



Overview
ソース: アレクサンダー S. ゴールド1, トーニャ M. コルピッツ1
1ボストン大学医学部微生物学科、国立新興感染症研究所、ボストン、マサチューセッツ州
トランスダクションは、バクテリオファージ(ファージ)を利用する細菌間の遺伝的交換の一形態であり、原核生物に排他的に感染するウイルスのクラスである。この形態のDNA移動は、ファージを通じてある細菌から別の細菌に移り、1951年にノートン・ジンダーとジョシュア・レデレルグによって発見された(1)。バクテリオファージは1915年に英国の細菌学者フレデリック・ツールトによって最初に発見され、その後1917年にフランス系カナダ人の微生物学者フェリックス・デレル(2)によって再び発見された。それ以来、これらのファージの構造と機能は広く特徴付けられており(3)、これらのファージを2つのクラスに分割する。これらのクラスの最初のクラスは、感染時に宿主細菌内で増殖し、細菌代謝を破壊し、細胞を溶解し、子孫ファージを放出する溶解ファージである(4)。この抗菌活性と抗生物質耐性細菌の有病率の増加の結果として、これらの溶解ファージは最近、抗生物質の代替治療として有用であることが証明された。これらのクラスの2番目は、溶解サイクルを介して宿主内で増殖するか、またはそのゲノムが宿主のゲノムが宿主のゲノムに統合される静止状態に入ることができるリソジェニックファージです(図1)。複数の後の世代(4)で誘発される生産。
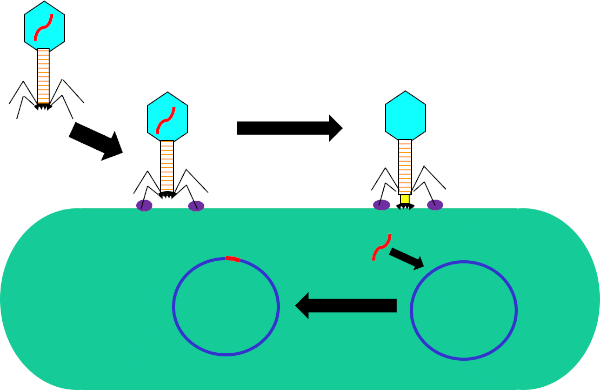
図1:バクテリオファージによる宿主細胞の感染尾線維と受容体(紫色)との相互作用を介して細菌細胞壁へのファージによる吸着。細胞表面に一度、ファージは収縮鞘(黄色)によって細胞壁に移動されるベースプレート(黒)を使用して細菌細胞に不可逆的に付着する。ファージゲノム(赤色)は、細胞に入り、宿主細胞ゲノムに統合する。
細菌の伝達は自然に起こるプロセスですが、現代の技術を使用して、実験室の設定で細菌に遺伝子を移用するために操作されています。ファージなどのリソジェニックファージのゲノムに目的の遺伝子を挿入することで、これらの遺伝子を細菌のゲノムに移し、その結果、これらの細胞内で発現することができます。形質転換などの他の遺伝子導入方法は、遺伝子の伝達および発現にプラスミドを使用するが、ファージゲノムをレシピエント細菌のそれに挿入することは、この細菌に新しい形質を与える可能性を有するだけでなく、自然に起こる突然変異および細胞環境の他の要因は、転移した遺伝子の機能を変化させる。
結合などの水平遺伝子導入の他の方法と比較して、トランスダクションはドナーおよびレシピエント細胞に必要な基準でかなり柔軟です。使用されているファージのゲノム内に収まる可能性のある任意の遺伝的要素は、ドナー細菌の任意の株から、両方がファージに寛容である限り、ドナー細菌の任意の株から、必要なファージ受容体の発現を必要とする。セル サーフェス。この遺伝子がドナーゲノムから移動し、ファージに包装されると、レシピエントに移すことができる。トランスダクションに続いて、目的の遺伝子を含むレシピエント細菌を選択する必要があります。これは、抗生物質耐性をコードする遺伝子の場合に、目的の遺伝子、または遺伝子の本質的な機能をマークするために、FLAGタグやポリヒスチジンタグなどの遺伝子マーカーを使用することによって行うことができます。さらに、PCRを使用して、トランスダクションの成功をさらに確認することができます。対象遺伝子内の領域にプライマーを使用し、シグナルを陽性対照と比較することにより、目的の遺伝子を有する細菌、および負の対照、ファージのない経度反応と同じステップを受けた細菌。細菌の伝達は分子生物学において有用なツールであるが、特に最近の抗生物質耐性の上昇に関して、細菌の進化において重要な役割を果たし続けている。
