



Overview
Fonte: Ewa Bukowska-Faniband1, Tilde Andersson1, Rolf Lood1
1 Departamento de Ciências Clínicas Lund, Divisão de Medicina de Infecção, Centro Biomédico, Universidade de Lund, 221 00 Lund, Suécia
O Planeta Terra é um habitat para milhões de espécies bacterianas, cada uma das quais tem características específicas. A identificação de espécies bacterianas é amplamente utilizada na ecologia microbiana para determinar a biodiversidade de amostras ambientais e microbiologia médica para diagnosticar pacientes infectados. As bactérias podem ser classificadas usando métodos convencionais de microbiologia, como microscopia, crescimento em mídia específica, testes bioquímicos e sorológicos e ensaios de sensibilidade a antibióticos. Nas últimas décadas, os métodos de microbiologia molecular revolucionaram a identificação bacteriana. Um método popular é o sequenciamento genético RNA ribossômico 16S (rRNA). Este método não só é mais rápido e mais preciso do que os métodos convencionais, mas também permite a identificação de cepas difíceis de crescer em condições laboratoriais. Além disso, a diferenciação das cepas no nível molecular permite a discriminação entre bactérias fenotipicamente idênticas (1-4).
16S rRNA une-se a um complexo de 19 proteínas para formar uma subunidade 30S do ribossomo bacteriano (5). É codificado pelo gene 16S rRNA, que está presente e altamente conservado em todas as bactérias devido à sua função essencial no conjunto ribossomo; no entanto, também contém regiões variáveis que podem servir como impressões digitais para determinadas espécies. Essas características tornaram o gene 16S rRNA um fragmento genético ideal a ser usado na identificação, comparação e classificação filogenética de bactérias (6).
O sequenciamento genético rRNA 16S é baseado na reação em cadeia de polimerase (PCR) (7-8) seguida pelo sequenciamento de DNA (9). PCR é um método de biologia molecular usado para amplificar fragmentos específicos de DNA através de uma série de ciclos que incluem:
i) Denaturação de um modelo de DNA duplo encalhado
ii) Ressaração de primers (oligonucleotídeos curtos) que são complementares ao modelo
iii) Extensão dos primers pela enzima dna polimerase, que sintetiza uma nova cadeia de DNA
Uma visão geral esquemática do método é mostrada na Figura 1.
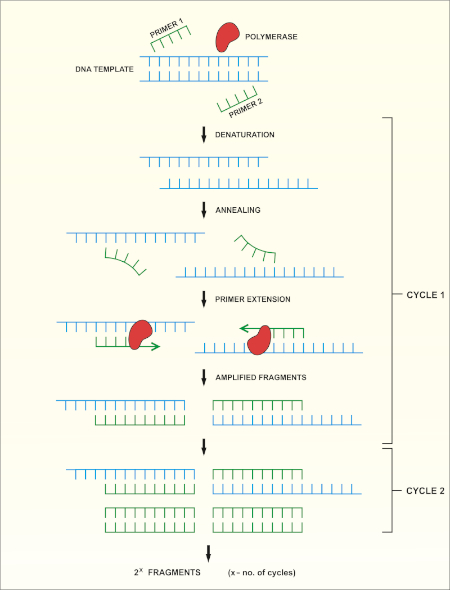
Figura 1: Visão geral esquemática da reação do PCR. Clique aqui para ver uma versão maior desta figura.
Existem vários fatores que são importantes para uma reação de sucesso do PCR, um dos quais é a qualidade do modelo de DNA. O isolamento do DNA cromossômico de bactérias pode ser realizado usando protocolos padrão ou kits comerciais. Deve-se tomar cuidado especial para obter DNA livre de contaminantes que podem inibir a reação da PCR.
Regiões conservadas do gene rRNA 16S permitem o desenho de pares de primer universais (um para a frente e um inverso) que podem se ligar e amplificar a região alvo em qualquer espécie bacteriana. A região alvo pode variar de tamanho. Enquanto alguns pares de primer podem amplificar a maior parte do gene 16S rRNA, outros amplificam apenas partes dele. Exemplos de primers comumente usados são mostrados na Tabela 1 e seus locais de vinculação são retratados na Figura 2.
| Nome do primer | Sequência (5'→3') | Para a frente/ para trás | Referência |
| 8F b) | AGAGTTTGATCCTGGCTCAG | encaminhar | -1 |
| 27F | AGAGTTTGATCMTGGCTCAG | encaminhar | -10 |
| 515F | GTGCCAGCCCCCGGGGTAA | encaminhar | -11 |
| 911R | GCCCCCGTCAATTCMTTTGA | inverter | -12 |
| 1391R | GACGGGCGGTGTGTRCA | inverter | -11 |
| 1492R | GGTTACCTTGTTACGACTT | inverter | -11 |
Tabela 1: Exemplos de oligonucleotídeos padrão utilizados na amplificação de genes de rRNA 16S a).
