



Overview
Fuente: Ewa Bukowska-Faniband1, Tilde Andersson1, Rolf Lood1
1 Departamento de Ciencias Clínicas Lund, División de Medicina de Infecciones, Centro Biomédico, Universidad de Lund, 221 00 Lund, Suecia
El planeta Tierra es un hábitat para millones de especies bacterianas, cada una de las cuales tiene características específicas. La identificación de especies bacterianas se utiliza ampliamente en la ecología microbiana para determinar la biodiversidad de muestras ambientales y microbiología médica para diagnosticar pacientes infectados. Las bacterias se pueden clasificar utilizando métodos de microbiología convencionales, como la microscopía, el crecimiento en medios específicos, las pruebas bioquímicas y serológicas y los ensayos de sensibilidad a los antibióticos. En las últimas décadas, los métodos de microbiología molecular han revolucionado la identificación bacteriana. Un método popular es la secuenciación del gen del ARN ribosomal 16S (ARN RRNA). Este método no sólo es más rápido y preciso que los métodos convencionales, sino que también permite la identificación de cepas que son difíciles de cultivar en condiciones de laboratorio. Además, la diferenciación de las cepas a nivel molecular permite la discriminación entre bacterias fenotípicamente idénticas (1-4).
El ARNR 16S se une a un complejo de 19 proteínas para formar una subunidad 30S del ribosoma bacteriano (5). Está codificado por el gen 16S rRNA, que está presente y altamente conservado en todas las bacterias debido a su función esencial en el ensamblaje ribosoma; sin embargo, también contiene regiones variables que pueden servir como huellas dactilares para determinadas especies. Estas características han hecho del gen rRNA 16S un fragmento genético ideal para ser utilizado en la identificación, comparación y clasificación filogenética de bacterias (6).
La secuenciación del gen rRNA 16S se basa en la reacción en cadena de la polimerasa (PCR) (7-8) seguida de la secuenciación del ADN (9). La PCR es un método de biología molecular utilizado para amplificar fragmentos específicos de ADN a través de una serie de ciclos que incluyen:
i) Desnaturalización de una plantilla de ADN de doble cadena
ii) Recocido de imprimaciones (oligonucleótidos cortos) que son complementarios a la plantilla
iii) Extensión de imprimaciones por la enzima ADN polimerasa, que sintetiza una nueva cadena de ADN
En la Figura 1se muestra una descripción esquemática del método.
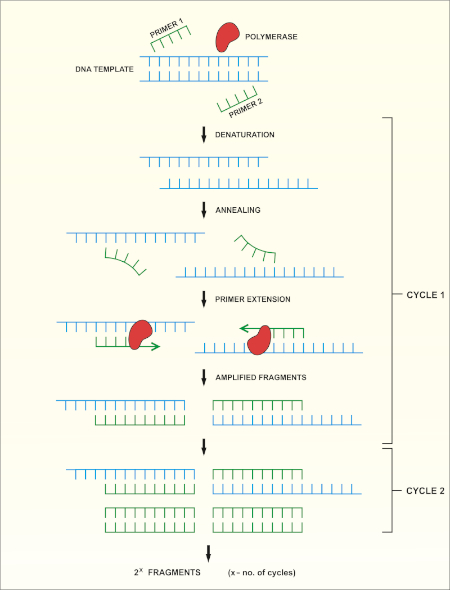
Figura 1: Descripción esquemática de la reacción PCR. Haga clic aquí para ver una versión más grande de esta figura.
Hay varios factores que son importantes para una reacción de PCR exitosa, uno de los cuales es la calidad de la plantilla de ADN. El aislamiento del ADN cromosómico de las bacterias se puede realizar utilizando protocolos estándar o kits comerciales. Se debe tener especial cuidado para obtener ADN que esté libre de contaminantes que puedan inhibir la reacción de PCR.
Las regiones conservadas del gen 16S rRNA permiten el diseño de pares de imprimación universales (uno hacia adelante y otro inverso) que pueden unirse y amplificar la región objetivo en cualquier especie bacteriana. La región de destino puede variar en tamaño. Mientras que algunos pares de imprimación pueden amplificar la mayor parte del gen rRNA 16S, otros amplifican sólo partes de él. En la Tabla 1 se muestran ejemplos de imprimaciones de uso común y sus sitios de enlace se muestran en la Figura 2.
| Primer nombre | Secuencia (5'-3') | Adelante/retroceso | Referencia |
| 8F b) | AGAGTTTGATCCTGGCTCAG | Adelante | -1 |
| 27F | AGAGTTTGATCMTGGCTCAG | Adelante | -10 |
| 515F | GTGCCAGCMGCCGCGGTAA | Adelante | -11 |
| 911R | GCCCCCGTCAATTCMTTTGA | Marcha atrás | -12 |
| 1391R | GACGGGCGGTGTARCA | Marcha atrás | -11 |
| 1492R | GGTTACCTTGTTACGACTT | Marcha atrás | -11 |
Tabla 1: Ejemplos de oligonucleótidos estándar utilizados en la amplificación de genes 16S rRNA a).
