generati da ciascun primer. Un cromatogramma ideale dovrebbe avere picchi uniformemente distanziati con segnali di fondo scarsi o nulli.
Se i cromatogrammi mostrano doppi picchi, più modelli di DNA potrebbero essere stati presenti nei prodotti PCR e la sequenza deve essere scartata. Se i cromatogrammi contenevano picchi di colori diversi nella stessa posizione, il software di sequenziamento probabilmente ha erroneamente chiamato nucleotidi. Questo errore può essere identificato e corretto manualmente nel file di testo. La presenza di ampi picchi nel cromatogramma indica una perdita di risoluzione, che causa un conteggio errato dei nucleotidi nelle regioni associate. Questo errore è difficile da correggere e le mancate corrispondenze in uno qualsiasi dei passaggi successivi devono essere considerate inaffidabili. La scarsa qualità di lettura del cromatogramma, indicata dalla presenza di picchi multipli, di solito si verifica alle cinque estremità prime e tre prime della sequenza. Alcuni programmi di sequenziazione rimuovono automaticamente queste sezioni di bassa qualità. Se la sequenza non è stata troncata automaticamente, identificare i frammenti di bassa qualità e rimuovere le rispettive basi dal file di testo.
Utilizzare un programma di assemblaggio del DNA per assemblare le due sequenze di primer in un'unica sequenza continua. Ricorda, le sequenze ottenute utilizzando primer avanti e indietro dovrebbero parzialmente sovrapporsi. Nel programma di assemblaggio del DNA, inserire le due sequenze in formato FASTA nell'apposita casella. Quindi, fai clic sul pulsante di invio e attendi che il programma restituisca i risultati.
Per visualizzare la sequenza assemblata, fate clic su Contigs nella scheda dei risultati. Quindi, per visualizzare i dettagli dell'allineamento, selezionate Dettagli assieme. Passare al sito Web per lo strumento di ricerca di allineamento locale di base, o BLAST, e selezionare lo strumento BLAST nucleotidico per confrontare la sequenza con il database. Immettere la sequenza nella casella di testo della sequenza di query e selezionare il database appropriato nel menu a scorrimento verso il basso. Infine, fai clic sul pulsante BLAST nella parte inferiore della pagina e attendi che lo strumento restituisca le sequenze più simili dal database.
In questo esempio, il primo hit è B. subtilis ceppo 168, che mostra l'identità al 100% con la sequenza nel database BLAST. Se il risultato principale non mostra l'identità al 100% della specie o del ceppo previsto, fai clic sulla sequenza che più si avvicina alla tua query per vedere i dettagli dell'allineamento. I nucleotidi allineati saranno uniti da brevi linee verticali e i nucleotidi non corrispondenti avranno spazi vuoti tra di loro. Concentrandosi sulle regioni non corrispondenti identificate, rivedere la sequenza e ripetere la ricerca BLAST, se lo si desidera.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
L'identificazione delle specie batteriche è importante per i diversi ricercatori, così come per quelli nel settore sanitario. Il sequenziamento dell'rRNA 16S è stato inizialmente utilizzato dai ricercatori per determinare le relazioni filogenetiche tra i batteri. Nel tempo, è stato implementato in studi metagenomici per determinare la biodiversità di campioni ambientali e nei laboratori clinici come metodo per identificare potenziali agenti patogeni. Consente un'identificazione rapida e accurata dei batteri presenti nei campioni clinici, facilitando la diagnosi precoce e il trattamento più rapido dei pazienti.
Subscription Required. Please recommend JoVE to your librarian.
References
- Weisburg, W.G., Barns, S.M., Pelletier, D.A. and Lane D.J. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 173 (2): 697-703. (1991)
- Drancourt, M., Bollet, C., Carlioz, A., Martelin, R., Gayral, J.P., Raoult D. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol. 38 (10):3623-3630. (2000)
- Woo, P.C., Lau, S.K., Teng, J.L., Tse, H., Yuen, K.Y. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect. 14 (10):908-934. (2008)
- Tang, Y.W., Ellis, N.M., Hopkins, M.K., Smith, D.H., Dodge, D.E., Persing, D.H. Comparison of phenotypic and genotypic techniques for identification of unusual aerobic pathogenic gram-negative bacilli. J Clin Microbiol. 36 (12):3674-3679. (1998)
- Tsiboli, P., Herfurth, E., Choli, T. Purification and characterization of the 30S ribosomal proteins from the bacterium Thermus thermophilus. Eur J Biochem. 226 (1):169-177. (1994)
- Woese, C.R. Bacterial evolution. Microbiol Rev. 51 (2):221-271. (1987)
- Bartlett, J.M., Stirling, D. A short history of the polymerase chain reaction. Methods Mol Biol. 226:3-6. (2003)
- Wilson, K.H., Blitchington, R.B., Greene, R.C. Amplification of bacterial 16S ribosomal DNA with polymerase chain reaction. J Clin Microbiol. 28 (9):1942-1946. (1990)
- Shendure, J., Balasubramanian, S., Church, G.M., Gilbert, W., Rogers, J., Schloss, J.A., Waterston, R.H. (2017) DNA sequencing at 40: past, present and future. Nature. 550:345-353.
- Lane, D.J. 16S/23S rRNA sequencing. (1991) In Nucleic acid techniques in bacterial systematics. (Goodfellow, M. and Stackebrandt, E., eds.) p.115-175. Wiley and Sons, Chichester, United Kingdom.
- Turner, S., Pryer, K.M., Miao, V.P., Palmer, J.D. (1999) Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryot Microbiol. 46:327-338.
- Fredricks, D.N., Relman, D.A. (1998) Improved amplification of microbial DNA from blood cultures by removal of the PCR inhibitor sodium polyanetholesulfonate. J Clin Microbiol. 36:2810-2816.
- Wilson, K. Preparation of genomic DNA from bacteria. (2001) Curr Protoc Mol Biol. Chapter 2:Unit 2.4.
- Wright, M. H., Adelskov, J., Greene, A.C. (2017) Bacterial DNA extraction using individual enzymes and phenol/chloroform separation. J Microbiol Biol Educ. 18:18.2.48.
- Huang, X., Madan, A. (1999). CAP3: A DNA sequence assembly program. Genome Res. 9:868-877.