a) Os comprimentos esperados do produto PCR gerados usando as diferentes combinações de primer podem ser estimados calculando-se a distância entre os locais de ligação para o primer para a frente e para o primer (ver Figura 2), por exemplo, o tamanho do produto PCR usando o par de primer 8F-1492R é de ~1500 bp, e para o par de primer 27F-911R ~900 bp.
b) também conhecido como fD1

Figura 2: Figura representativa da sequência de rRNA 16S e dos sites de vinculação de primer. As regiões conservadas são coloridas em cinza e regiões variáveis são preenchidas com linhas diagonais. Para permitir a maior resolução, o primer 8F e 1492R (nome baseado na localização na sequência de rRNA) são usados para amplificar toda a sequência, permitindo o sequenciamento de várias regiões variáveis do gene. Clique aqui para ver uma versão maior desta figura.
As condições de ciclismo para PCR (ou seja, a temperatura e o tempo necessários para que o DNA seja desnaturado, enrosco com primers e sintetizado) dependem do tipo de polimerase que é usado e das propriedades dos primers. Recomenda-se seguir as diretrizes do fabricante para uma determinada polimerase.
Após a conclusão do programa PCR, os produtos são analisados por eletroforese de gel agarose. Um PCR bem sucedido rende uma única faixa de tamanho esperado. O produto deve ser purificado antes do sequenciamento para remover primers residuais, desoxiribonucleotídeos, polimerase e tampão que estavam presentes na reação pcr. Os fragmentos de DNA purificados são geralmente enviados para sequenciamento para serviços de sequenciamento comercial; no entanto, algumas instituições realizam sequenciamento de DNA em suas próprias instalações principais.
A sequência de DNA é gerada automaticamente a partir de um cromatgrama de DNA por um computador e deve ser cuidadosamente verificada para a qualidade, pois a edição manual às vezes é necessária. Após esta etapa, a sequência genética é comparada com sequências depositadas no banco de dados de rRNA 16S. As regiões de semelhança são identificadas, e as sequências mais semelhantes são entregues.
Procedure
1. Configurar
- Ao manusear microrganismos, é necessário seguir uma boa prática microbiológica. Todos os microrganismos, especialmente amostras desconhecidas, devem ser tratados como patógenos potenciais. Siga a técnica asséptica para evitar contaminar as amostras, pesquisadores ou o laboratório. Lave as mãos antes e depois de manusear bactérias, use luvas e use roupas de proteção.
- Realizar uma avaliação de risco para o protocolo experimental para o isolamento genômico de DNA e purificação do produto PCR. Alguns reagentes podem ser prejudiciais!
- A cultura pura é essencial para o sequenciamento de rRNA 16S. Antes de proceder ao isolamento do DNA genômico, certifique-se de que o material inicial é totalmente puro. Isso pode ser feito por uma série de listras para isolar colônias individuais. Estes podem ser ainda mais crescidos em placas individualmente, ou em caldo, se necessário.
- Equipamento de laboratório necessário:
- Ciclofadão térmico para PCR. A função do cicloviário térmico é aumentar e baixar a temperatura de acordo com um programa definido. Ao criar o programa, você será solicitado a inserir os valores de temperatura e tempo para cada etapa do PCR, bem como o número total de ciclos.
- Sistema de eletroforese de gel agarose. É usado para separar fragmentos de DNA com base em seu tamanho e carga. Neste protocolo, a eletroforese de gel agarose será usada para visualizar a qualidade de produtos isolados de DNA genômico e PCR.
2. Protocolo
Nota: O protocolo demonstrado se aplica ao sequenciamento genético de 16S rRNA de uma cultura pura de bactérias. Não se aplica a estudos metagenômicos.
-
Culturing bactérias para isolamento do DNA genômico (gDNA).
- Cresça seu microrganismo em um meio adequado. Tanto as mídias líquidas quanto as sólidas podem ser usadas nesta etapa. Escolha condições que produzam o melhor crescimento. Ao planejar o experimento, tenha em mente que as bactérias de crescimento lento podem precisar de vários dias para chegar à fase de crescimento estacionário/de tronco tardio. Neste protocolo, o Bacillus subtilis 168 foi cultivado em caldo de lysogenia (LB) durante a noite em uma incubadora de agitação fixada a 200 rpm, 37°C.