a) Las longitudes esperadas del producto PCR generado utilizando las diferentes combinaciones de imprimación se pueden estimar calculando la distancia entre los sitios de unión para la imprimación delantera y la inversa (véase la figura 2), por ejemplo, el tamaño del PCR producto con el par de imprimación 8F-1492R es de 1500 bp, y para el par de imprimación 27F-911R a 900 bp.
b) también conocido como fD1

Figura 2: Figura representativa de la secuencia de rRNA 16S y los sitios de enlace de imprimación. Las regiones conservadas se colorean en gris y las regiones variables se rellenan con líneas diagonales. Para permitir la resolución más alta, la imprimación 8F y 1492R (nombre basado en la ubicación en la secuencia rRNA) se utilizan para amplificar toda la secuencia, permitiendo la secuencia de varias regiones variables del gen. Haga clic aquí para ver una versión más grande de esta figura.
Las condiciones de ciclismo para pcR(es decir, la temperatura y el tiempo necesarios para que el ADN sea desnaturalizado, recocido con imprimadores y sintetizado) dependen del tipo de polimerasa que se utiliza y de las propiedades de las imprimaciones. Se recomienda seguir las pautas del fabricante para una polimerasa en particular.
Una vez completado el programa PCR, los productos son analizados por electroforesis de gel de agarosa. Un PCR exitoso produce una sola banda de tamaño esperado. El producto debe purificarse antes de la secuenciación para eliminar las imprimaciones residuales, los desoxirribonucleótidos, la polimerasa y el tampón que estaban presentes en la reacción de PCR. Los fragmentos de ADN purificados generalmente se envían para la secuenciación a servicios de secuenciación comercial; sin embargo, algunas instituciones realizan la secuenciación del ADN en sus propias instalaciones principales.
La secuencia de ADN se genera automáticamente a partir de un cromatograma de ADN por una computadora y debe ser cuidadosamente revisada para la calidad, ya que a veces se necesita la edición manual. Siguiendo este paso, la secuencia genética se compara con las secuencias depositadas en la base de datos 16S rRNA. Se identifican las regiones de similitud y se entregan las secuencias más similares.
Procedure
1. Configurar
- Mientras se manipulan microorganismos, se requiere seguir buenas prácticas microbiológicas. Todos los microorganismos, especialmente las muestras desconocidas, deben tratarse como patógenos potenciales. Siga la técnica aséptica para evitar contaminar las muestras, los investigadores o el laboratorio. Lávese las manos antes y después de manipular las bacterias, use guantes y use ropa protectora.
- Llevar a cabo una evaluación de riesgos para el protocolo experimental para el aislamiento del ADN genómico y la purificación del producto PCR. ¡Algunos reactivos pueden ser dañinos!
- El cultivo puro es esencial para la secuenciación de rRNA 16S. Antes de proceder al aislamiento del ADN genómico, asegúrese de que el material de partida sea completamente puro. Esto se puede hacer mediante el revestimiento de rayas para aislar colonias individuales. Estos pueden ser más cultivados rayados en platos individualmente, o en caldo, si es necesario.
- Equipo de laboratorio requerido:
- Ciclor térmico para PCR. La función del ciclor térmico es subir y bajar la temperatura según un programa establecido. Al crear el programa se le pedirá que introduzca los valores de temperatura y tiempo para cada paso de PCR, así como el número total de ciclos.
- Sistema de electroforesis de gel de agarosa. Se utiliza para separar fragmentos de ADN en función de su tamaño y carga. En este protocolo, la electroforesis de gel de agarosa se utilizará para visualizar la calidad de los productos aislados de ADN genómico y PCR.
2. Protocolo
Nota: El protocolo demostrado se aplica a la secuenciación del gen rRNA 16S de un cultivo puro de bacterias. No se aplica a estudios metagenómicos.
-
Bacterias de cultivo para el aislamiento del ADN genómico (ADN)
- Haga crecer su microorganismo en un medio adecuado. Tanto los medios líquidos como los sólidos se pueden utilizar en este paso. Elija condiciones que produzcan el mejor crecimiento. Al planear el experimento, tenga en cuenta que las bacterias de crecimiento lento pueden necesitar varios días para llegar a la fase de crecimiento estacionario o registro tardío. En este protocolo, Bacillus subtilis 168 fue cultivado en caldo de lisabagenia (LB) durante la noche en una incubadora de agitación establecida a 200 rpm, 37oC.
-
Aislamiento de gDNA.