この実験では、細菌伝達を用いて、抗生物質アンピシリンに対する耐性をコードする遺伝子を、大腸菌のW3110株からP1バクテリオファージ(5)を介してJ53株に伝達した。この実験は、2つの主要なステップで構成されていました。まず、ドナー株からアンピシリン耐性遺伝子を含むP1ファージの調製。第2に、この遺伝子をP1ファージによるトランスダクションによりレシピエント株に転移させる(図1)。いったん実施すると、アンピシリン耐性遺伝子の正常な転写はqPCRによって決定され得る(図2)。経転移が成功した場合、大腸菌のJ53株はアンピシリンに対して耐性であり、この耐性を与える遺伝子はqPCRによって検出可能である。もし失敗した場合、アンピシリン耐性遺伝子の検出はなく、アンピシリンはJ53株に対する有効な抗生物質として機能し続けるであろう。
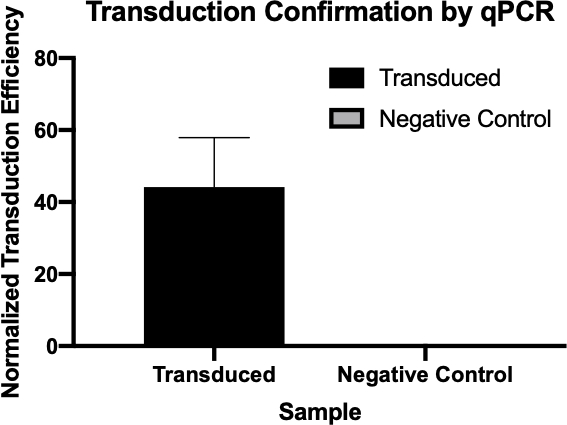
図2:qPCRによるトランスダクションの成功の確認トランスダクション反応と陰性対照反応から対象遺伝子に対して検出されたCq値を比較し、これらの値をハウスキーピング遺伝子に対して正規化することで、細菌の伝達が成功したことを確認することができました。
Procedure
1. セットアップ
- 微生物を含む任意の作業を開始する前に、70%エタノールを使用してワークスペースを殺菌します。常に必要なPPE(ラボコートと手袋)を使用してください。
- 1xアンピシリンを含むLB寒天プレート、市販のP1ファージ溶解液、クロロホルム、1Mクエン酸ナトリウム、グリセロール、および殺菌前のプラスチックピペットチップとセル拡散機の箱が近くにあることを確認してください。
- オートクレーブによって滅菌LBを準備し、LB塩溶液の3つの1 mLアリコートを作るためにそれを使用します。
- mM MgCl2 (952.11-2.3803 μg), 5 mM CaCl2 (11.098 mg), 0.1-0.2% ブドウ糖 (100-200 μg)
- mM MgSO4 (12.0366 mg), 5 mM CaCl2 (11.098 mg)
- mM ク硝酸ナトリウム (25.806 mg)
- 完成したら、70%のエタノールで手袋と同様に、すべての表面を殺菌し、手を洗います。
2. プロトコル
- ドナーファージリサート製剤
- LBでドナーW3110株大腸菌の1mL培養を37°Cで一晩増殖させ、220rpmで振盪を起こす。
- この一晩培養 1:100 を 1 mL の新鮮な LB 1 mL で希釈し、10-25 mM MgCl2、5mM CaCl2、および 0.1-0.2% グルコースを補充した。
- この細菌希釈を37°Cで2時間、通気と220rpmで振ります。
- これらの細胞が早期対数成長期(顕著な成長とわずかな濁り)に達したら、市販のP1ファージの40 μLを追加し、通気と220rpmで振って37°Cで残します。
- ファージ添加の前に、これらの細胞の600nmで測定された光学密度は、約0.4(6)でなければなりません。
- 培養がリズするまで1~3時間細胞を監視します。
- Lysisは、細胞の破片の増加だけでなく、濁度の顕著な減少をもたらす(すなわち、細胞は、培養を通して見ることができたら、細胞がlyslysと見なされます)。
- 塩分にクロロホルムの数滴(50-100 μL)を加え、渦で混合します。
- クロロホルムは、残りのドナー細胞を殺すことによってファージリサートを殺菌し、ファージだけを残し、このリサートのツチエーターを増加させる。
- 遠心分離機は2分間14,000rpmでライサートし、破片を除去し、上清を新鮮なチューブに移します。
- クロロホルムを数滴加え、1日以上4°Cに保存します。
- 伝達
- レシピエントJ53株大腸菌の1mL培養を37°CでLBで一晩成長させ、220rpmで通気および振盪を行う。
- ドナーファージライザート(2.1)の100 μLを1.5mLマイクロフュージチューブに移し、キャップを37°Cで30分間開けてインキュベートします。