-
Isolamento do GDNA.
- Se as bactérias foram cultivadas em meio sólido, raspe algumas células usando um laço estéril e resuspensá-las em 1 mL de água destilada
- Se as bactérias foram cultivadas em meio líquido, utilize aproximadamente 1,5 mL de uma cultura durante a noite.
- Pelotar as células por centrifugação (1 minuto, 12.000 - 16.000 × g), remover o supernante e usar as células para isolamento gDNA usando um kit comercial ou protocolos padrão [por exemplo, preparação total de DNA CTAB (13) ou extração de fenol-clorofórmio (14)]. Aqui, um kit comercial foi utilizado para isolar gDNA de 1,5 mL de B. subtilis 168 overnight culture, OD600 = 1.5.
Nota 1: Para algumas bactérias Gram-negativas esta etapa pode ser omitida e substituída pela simples liberação de DNA das células por ebulição. Resuspenque a pelota bacteriana em água destilada e incuba em um bloco de aquecimento a 100 °C por 10 minutos.
Nota 2: Células bacterianas gram-positivas são difíceis de interromper. Recomenda-se, portanto, escolher um método de isolamento gDNA ou kit dedicado ao isolamento desse grupo de bactérias.
-
verificação de qualidade gDNA.
- Verifique a qualidade do gDNA isolado por eletroforese de gel agarose. Primeiro, misture 5 μL do gDNA isolado com 1 μL do corante de carregamento (6x), e carregue a amostra em um gel de 0,8% de agarose que contenha um reagente de coloração de DNA.
- Carregue um padrão de massa molecular e execute a eletroforese até que a frente de corante atinja a parte inferior do gel.
- Uma vez que a eletroforese esteja concluída, visualize o gel em um transilluminador adequado (UV ou luz azul). gDNA aparece como uma faixa molecular grossa (acima de 10 kb). Um exemplo da verificação de qualidade gDNA é mostrado na Figura 3.
- Se o gDNA passar pelo controle de qualidade (ou seja,a banda molecular alta está presente e há pouca ou nenhuma mancha do gDNA), diluir seu gDNA serialmente pela primeira rotulagem de 3 tubos de microcentrifuuge da seguinte forma: "10x", "100x" e "1000x".
- Pipeta 90 μL de água destilada estéril em cada um dos 3 tubos.
- Pegue 10 μL da solução gDNA e adicione-a ao tubo marcado como "10x".
- Em pipetar todo o volume(ou seja, 100 μL) para cima e para baixo completamente para garantir que a solução seja misturada uniformemente. Em seguida, pegue 10 μL da solução deste tubo e transfira-a para o tubo marcado como "100x".
- Misture como descrito antes e repita o mesmo procedimento transferindo 10 μL da solução do tubo "100x" para o tubo "1000x". Essas diluições serão usadas como modelo na reação do PCR.

Figura 3: Eletroforese de gel agarose de gDNA isolada de Bacillus subtilis. Pista 1: M - marcador de massa molecular (de cima a baixo: 10000 bp, 8000 bp, 6000 bp, 5000 bp, 4000 bp, 3500 bp, 3000 bp, 2500 bp, 2000 bp, 1500 bp, 1000 bp). Raia 2: gDNA - DNA genômico isolado do Bacillus subtilis. Clique aqui para ver uma versão maior desta figura.
-
Amplificação do gene 16S rRNA por PCR.
Nota: O protocolo PCR abaixo é otimizado para um determinado polimerase de DNA e par de primer 8F - 1492R (ver Tabela 1). A otimização do protocolo é necessária para cada par de polimerase e primer.- Descongele todos os reagentes no gelo.
- Prepare o mix mestre pcr como mostrado na Tabela 2. Uma vez que a polimerase de DNA está ativa à temperatura ambiente, a configuração de reação deve ser realizada no gelo, ou seja, os tubos PCR e os componentes de reação devem ser mantidos no gelo o tempo todo. Prepare uma reação por cada amostra gDNA e uma reação para controle negativo. O controle negativo é uma mistura pcr sem o modelo gDNA e é usado para garantir que os outros componentes da reação não estejam contaminados.
Nota: No caso de múltiplas amostras, uma mistura mestra é comumente preparada. Master mix é uma solução que contém todos os componentes de reação, exceto o modelo. Ajuda a omitir a tubulação repetitiva, evitar erros de pipetação e garante alta consistência entre as amostras. Para preparar a mistura mestre, multiplique o volume de cada componente (exceto o modelo de DNA) pelo número de amostras testadas. Misture todos os componentes no tubo de microcentrifusagem e encorregue todo o volume várias vezes. - Alíquota de 49 μL da mistura mestre nos tubos PCR individuais.