- Si las bacterias se cultivaban en un medio sólido, raspar algunas células usando un lazo estéril y resuspenderlas en 1 ml de agua destilada
- Si las bacterias se cultivan en medio líquido, utilice aproximadamente 1,5 ml de un cultivo nocturno.
- Reventar las células por centrifugación (1 minuto, 12.000 - 16.000 g), eliminar el sobrenadante y utilizar las células para el aislamiento de ADNes utilizando un kit comercial o protocolos estándar[ por ejemplo, preparación total del ADN CTAB (13) o extracción de fenol-cloroformo (14)]. Aquí, se utilizó un kit comercial para aislar el ADNr de 1,5 ml de cultivo nocturno De B. subtilis 168, OD600 a 1,5.
Nota 1: Para algunas bacterias Gram-negativas este paso puede ser omitido y reemplazado por la simple liberación de ADN de las células por ebullición. Resuspender el pellet bacteriano en agua destilada e incubar en un bloque de calentamiento establecido a 100 oC durante 10 minutos.
Nota 2: Las células bacterianas Gram-positivas son difíciles de interrumpir. Por lo tanto, se recomienda elegir un método o kit de aislamiento de ADNado que se dedique al aislamiento de este grupo de bacterias.
-
control de calidad gDNA.
- Compruebe la calidad del ADNamediante aislado mediante electroforesis de gel de agarosa. En primer lugar, mezcle 5 ml del ADNN aislado con 1 l del tinte de carga (6x) y cargue la muestra en un gel de agarosa del 0,8% que contenga un reactivo de tinción de ADN.
- Cargue un estándar de masa molecular y ejecute la electroforesis hasta que el frente del tinte alcance la parte inferior del gel.
- Una vez completada la electroforesis, visualice el gel en un transiluminador adecuado (luz UV o azul). gDNA aparece como una banda molecular alta gruesa (por encima de 10 kb). En la Figura 3se muestra un ejemplo de la comprobación de calidad de gDNA.
- Si el gDNA pasa el control de calidad(es decir,la banda molecular alta está presente y hay poco o ningún frotis del ADN) , diluir su gDNA en serie mediante el primer etiquetado de 3 tubos de microcentrífuga de la siguiente manera: "10x", "100x" y "1000x".
- Pipetear 90 l de agua destilada estéril en cada uno de los 3 tubos.
- Tomar 10 sde la solución de ADNG y añadirla al tubo marcado como "10x".
- Encinerñe todo el volumen(es decir, 100 l) hacia arriba y hacia abajo a fondo para asegurarse de que la solución se mezcla uniformemente. A continuación, tome 10 s de la solución de este tubo y transfiérala al tubo marcado como "100x".
- Mezclar como se ha descrito anteriormente y repetir el mismo procedimiento transfiriendo 10 l de la solución del tubo "100x" al tubo "1000x". Estas diluciones se utilizarán como plantilla en la reacción PCR.

Figura 3: Electroforesis de gel de agarosa de ADNaaislado de Bacillus subtilis. Carril 1: M - marcador de masa molecular (de arriba a abajo: 10000 bp, 8000 bp, 6000 bp, 5000 bp, 4000 bp, 3500 bp, 3000 bp, 2500 bp, 2000 bp, 1500 bp, 1000 bp). Carril 2: gDNA - ADN genómico aislado de Bacillus subtilis. Haga clic aquí para ver una versión más grande de esta figura.
-
Amplificación del gen rRNA 16S por PCR.
Nota: El protocolo PCR que figura a continuación está optimizado para un par de imprimación y polimera sin ADN en particular 8F - 1492R (véase el cuadro 1). Se requiere la optimización del protocolo para cada par de polimerasa e imprimación.- Descongela todos los reactivos en hielo.
- Prepare la mezcla maestra PCR tal y como se muestra en del Cuadro 2. Dado que la polimerasa de ADN está activa a temperatura ambiente, la configuración de reacción debe realizarse en hielo, es decir, los tubos PCR y los componentes de reacción deben mantenerse en hielo todo el tiempo. Prepare una reacción por cada muestra de ADNr y una reacción para un control negativo. El control negativo es una mezcla de PCR sin la plantilla de ADNr y se utiliza para garantizar que los demás componentes de la reacción no estén contaminados.
Nota: En el caso de varias muestras, se prepara comúnmente una mezcla maestra. La mezcla maestra es una solución que contiene todos los componentes de reacción excepto la plantilla. Ayuda a omitir el pipeteo repetitivo, evitar errores de pipeteo y garantiza una alta consistencia entre las muestras. Para preparar la mezcla maestra, multiplique el volumen de cada componente (excepto la plantilla de ADN) por el número de muestras analizadas. Mezclar todos los componentes en el tubo de microcentrífuga y canalizar todo el volumen hacia arriba y hacia abajo varias veces. - Aliquot 49 l de la mezcla maestra en los tubos PCR individuales.