- このインキュベーションは、P1溶解液中の残りのクロロホルムをレシピエント細胞に添加する前に蒸発することを可能にする。
- 6,000rpmで5分間遠心分離によるペレットレシピエント株細胞。
- 100 mM MgSO4および 5 mM CaCl2を含む新鮮な LB の 300 μL でこれらの細胞を再中断します。(P1ファージは、感染するカルシウムを必要とします)。
- レシピエント細菌細胞を用いて2つの反応を設定し、ドナーファージライセートを調製:1)100μLレシピエントJ53株大腸菌と100μLドナーファージリサートを組み合わせた経度反応、および2)100μLレシピエントJ53株を組み合わせた陰性対照大腸菌および100 mM MgSO4および5 mM CaCl2を含有するLBの100 μL。
- 37°Cで30分間インキュベートし、220rpmで振ります。
- 1 mL LB および 200 μL 1M クエン酸ナトリウム (pH=5.5) を追加し、220 rpm で振盪を起こす 37 °C で 1 時間インキュベートします。
- クレートは、カルシウムでキレートすることによりP1の感染率を減少させるために使用され、レシピエント細菌のリシスを防止する。
- この溶液のインキュベーションは、アンピシリン耐性マーカーの発現を可能にする。
- ペレット細胞を6,000rpmで5分間遠心分離する。
- 100 mMクエン酸ナトリウム(pH 5.5)でLBの100 μLで細胞ペレットを再ステースします。2つのLB寒天プレート上の両方の反応のための渦およびプレート全体の溶液。
- LBプレートは、変換されたサンプルに対して1xアンペアを持ち、負のコントロールにはアンプを持たない必要があります。
- このプレート上のP1ファージ汚染は、冷凍庫の在庫を準備する前に再ストリークを必要とします。
- ファージを除去しない場合、これらのコロニーから成長した培養物は、カルシウムキレートの存在下でなければ成長しません。
- 両方のプレートから約3-4コロニーを選び、クエン酸ナトリウム100μL(pH=5.5)で広がる2つのLB寒天プレートに再びストリークします。
- LBプレートは、変換されたサンプルに対して1xアンペアを持ち、負のコントロールにはアンプを持たない必要があります。
- 一晩37°Cでプレートをインキュベートし、ファージを含まないコロニーが成長できるようにします。
- 両方のプレートからコロニーを選び、37°CでLBの5 mLで一晩培養し、通気と220rpmで振ります。
- 総培養量の4.5mLを用いてDNAミニプレップによりこれらの培養物からDNAを分離する。
- ヌクレアーゼフリー水の35μLを用いて溶出DNA。
- ナノドロップで得られた濃度を測定します。純DNAは約1.8の吸光度比(A260/280)を生成します。
- 各培養の残りの0.5 mLを使用して、100%グリセロールと細菌培養の1:1混合物を作ることによって1 mLグリセロールストックを調製します。
- 細菌のグリセロールストックを-80 °Cで保存します。
3. データ分析と結果
- qPCRによるトランスダクションの確認
- 6つのqPCR反応に対して2つのqPCRマスターミックスを準備し、3つはアンピシリン耐性遺伝子にqPCRプライマーを使用し、他の3つはハウスキーピング遺伝子にqPCRプライマーを使用します(反応あたり14.5 μL):12.5 μL qPCRバッファミックス+1μLフォワードプライマー+1μLリバースプライマー。
- この実験では、SYBRグリーンマスターミックスを用いて使用した。
- ハウスキーピング遺伝子プライマーは、DNAジャイラゼB(7)をコードする細菌遺伝子内のDNAのセグメントを増幅するように設計された。
- 各qPCR反応について、各反応(10.5 μL)のDNAの100 μgと14.5 μLのqPCRマスターミックスを組み合わせます。