- Adicione 1 modelo μL em tubos com mix mestre. Para controle negativo adicione 1 μL de água estéril. Para garantir que os componentes estejam bem misturados, encobre suavemente a mistura para cima e para baixo ~10 vezes com uma pipeta definida para 30-50μL.
- Defina a máquina PCR com o programa mostrado na Tabela 3.
- Coloque os tubos na máquina PCR e inicie o programa.
- Uma vez concluído o programa, examine a qualidade do seu produto PCR por eletroforese de gel agarose.
- Uma reação de PCR bem sucedida usando o par de primer 8F-1492R rende uma única faixa de aproximadamente 1,5 kb(Figura 4). Se outras bandas (ou seja, produtos inespecíficos) estiverem presentes, otimize o programa PCR ajustando a temperatura de reclusão. Se uma única faixa de tamanho esperado estiver presente, proceda para o próximo passo. Aqui, a reação do PCR com modelo gDNA diluído de 100x rendeu o melhor produto, pois tinha uma faixa acentuada de tamanho esperado e não tinha produtos inespecíficos. Por isso, foi escolhido para ser purificado e enviado para sequenciamento.
- Antes do sequenciamento, o produto deve ser limpo a partir de primers residuais, desoxyribonucleotídeos, polimerase e tampão que estavam presentes na reação pcr. Os produtos PCR podem ser isolados usando um kit comercial de purificação pcr. A reação do PCR é carregada em uma coluna que contém uma matriz de ligação de DNA. O produto PCR se liga à coluna, enquanto outros componentes fluem através da coluna. A coluna é então lavada usando tampão de lavagem, e, finalmente, o DNA é elucido no buffer de escolha. Confirme se o tampão de elução que é complementado com o kit é compatível com sequenciamento.
- Envie o produto PCR purificado para sequenciamento de DNA. Siga as orientações para a apresentação de amostras de sequenciamento na instalação de sequenciamento escolhida. Para obter a melhor cobertura de sequência, use os primers de amplificação PCR (o mesmo usado na seção 2.4.1) como primers de sequenciamento. Aqui, foram utilizados primers 8F e 1492R para sequenciar o produto PCR.
| Componente | Concentração final | Volume por reação | Volume por x reações (mix mestre) |
| Tampão de reação 5x | 1x | 10 μL | 10 μL × x |
| DNTPs de 10 mM | 200 μM | 1 μL | 1 μL × x |
| Primer 8F de 10 μM | 0,5 μM | 2,5 μL | 2,5 μL × x |
| Primer 10 μM 1492R | 0,5 μM | 2,5 μL | 2,5 μL × x |
| Polimerase de phusion | 1 unidade | 0,5 μL | 0,5 μL × x |
| DNA de modelo * | - | 1 μL | - |
| ddH2O | - | 32,5 μL | 32,5 μL × x |
| Volume total | 50 μL | 49 μL × x |
Tabela 2: Componentes de reação do PCR. * use o gDNA diluído de 10x, 100x ou 1000x a partir da etapa 2.3.
| Passo | Temperatura | Hora | Ciclos |
| Desnaturação inicial | 98°C | 30 segundos | |
| Desnaturação | 98°C | 10 segundos | 25-30 |
| Recozimento | 60°C | 30 segundos | |
| Extensão | 72°C | 45 seg | |
| Extensão final | 72°C | 7 min. | |
| Segurar | 4°C | ∞ |
Tabela 3: Programa PCR para a amplificação do gene rRNA 16S.

Figura 4: Eletroforese de gel agarose de produtos PCR amplificados usando primers 8F e 1492R e gDNA como modelo. A amostra de gDNA de B. subtilis (ver Figura 3) foi diluída 10, 100 e 1000 vezes para testar o melhor resultado. Raia 1: M - marcador de massa molecular (de cima a baixo: 10000 bp, 8000 bp, 6000 bp, 5000 bp, 4000 bp, 3500 bp, 3000 bp, 2500 bp, 2000 bp, 1500 bp, 1000bp, 750 bp, 500 bp, 250 bp). Faixa 2: Reação pcr com modelo diluído de 10x. Pista 3: Reação pcr com modelo diluído de 100x. Pista 4: Reação pcr com modelo diluído de 1000x. Raia 5: (C-) - controle negativo (reação sem o modelo de DNA). Clique aqui para ver uma versão maior desta figura.