- Añadir plantilla de 1 L en tubos con mezcla maestra. Para un control negativo, agregue 1 l de agua estéril. Para asegurarse de que los componentes estén bien mezclados, entube suavemente la mezcla hacia arriba y hacia abajo 10 veces con una pipeta establecida en 30-50 l.
- Ajuste la máquina PCR con el programa que se muestra en la Tabla 3.
- Coloque los tubos en la máquina PCR e inicie el programa.
- Una vez completado el programa, examine la calidad de su producto PCR mediante la electroforesis de gel de agarosa.
- Una reacción de PCR exitosa utilizando el par de imprimación 8F-1492R produce una sola banda de aproximadamente 1,5 kb(Figura 4). Si hay otras bandas (es decir, productos inespecíficos), optimice el programa de PCR ajustando la temperatura de recocido. Si hay una sola banda del tamaño esperado, continúe con el paso siguiente. Aquí, la reacción de PCR con plantilla de ADNado diluido 100x produjo el mejor producto, ya que tenía una banda afilada de tamaño esperado y carecía de productos inespecíficos. Por lo tanto, fue elegido para ser purificado y enviado para la secuenciación.
- Antes de la secuenciación, el producto debe limpiarse de imprimaciones residuales, desoxirribonucleótidos, polimerasa y tampón que estaban presentes en la reacción de PCR. Los productos PCR se pueden aislar utilizando un kit de purificación de PCR comercial. La reacción PCR se carga en una columna que contiene una matriz de enlace de ADN. El producto PCR se enlaza a la columna, mientras que otros componentes fluyen a través de la columna. La columna se lava con tampón de lavado, y finalmente, el ADN se eluye en el tampón de elección. Confirme que el búfer de elución que se complementa con el kit es compatible con la secuenciación.
- Envíe el producto PCR purificado para la secuenciación de ADN. Siga las directrices para la presentación de muestras de secuenciación en la instalación de secuenciación elegida. Para obtener la mejor cobertura de secuencia, utilice las imprimaciones de amplificación de PCR (las mismas que se utilizan en la sección 2.4.1) que las imprimaciones de secuenciación. Aquí, las imprimaciones 8F y 1492R se utilizaron para secuenciar el producto PCR.
| Componente | Concentración final | Volumen por reacción | Volumen por x reacciones (mezcla maestra) |
| Búfer de reacción 5x | 1x | 10 l | 10 l x x |
| DNTP de 10 mM | 200 M | 1 L | 1 l x x |
| 10 M Primer 8F | 0,5 m | 2,5 l | 2,5 sL x x |
| 10 M Primer 1492R | 0,5 m | 2,5 l | 2,5 sL x x |
| Phusion polimerasa | 1 unidad | 0,5 l | 0,5 sL x x |
| ADN de plantilla * | - | 1 L | - |
| ddH2O | - | 32,5 l | 32,5 l x x |
| Volumen total | 50 l | 49 l x x |
Cuadro 2: Componentes de reacción PCR. * Utilice el ADNado diluido de 10x, 100x o 1000x del paso 2.3.
| Paso | Temperatura | hora | Ciclos |
| Desnaturalización inicial | 98oC | 30 seg | |
| Desnaturalización | 98oC | 10 seg | 25-30 |
| Recocido | 60oC | 30 seg | |
| Extensión | 72oC | 45 seg | |
| Extensión final | 72oC | 7 min | |
| Mantener | 4oC | ∞ |
Cuadro 3: Programa PCR para la amplificación del gen rRNA 16S.

Figura 4: Electroforesis de gel de agarosa de productos PCR amplificadas utilizando imprimaciones 8F y 1492R y gDNA como plantilla. La muestra de ADN g de B. subtilis (ver figura 3) se diluyó 10, 100 y 1000 veces con el fin de probar el mejor resultado. Carril 1: M - marcador de masa molecular (de arriba a abajo: 10000 bp, 8000 bp, 6000 bp, 5000 bp, 4000 bp, 3500 bp, 3000 bp, 2500 bp, 2000 bp, 1500 bp, 1000bp, 750 bp, 500 bp, 250 bp). Carril 2: Reacción PCR con plantilla diluida 10x. Carril 3: Reacción PCR con plantilla diluida de 100x. Carril 4: Reacción PCR con plantilla diluida 1000x. Carril 5: (C-) - control negativo (reacción sin la plantilla de ADN). Haga clic aquí para ver una versión más grande de esta figura.
3. Análisis de datos y resultados
Nota: El producto PCR se secuencia utilizando las imprimaciones delanteras (aquí 8F) y inversas (aquí 1492R). Por lo tanto, se generan dos conjuntos de secuencia de datos, uno para el avance y otro para el primer inverso. Para cada secuencia se generan al menos dos tipos de archivo: i) un archivo de texto que contiene la secuencia de ADN y ii) un cromatograma de ADN, que muestra la calidad de la ejecución de secuenciación.
- Para la imprimación delantera, abra el cromatograma y examine cuidadosamente la secuencia. Un cromatograma ideal para una secuencia de calidad debe tener picos espaciados uniformemente y pocas o ninguna señal de fondo(Figura 5A).
- Si el cromatograma no es de alta calidad, la secuencia debe descartarse o el archivo de texto de secuencia debe revisarse de acuerdo con lo siguiente:
- La presencia de picos dobles a lo largo del cromatograma indica la presencia de múltiples plantillas de ADN. Esto puede ser el caso si el cultivo bacteriano no era puro. Dicha secuencia debe descartarse(Figura 5B).
- Un cromatograma ambiguo puede surgir de la presencia de picos de diferentes colores en la misma ubicación. Uno de los errores más comunes es la presencia de dos picos de colores diferentes en la misma posición y la asignación incorrecta de las bases por el software de secuenciación(Figura 5C). Corrija manualmente los nucleótidos asignados incorrectamente y edítelos en el archivo de texto.
- Los cromatogramas de baja resolución pueden dar lugar a "picos amplios" que a menudo causan recuentos erróneos de los nucleótidos en estas regiones(Figura 5D). Este error es difícil de corregir y, por lo tanto, las posibles discrepancias en el paso de alineación posterior no deben tratarse como fiables.
- La mala calidad de lectura de cromatogramas y la presencia de múltiples picos se ve comúnmente en los extremos de 5' y 3' de la secuencia. Algunos software secuenciador eliminan estos fragmentos de baja calidad automáticamente(Figura 5E),y los nucleótidos no se incluyen en el archivo de texto. Si la secuencia no se truncó automáticamente, determine los fragmentos de baja calidad(por ejemplo, señal débil, picos superpuestos, pérdida de resolución) en los extremos y elimine las bases respectivas del archivo de texto.

Figura 5: Ejemplos de solución de problemas de secuenciación de ADN. A) Un ejemplo de una secuencia de cromatogramas de calidad (picos uniformemente espaciados e inequívocos). B) Secuencia de mala calidad que generalmente ocurre al principio del cromatograma. El área de la zona gris se considera de baja calidad y se elimina automáticamente por el software de secuenciación. Más bases se pueden recortar manualmente. C) Presencia de picos dobles (indicados por flechas). Un nucleótido que se indica mediante la flecha roja ha sido leído por el secuenciador como "T" (pico rojo), pero el pico azul es más fuerte, y también se puede interpretar como "C". D) Los picos superpuestos indican contaminación del ADN(es decir, más de una plantilla). E) Pérdida de resolución y los llamados "picos anchos" (marcados por rectángulo) que impiden la llamada base confiable. Haga clic aquí para ver una versión más grande de esta figura.
- Repita 3.1 y 3.2 para la imprimación inversa.
- Por último, ensamble las secuencias de avance e retroceso en una secuencia contigua. Una buena ejecución de secuenciación produce una secuencia de hasta 1100 bp. Teniendo en cuenta que el producto PCR tiene 1500 bp de largo, las secuencias obtenidas utilizando imprimaciones hacia delante y hacia atrás deben superponerse parcialmente.
- Combine las dos secuencias utilizando el programa de ensamblaje de secuencia de ADN, por ejemplo, una herramienta gratuita como CAP3 (http://doua.prabi.fr/software/cap3) (15).
- Inserte las dos secuencias en formato FASTA en el cuadro indicado. Haga clic en el botón "Enviar" y espere a que los resultados vuelvan.
- Para ver la secuencia montada pulse "Contigs" en la pestaña de resultados. Para ver los detalles de la alineación pulse "Detalles de ensamblaje".
Nota 1: Si el software CAP3 se utiliza para el ensamblaje contig, no es necesario convertir la secuencia de imprimación inversa en complementaria inversa; sin embargo, este paso podría ser necesario si se utiliza otro programa.
Nota 2: El formato FASTA es un formato basado en texto para representar la secuencia de nucleótidos. La primera línea (la línea de descripción) de un archivo FASTA comienza con un símbolo ">" seguido del nombre o un identificador único de la secuencia. Siguiendo la línea de descripción está la secuencia de nucleótidos. Pegue las secuencias en el siguiente formato:
>sequence_frw_primer
pegar su secuencia desde el archivo de texto aquí
>sequence_rvs_primer
pegar su secuencia desde el archivo de texto aquí
- Realice una búsqueda en la base de datos visitando el sitio web para la Herramienta básica de búsqueda de alineación local (BLAST; https://blast.ncbi.nlm.nih.gov/Blast.cgi).