3. Análise de dados e resultados
Nota: O produto PCR é sequenciado usando os primers para a frente (aqui 8F) e o inverso (aqui 1492R). Portanto, são gerados dois conjuntos de sequência de dados, um para o atacante e outro para o primer reverso. Para cada sequência são gerados pelo menos dois tipos de arquivo: i) um arquivo de texto contendo a sequência de DNA e ii) um cromatógrafo de DNA, que mostra a qualidade da execução de sequenciamento.
- Para o primer dianteiro, abra o cromatograma e examine cuidadosamente a sequência. Um cromatógrafo ideal para uma sequência de qualidade deve ter picos espaçados uniformemente e pouco ou nenhum sinal de fundo(Figura 5A).
- Se o cromatógrafo não for de alta qualidade, a sequência deve ser descartada ou o arquivo de texto sequencial deve ser revisado de acordo com o seguinte:
- A presença de picos duplos ao longo do cromatograma indica a presença de múltiplos modelos de DNA. Este pode ser o caso se a cultura bacteriana não fosse pura. Tal sequência deve ser descartada(Figura 5B).
- Um cromatógrama ambíguo pode surgir da presença de diferentes picos coloridos no mesmo local. Um dos erros mais comuns é a presença de dois picos coloridos diferentes na mesma posição e a atribuição inadequada das bases pelo software de sequenciamento(Figura 5C). Corrija manualmente nucleotídeos atribuídos incorretamente e edite-os no arquivo de texto.
- Cromatógramas de baixa resolução podem resultar em "picos amplos" que muitas vezes causam má contagem dos nucleotídeos nessas regiões(Figura 5D). Este erro é difícil de corrigir e, portanto, possíveis desencontros na etapa de alinhamento adicional não devem ser tratados como confiáveis.
- A má qualidade de leitura do cromatograma e a presença de múltiplos picos são comumente vistas nas extremidades de 5' e 3' da sequência. Alguns softwares sequenciadores removem esses fragmentos de baixa qualidade automaticamente(Figura 5E),e os nucleotídeos não estão incluídos no arquivo de texto. Se sua sequência não foi truncada automaticamente, determine os fragmentos de baixa qualidade (por exemplo, sinal fraco, picos sobrepostos, perda de resolução) nas extremidades e remova as respectivas bases do arquivo de texto.

Figura 5: Exemplos de sequenciação de DNA. A) Um exemplo de uma sequência de cromatograma de qualidade (picos uniformemente espaçados e inequívocos). B) Sequência de baixa qualidade que geralmente ocorre no início do cromatograma. A área de zona cinzenta é considerada de baixa qualidade e automaticamente removida pelo software de sequenciamento. Mais bases podem ser aparadas manualmente. C) Presença de picos duplos (indicados por setas). Um nucleotídeo que é indicado pela seta vermelha foi lido pelo sequenciador como "T" (pico vermelho), mas o pico azul é mais forte, e também pode ser interpretado como "C". D) Picos sobrepostos indicam contaminação de DNA(ou seja, mais de um modelo). E) Perda de resolução e os chamados "picos amplos" (marcados por retângulo) que impedem a chamada de base confiável. Clique aqui para ver uma versão maior desta figura.
- Repita 3.1 e 3.2 para o primer reverso.
- Finalmente, monte as sequências para frente e invertida em uma sequência contígua. Uma boa corrida de sequenciamento rende uma sequência de até 1100 bp. Considerando que o produto PCR tem ~1500 bp de comprimento, as sequências obtidas usando primers dianteiros e invertidos devem se sobrepor parcialmente.
- Mescle as duas sequências usando o programa de montagem de sequência de DNA, por exemplo, uma ferramenta gratuita como CAP3 (http://doua.prabi.fr/software/cap3) (15).
- Insira as duas sequências no formato FASTA na caixa indicada. Clique no botão "Enviar" e aguarde o retorno dos resultados.
- Para visualizar a sequência montada pressione "Contigs" na guia de resultado. Para ver os detalhes do alinhamento pressione "Detalhes da montagem".
Nota 1: Se o software CAP3 for usado para montagem de contig, não há necessidade de converter a sequência de primer reverso em reverso-complementar; no entanto, esse passo pode ser necessário se outro programa for usado.
Nota 2: O formato FASTA é um formato baseado em texto para representar a sequência nucleotídea. A primeira linha (a linha de descrição) em um arquivo FASTA começa com um símbolo ">" seguido pelo nome ou um identificador exclusivo da sequência. Seguindo a linha de descrição está a sequência de nucleotídeos. Cole suas sequências no seguinte formato:
>sequence_frw_primer
cole sua sequência do arquivo de texto aqui
>sequence_rvs_primer
cole sua sequência do arquivo de texto aqui
- Realize uma pesquisa no banco de dados visitando o site da Ferramenta de Pesquisa de Alinhamento Local Básico (BLAST; https://blast.ncbi.nlm.nih.gov/Blast.cgi).