- Seleccione la herramienta "Nucleotide BLAST" para comparar su secuencia con la base de datos.
- Introduzca su secuencia (la contig ensamblada en 3.5) en el cuadro de texto "Secuencia de consulta" y, a continuación, seleccione la base de datos "Secuencias rRNA 16S (Bacterias y Archea)" en el menú de desplazamiento hacia abajo.
- Pulse el botón "BLAST" en la parte inferior de la página. Se devolverán las secuencias más similares. En la Figura 6se muestra un ejemplo de resultado BLAST. En el experimento presentado el éxito más alto es B. subtilis strain 168, mostrando 100% de identidad con la secuencia disponible en la base de datos BLAST.
- Si el hit superior no muestra la identidad del 100%, vaya a la alineación y compruebe si hay discrepancias. Al hacer clic en el golpe superior, se le dirigirá a los detalles de la alineación. Los nucleótidos alineados se unirán por líneas verticales cortas, mientras que los nucleótidos no coincidentes tienen un espacio entre ellos. Vuelva al cromatograma que recibió de la empresa de secuenciación y revise la secuencia una vez más con el foco en la región no coincidente. Corrija la secuencia si se encuentra algún error adicional. Ejecute BLAST de nuevo utilizando la secuencia corregida.

Figura 6: Ejemplo del resultado de la BLAST de nucleótidos. La secuencia del gen 16S rRNA del cultivo puro de B. subtilis 168 se utilizó como secuencia de consulta. El primer éxito muestra 100% de identidad (subrayado) a la cepa B. subtilis 168, como se esperaba. Haga clic aquí para ver una versión más grande de esta figura.
La Tierra es el hogar de millones de especies bacterianas, cada una con características únicas. La identificación de estas especies es fundamental en la evaluación de muestras ambientales. Los médicos también necesitan distinguir diferentes especies bacterianas para diagnosticar pacientes infectados.
Para identificar bacterias, se pueden emplear una variedad de técnicas, incluyendo la observación microscópica de morfología o crecimiento en un medio específico para observar la morfología de la colonia. El análisis genético, otra técnica para identificar bacterias ha crecido en popularidad en los últimos años, debido en parte a la secuenciación del gen del ARN ribosomal 16S.
El ribosoma bacteriano es un complejo de ARN proteico que consta de dos subunidades. La subunidad 30S, la más pequeña de estas dos subunidades, contiene 16S rRNA, que está codificado por el gen rRNA 16S contenido en el ADN genómico. Las regiones específicas de 16S rRNA están altamente conservadas, debido a su función esencial en el montaje ribosoma. Mientras que otras regiones, menos críticas para funcionar, pueden variar entre las especies bacterianas. Las regiones variables en 16S rRNA, pueden servir como huellas moleculares únicas para las especies bacterianas, lo que nos permite distinguir cepas fenotípicamente idénticas.
Después de obtener una muestra de calidad de gDNA, pcR del gen de codificación 16S rRNA puede comenzar. La PCR es un método de biología molecular comúnmente utilizado, que consiste en ciclos de desnaturalización de la plantilla de ADN de doble cadena, recocido de pares de imprimación universales, que amplifican regiones altamente conservadas del gen, y la extensión de imprimaciones por ADN polimerasa. Mientras que algunos imprimadores amplifican la mayor parte del gen de codificación 16S rRNA, otros sólo amplifican fragmentos de él. Después de pcR, los productos se pueden analizar a través de electroforesis de gel de agarosa. Si la amplificación fue exitosa, el gel debe contener una sola banda de un tamaño esperado, dependiendo del par de imprimación utilizado, hasta 1500bp, la longitud aproximada del gen rRNA 16S.
Después de la purificación y la secuenciación, las secuencias obtenidas se pueden introducir en la base de datos BLAST, donde se pueden comparar con las secuencias rRNA de referencia 16S. Como esta base de datos devuelve coincidencias basadas en la mayor similitud, esto permite la confirmación de la identidad de las bacterias de interés. En este vídeo, observará la secuenciación del gen rRNA 16S, incluyendo PCR, análisis y edición de secuencia de ADN, montaje de secuencia según la base de datos.
Al manipular microorganismos, es esencial seguir buenas prácticas microbiológicas, incluyendo el uso de una técnica aséptica y el uso de equipos de protección personal adecuados. Después de realizar una evaluación de riesgos adecuada para el microorganismo o la muestra de interés ambiental, obtenga un cultivo de prueba. En este ejemplo, se utiliza una cultura pura de Bacillus subtilis.
Para empezar, haz crecer tu microorganismo en un medio adecuado en las condiciones adecuadas. En este ejemplo, Bacillus subtilis 168 se cultiva en caldo LB durante la noche en una incubadora de agitación establecida a 200 rpm a 37 grados centígrados. A continuación, utilice un kit disponible comercialmente para aislar el ADN genómico o el ADN electrónico de 1,5 mililitros del cultivo nocturno de B. subtilis.
Para comprobar la calidad del ADN aislado, primero mezcle cinco microlitros del ADN gDNA aislado con un microlitro de color ante la carga de gel de ADN. A continuación, cargue la muestra en un gel de agarosa del 0,8%, que contenga un reactivo de tinción de ADN, como SYBR seguro o EtBr. Después de esto, cargue un estándar de masa molecular de una kilobase en el gel, y ejecute la electroforesis hasta que el tinte frontal esté aproximadamente a 0,5 centímetros de la parte inferior del gel. Una vez que la electroforesis de gel esté completa, visualice el gel en un transiluminador de luz azul. El ADNes debe aparecer como una banda gruesa, por encima de 10 kilobase de tamaño y tener un mínimo de manchas.
Después de esto, para crear diluciones en serie del ADN, etiquete tres tubos de microcentrífuga como 10X, 100X y 1000X. A continuación, utilice una pipeta para dispensar 90 microlitros de agua destilada estéril en cada uno de los tubos. A continuación, agregue 10 microlitros de la solución de ADN-a g al tubo 10X. Pipetear todo el volumen hacia arriba y hacia abajo para asegurarse de que la solución se mezcla a fondo. A continuación, retire 10 microlitros de la solución del tubo 10X y transfiera esto al tubo 100X. Mezcle la solución como se describió anteriormente. Finalmente, transfiera 10 microlitros de la solución en el tubo 100X, al tubo 1000X.
Para comenzar el protocolo PCR, descongelar los reactivos necesarios en el hielo. A continuación, prepare la mezcla maestra de PCR. Dado que la polimerasa de ADN está activa a temperatura ambiente, la reacción establecida debe producirse en el hielo. Aliquot 49 microlitros de la mezcla maestra en cada uno de los tubos PCR. Luego, agregue un microlitro de plantilla a cada uno de los tubos experimentales y un microlitro de agua estéril al tubo de control negativo, pipeteando hacia arriba y hacia abajo para mezclar. Después de esto, configure la máquina PCR de acuerdo con el programa descrito en la tabla. Coloque los tubos en el termociclador e inicie el programa.
Una vez completado el programa, examine la calidad de su producto a través de la electroforesis de gel de agarosa, como se ha demostrado anteriormente. Una reacción exitosa utilizando el protocolo descrito debe producir una sola banda de aproximadamente 1,5 kilobase. En este ejemplo, la muestra que contiene 100X de ADN adobloca diluido produjo un producto de la más alta calidad. A continuación, purificar el mejor producto PCR, en este caso, el 100X gDNA, con un kit disponible comercialmente. Ahora el producto PCR se puede enviar para la secuenciación.
En este ejemplo, el producto PCR se secuencia mediante imprimaciones directas e inversas. Así, se generan dos conjuntos de datos, cada uno con una secuencia de ADN y un cromatograma de ADN: uno para la imprimación delantera y el otro para la imprimación inversa. En primer lugar, examine los cromatogramas generados a partir de cada imprimación. Un cromatograma ideal debe tener picos espaciados uniformemente con pocas o ninguna señal de fondo.
Si los cromatogramas muestran picos dobles, es posible que haya varias plantillas de ADN presentes en los productos PCR y la secuencia debe descartarse. Si los cromatogramas contenían picos de diferentes colores en la misma ubicación, es probable que el software de secuenciación haya llamado mal los nucleótidos. Este error se puede identificar y corregir manualmente en el archivo de texto. La presencia de picos amplios en el cromatograma indica una pérdida de resolución, lo que provoca recuentos erróneos de los nucleótidos en las regiones asociadas. Este error es difícil de corregir y las discrepancias en cualquiera de los pasos subsiguientes deben tratarse como poco fiables. La mala calidad de lectura del cromatograma, indicada por la presencia de múltiples picos, generalmente ocurre en los cinco extremos primos y tres extremos principales de la secuencia. Algunos programas de secuenciación eliminan estas secciones de baja calidad automáticamente. Si la secuencia no se truncó automáticamente, identifique los fragmentos de baja calidad y elimine sus respectivas bases del archivo de texto.
Utilice un programa de ensamblaje de ADN para ensamblar las dos secuencias de imprimación en una secuencia continua. Recuerde, las secuencias obtenidas utilizando imprimaciones hacia adelante e inversas deben superponerse parcialmente. En el programa de ensamblaje de ADN, inserte las dos secuencias en formato FASTA en el cuadro correspondiente. A continuación, haga clic en el botón Enviar y espere a que el programa devuelva los resultados.