- Selecione a ferramenta "Nucleotide BLAST" para comparar sua sequência com o banco de dados.
- Digite sua sequência (a contig montada em 3.5) na caixa de texto "Sequência de consulta" e selecione o banco de dados "sequências de rRNA 16S (Bactérias e Archea)" no menu scroll down.
- Pressione o botão "BLAST" na parte inferior da página. As sequências mais semelhantes serão devolvidas. Um exemplo de resultado BLAST é mostrado na Figura 6. No experimento apresentado, o principal hit é b. subtilis strain 168, mostrando 100% de identidade com a sequência disponível no banco de dados BLAST.
- Se o topo não mostrar 100% de identidade, vá para o alinhamento e verifique se há desencontros. Ao clicar no topo, você será direcionado para os detalhes do alinhamento. Nucleotídeos alinhados serão unidos por linhas verticais curtas, enquanto nucleotídeos incompatíveis têm uma lacuna entre eles. Retorne ao cromatógrafo que recebeu da empresa de sequenciamento e revise a sequência mais uma vez com o foco na região incompatível. Corrija a sequência se forem encontrados erros adicionais. Execute blast novamente usando a sequência corrigida.

Figura 6: Exemplo do resultado blast nucleotídeo. 16S sequência genética rRNA a partir da cultura pura de B. subtilis 168 foi usada como uma sequência de consulta. O sucesso superior mostra 100% de identidade (sublinhada) para a cepa B. subtilis 168, como esperado. Clique aqui para ver uma versão maior desta figura.
A Terra abriga milhões de espécies bacterianas, cada uma com características únicas. Identificar essas espécies é fundamental na avaliação de amostras ambientais. Os médicos também precisam distinguir diferentes espécies bacterianas para diagnosticar pacientes infectados.
Para identificar bactérias, uma variedade de técnicas pode ser empregada, incluindo observação microscópica de morfologia ou crescimento em uma mídia específica para observar a morfologia das colônias. Análise genética, outra técnica de identificação de bactérias tem crescido em popularidade nos últimos anos, devido em parte ao sequenciamento de genes RNA ribossômico 16S.
O ribossomo bacteriano é um complexo de RNA proteico composto por duas subunidades. A subunidade 30S, a menor dessas duas subunidades, contém rRNA 16S, que é codificado pelo gene de rRNA 16S contido no DNA genômico. Regiões específicas de rRNA 16S são altamente conservadas, devido à sua função essencial na montagem ribossosome. Enquanto outras regiões, menos críticas para funcionar, podem variar entre espécies bacterianas. As regiões variáveis em rRNA 16S, podem servir como impressões digitais moleculares únicas para espécies bacterianas, permitindo-nos distinguir cepas fenotipicamente idênticas.
Após a obtenção de uma amostra de qualidade de gDNA, o PCR do gene de codificação de rRNA 16S pode começar. PCR é um método de biologia molecular comumente usado, consistindo de ciclos de desnaturação do modelo de DNA de duplaridade, ressarcido de pares de primer universais, que amplificam regiões altamente conservadas do gene, e a extensão de primers por polimerase de DNA. Enquanto alguns primers amplificam a maior parte do gene de codificação de rRNA 16S, outros apenas amplificam fragmentos dele. Após pcr, os produtos podem ser analisados via eletroforese de gel agarose. Se a amplificação foi bem sucedida, o gel deve conter uma única faixa de um tamanho esperado, dependendo do par de primer usado, até 1500bp, o comprimento aproximado do gene 16S rRNA.
Após a purificação e sequenciamento, as sequências obtidas podem então ser inseridas no banco de dados BLAST, onde podem ser comparadas com sequências de rRNA de referência 16S. Como este banco de dados retorna corresponde à maior similaridade, isso permite a confirmação da identidade das bactérias de interesse. Neste vídeo, você observará o sequenciamento genético de 16S rRNA, incluindo PCR, análise de sequência de DNA e edição, montagem de sequência e pesquisa de banco de dados.
Ao manusear microrganismos, é essencial seguir uma boa prática microbiológica, incluindo o uso de técnica asséptica e o uso de equipamentos de proteção individual adequados. Após a realização de uma avaliação de risco adequada para o microrganismo ou amostra ambiental de interesse, obtenha uma cultura de teste. Neste exemplo, é utilizada uma cultura pura de Bacillus subtilis.