Para ver la secuencia ensamblada, haga clic en Contigs en la pestaña de resultados. A continuación, para ver los detalles de la alineación, seleccione detalles de ensamblaje. Navegue hasta el sitio web para obtener la herramienta básica de búsqueda de alineación local, o BLAST, y seleccione la herramienta blast de nucleótidos para comparar la secuencia con la base de datos. Escriba la secuencia en el cuadro de texto de la secuencia de consulta y seleccione la base de datos adecuada en el menú desplegable. Por último, haga clic en el botón BLAST en la parte inferior de la página y espere a que la herramienta devuelva las secuencias más similares de la base de datos.
En este ejemplo, el hit más alto es B. subtilis strain 168, mostrando 100% de identidad con la secuencia en la base de datos BLAST. Si el hit superior no muestra 100% de identidad a su especie o cepa esperada, haga clic en la secuencia que coincida más estrechamente con su consulta para ver los detalles de la alineación. Los nucleótidos alineados se unirán por líneas verticales cortas y los nucleótidos no coincidentes tendrán huecos entre ellos. Centrándose en las regiones no coincidentes identificadas, revise la secuencia y repita la búsqueda BLAST si lo desea.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
La identificación de especies bacterianas es importante para diferentes investigadores, así como para aquellos en la atención médica. La secuenciación de arNM 16S fue utilizada inicialmente por los investigadores para determinar las relaciones filogenéticas entre las bacterias. Con el tiempo, se ha implementado en estudios metagenómicos para determinar la biodiversidad de muestras ambientales y en laboratorios clínicos como un método para identificar patógenos potenciales. Permite una identificación rápida y precisa de las bacterias presentes en las muestras clínicas, facilitando un diagnóstico más temprano y un tratamiento más rápido de los pacientes.
Subscription Required. Please recommend JoVE to your librarian.
References
- Weisburg, W.G., Barns, S.M., Pelletier, D.A. and Lane D.J. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 173 (2): 697-703. (1991)
- Drancourt, M., Bollet, C., Carlioz, A., Martelin, R., Gayral, J.P., Raoult D. 16S ribosomal DNA sequence analysis of a large collection of environmental and clinical unidentifiable bacterial isolates. J Clin Microbiol. 38 (10):3623-3630. (2000)
- Woo, P.C., Lau, S.K., Teng, J.L., Tse, H., Yuen, K.Y. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect. 14 (10):908-934. (2008)
- Tang, Y.W., Ellis, N.M., Hopkins, M.K., Smith, D.H., Dodge, D.E., Persing, D.H. Comparison of phenotypic and genotypic techniques for identification of unusual aerobic pathogenic gram-negative bacilli. J Clin Microbiol. 36 (12):3674-3679. (1998)
- Tsiboli, P., Herfurth, E., Choli, T. Purification and characterization of the 30S ribosomal proteins from the bacterium Thermus thermophilus. Eur J Biochem. 226 (1):169-177. (1994)
- Woese, C.R. Bacterial evolution. Microbiol Rev. 51 (2):221-271. (1987)
- Bartlett, J.M., Stirling, D. A short history of the polymerase chain reaction. Methods Mol Biol. 226:3-6. (2003)
- Wilson, K.H., Blitchington, R.B., Greene, R.C. Amplification of bacterial 16S ribosomal DNA with polymerase chain reaction. J Clin Microbiol. 28 (9):1942-1946. (1990)
- Shendure, J., Balasubramanian, S., Church, G.M., Gilbert, W., Rogers, J., Schloss, J.A., Waterston, R.H. (2017) DNA sequencing at 40: past, present and future. Nature. 550:345-353.
- Lane, D.J. 16S/23S rRNA sequencing. (1991) In Nucleic acid techniques in bacterial systematics. (Goodfellow, M. and Stackebrandt, E., eds.) p.115-175. Wiley and Sons, Chichester, United Kingdom.
- Turner, S., Pryer, K.M., Miao, V.P., Palmer, J.D. (1999) Investigating deep phylogenetic relationships among cyanobacteria and plastids by small subunit rRNA sequence analysis. J Eukaryot Microbiol. 46:327-338.
- Fredricks, D.N., Relman, D.A. (1998) Improved amplification of microbial DNA from blood cultures by removal of the PCR inhibitor sodium polyanetholesulfonate. J Clin Microbiol. 36:2810-2816.
- Wilson, K. Preparation of genomic DNA from bacteria. (2001) Curr Protoc Mol Biol. Chapter 2:Unit 2.4.
- Wright, M. H., Adelskov, J., Greene, A.C. (2017) Bacterial DNA extraction using individual enzymes and phenol/chloroform separation. J Microbiol Biol Educ. 18:18.2.48.
- Huang, X., Madan, A. (1999). CAP3: A DNA sequence assembly program. Genome Res. 9:868-877.