Para começar, cresça seu microrganismo em um meio adequado nas condições apropriadas. Neste exemplo, o Bacillus subtilis 168 é cultivado em caldo LB durante a noite em uma incubadora de agitação programada para 200 rpm a 37 graus Celsius. Em seguida, use um kit comercialmente disponível para isolar DNA genômico ou gDNA de 1,5 mililitros da cultura B. subtilis overnight.
Para verificar a qualidade do DNA isolado, misture primeiro cinco microlitrais do gDNA isolado com um microliter de corante de carregamento de gel de DNA. Em seguida, carregue a amostra em um gel de 0,8%, contendo reagente de coloração de DNA, como seguro SYBR ou EtBr. Depois disso, carregue um padrão de massa molecular de uma quilobase no gel, e execute a eletroforese até que o corante frontal esteja a aproximadamente 0,5 centímetros do fundo do gel. Uma vez que a eletroforese de gel esteja completa, visualize o gel em um transilluminator de luz azul. O gDNA deve aparecer como uma faixa grossa, acima de 10 quilobases em tamanho e ter manchas mínimas.
Depois disso, para criar diluições seriais do gDNA, rotule três tubos de microcentrifuuge como 10X, 100X e 1000X. Em seguida, use uma pipeta para distribuir 90 microliters de água destilada estéril em cada um dos tubos. Em seguida, adicione 10 microliters da solução gDNA ao tubo 10X. Pipeta todo o volume para cima e para baixo para garantir que a solução seja completamente misturada. Em seguida, remova 10 microliters da solução do tubo 10X e transfira isso para o tubo 100X. Misture a solução como descrito anteriormente. Por fim, transfira 10 microliters da solução no tubo 100X, para o tubo 1000X.
Para iniciar o protocolo PCR, descongele os reagentes necessários no gelo. Em seguida, prepare a mistura mestre pcr. Uma vez que a polimerase de DNA está ativa à temperatura ambiente, a reação configurada deve ocorrer no gelo. Alíquota 49 microliters da mistura mestre em cada um dos tubos PCR. Em seguida, adicione um microliter de modelo a cada um dos tubos experimentais e um microliter de água estéril ao tubo de controle negativo, tubulação para cima e para baixo para misturar. Depois disso, defina a máquina PCR de acordo com o programa descrito na tabela. Coloque os tubos no termociclador e inicie o programa.
Uma vez que o programa esteja concluído, examine a qualidade do seu produto através da eletroforese do gel agarose, como demonstrado anteriormente. Uma reação bem sucedida usando o protocolo descrito deve produzir uma única faixa de aproximadamente 1,5 quilobase. Neste exemplo, a amostra contendo gDNA diluída de 100X produziu o produto de maior qualidade. Em seguida, purifique o melhor produto PCR, neste caso, o gDNA 100X, com um kit comercialmente disponível. Agora, o produto PCR pode ser enviado para sequenciamento.
Neste exemplo, o produto PCR é sequenciado usando primers para frente e para trás. Assim, são gerados dois conjuntos de dados, cada um contendo uma sequência de DNA e um cromatógrafo de DNA: um para o primer dianteiro e outro para o primer reverso. Primeiro, examine os cromatogramas gerados a partir de cada primer. Um cromatograma ideal deve ter picos espaçados uniformemente com pouco ou nenhum sinal de fundo.
Se os cromatogramas apresentarem picos duplos, vários modelos de DNA podem ter estado presentes nos produtos PCR e a sequência deve ser descartada. Se os cromatógramas contiveram picos de cores diferentes no mesmo local, o software de sequenciamento provavelmente chamou mal nucleotídeos. Esse erro pode ser identificado manualmente e corrigido no arquivo de texto. A presença de picos amplos no cromatógrama indica perda de resolução, o que causa má contagem dos nucleotídeos nas regiões associadas. Este erro é difícil de corrigir e os desencontros em qualquer uma das etapas subsequentes devem ser tratados como não confiáveis. A má qualidade de leitura do cromatograma, indicada pela presença de múltiplos picos, geralmente ocorre nas cinco extremidades primos e três principais da sequência. Alguns programas de sequenciamento removem essas seções de baixa qualidade automaticamente. Se sua sequência não estiver truncada automaticamente, identifique os fragmentos de baixa qualidade e remova suas respectivas bases do arquivo de texto.
Use um programa de montagem de DNA para montar as duas sequências de primer em uma sequência contínua. Lembre-se, as sequências obtidas usando primers para frente e para trás devem se sobrepor parcialmente. No programa de montagem de DNA, insira as duas sequências no formato FASTA na caixa apropriada. Em seguida, clique no botão enviar e aguarde que o programa retorne os resultados.
Para ver a sequência montada, clique em Contigs na guia resultados. Em seguida, para ver os detalhes do alinhamento, selecione os detalhes do conjunto. Navegue até o site para obter a ferramenta básica de pesquisa de alinhamento local, ou BLAST, e selecione a ferramenta BLAST nucleotídeo para comparar sua sequência com o banco de dados. Digite sua sequência na caixa de texto sequência de consulta e selecione o banco de dados apropriado no menu rolar para baixo. Por fim, clique no botão BLAST na parte inferior da página e aguarde que a ferramenta retorne as sequências mais semelhantes do banco de dados.
Neste exemplo, o topo é b. subtilis strain 168, mostrando 100% de identidade com a sequência no banco de dados BLAST. Se o hit superior não mostrar 100% de identidade para sua espécie ou cepa esperada, clique na sequência que mais combina com sua consulta para ver os detalhes do alinhamento. Nucleotídeos alinhados serão unidos por linhas verticais curtas e nucleotídeos incompatíveis terão lacunas entre eles. Focando nas regiões incompatíveis identificadas, revise a sequência e repita a pesquisa BLAST, se desejar.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
Identificar espécies bacterianas é importante para diferentes pesquisadores, bem como para aqueles em saúde. O sequenciamento de rRNA 16S foi inicialmente utilizado por pesquisadores para determinar as relações filogenéticas entre bactérias. Com o tempo, foi implementado em estudos metagenômicos para determinar a biodiversidade de amostras ambientais e em laboratórios clínicos como método para identificar potenciais patógenos. Permite uma identificação rápida e precisa das bactérias presentes em amostras clínicas, facilitando o diagnóstico precoce e o tratamento mais rápido dos pacientes.
Subscription Required. Please recommend JoVE to your librarian.
References
- Weisburg, W.G., Barns, S.M., Pelletier, D.A. and Lane D.J. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 173 (2): 697-703. (1991)
- Drancourt, M., Bollet, C., Carlioz, A., Martelin, R., Gayral, J.P., Raoult D. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol. 38 (10):3623-3630. (2000)
- Woo, P.C., Lau, S.K., Teng, J.L., Tse, H., Yuen, K.Y. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect. 14 (10):908-934. (2008)
- Tang, Y.W., Ellis, N.M., Hopkins, M.K., Smith, D.H., Dodge, D.E., Persing, D.H. Comparison of phenotypic and genotypic techniques for identification of unusual aerobic pathogenic gram-negative bacilli. J Clin Microbiol. 36 (12):3674-3679. (1998)
- Tsiboli, P., Herfurth, E., Choli, T. Purification and characterization of the 30S ribosomal proteins from the bacterium Thermus thermophilus. Eur J Biochem. 226 (1):169-177. (1994)
- Woese, C.R. Bacterial evolution. Microbiol Rev. 51 (2):221-271. (1987)
- Bartlett, J.M., Stirling, D. A short history of the polymerase chain reaction. Methods Mol Biol. 226:3-6. (2003)
- Wilson, K.H., Blitchington, R.B., Greene, R.C. Amplification of bacterial 16S ribosomal DNA with polymerase chain reaction. J Clin Microbiol. 28 (9):1942-1946. (1990)
- Shendure, J., Balasubramanian, S., Church, G.M., Gilbert, W., Rogers, J., Schloss, J.A., Waterston, R.H. (2017) DNA sequencing at 40: past, present and future. Nature. 550:345-353.
- Lane, D.J. 16S/23S rRNA sequencing. (1991) In Nucleic acid techniques in bacterial systematics. (Goodfellow, M. and Stackebrandt, E., eds.) p.115-175. Wiley and Sons, Chichester, United Kingdom.
- Turner, S., Pryer, K.M., Miao, V.P., Palmer, J.D. (1999) Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryot Microbiol. 46:327-338.
- Fredricks, D.N., Relman, D.A. (1998) Improved amplification of microbial DNA from blood cultures by removal of the PCR inhibitor sodium polyanetholesulfonate. J Clin Microbiol. 36:2810-2816.
- Wilson, K. Preparation of genomic DNA from bacteria. (2001) Curr Protoc Mol Biol. Chapter 2:Unit 2.4.
- Wright, M. H., Adelskov, J., Greene, A.C. (2017) Bacterial DNA extraction using individual enzymes and phenol/chloroform separation. J Microbiol Biol Educ. 18:18.2.48.
- Huang, X., Madan, A. (1999). CAP3: A DNA sequence assembly program. Genome Res. 9:868-877.