- 表1に記載されているqPCRマシンとサーモサイクリングプロトコルを用いて、6つの反応すべてに対するアンピシリン耐性およびハウスキーピング遺伝子の増幅を測定した。
- qPCRによって生成されたCq値は、アンピシリン耐性遺伝子の正規化されたトランスダクション効率を計算するために使用され(図3)、アンピシリン耐性遺伝子の転写に成功したことを確認した。
- サンプルのCq値(サイクル定量値)は、信号がバックグラウンドしきい値を超えた最も早いPCRサイクル番号です。低い Cq 値は、より多くのターゲット シーケンスに対応しますが、その逆も同様です。
- サンプル内の遺伝子の正規化されたトランスダクション効率は、これらのCq値を使用して、標的遺伝子の値からハウスキーピング遺伝子の値を引くことによって計算することができ、正規化されたトランスダクションの計算に使用できるΔCq値を生成する。効率 2(-ΔCq)
- 6つのqPCR反応に対して2つのqPCRマスターミックスを準備し、3つはアンピシリン耐性遺伝子にqPCRプライマーを使用し、他の3つはハウスキーピング遺伝子にqPCRプライマーを使用します(反応あたり14.5 μL):12.5 μL qPCRバッファミックス+1μLフォワードプライマー+1μLリバースプライマー。
| 温度 | 時間 | |
| 変性 | 94 °C | 2 分 |
| 40サイクル: | ||
| 変性 | 94 °C | 15秒 |
| アニーリング、延長、蛍光読み取り | 最低プライマーTm以下60 °Cまたは5 °C | 1 分 |
表 1: qPCR サーモサイクリング プロトコル
細菌は、遺伝物質を交換することによって、急速に変化する環境に迅速に適応することができ、彼らはそれを行うことができる一つの方法は、トランスダクション、細菌ウイルスによって媒介される遺伝物質の交換です。バクテリオファージは、しばしばファージと略されるが、最初に宿主の表面に付着し、そのDNAを細菌細胞に注入することによって細菌に感染するウイルスの一種である。その後、宿主細胞自身のDNAを分解し、ウイルスゲノムを複製し、細胞の機械を乗っ取ってタンパク質の多くのコピーを合成します。これらのファージタンパク質は、自己組み立て、複数の子孫を形成するためにファージゲノムをパッケージ化します。しかし、DNA包装機構の忠実度が低いため、時折、ファージは細菌DNAの断片をファージカプシドにパッケージ化する。宿主の逆化を誘導した後、ファージ子孫が放出され、そのようなファージが別の宿主細胞に感染すると、前の宿主のDNA断片が伝達される。これは、再結合し、新しい宿主の染色体に永久に組み込まれ、それによって2つの細菌間の遺伝子伝達を媒次化することができる。
実験室でファージトランスダクションを行うには、目的の遺伝子を含むドナー株、それを欠くレシピエント株、両方の菌株に感染しうるファージ、およびトランスメーション細菌を選択する方法が必要です。ほとんどの場合、これは、トランスネクスト細菌の増殖をサポートするが、非トランスネクストされたものの成長を阻害する選択的固体成長培地になります。まず、目的の遺伝子を含むドナー株は、液体成長培地で培養される。すべての細菌が成長のログ段階で活発に分裂している場合、培養は標的ファージに接種される。インキュベーションの3〜4時間後、ほぼすべての細菌がファージ粒子をlyslyseおよび放出した場合、ドナーファージリサートは、レシピエント細菌株の新たに成長した培養物に接種される。1時間の短いインキュベーションの後、培養物は、現在、トランスネクストおよび非トランスネクスト細菌細胞の混合物を含むべきであり、これは適切な選択的固形成長培地に懸濁液の一部を広げることによって、トランスネク下細胞についてスクリーニングされる。さらなるインキュベーションの際に、トランスネクテーションされた細胞は成長し、増加し、目に見えるコロニーを得る必要があります。これらのコロニーは、コロニーPCR、DNAシーケンシング、定量PCRなどの正常な伝達をさらに確認するために、様々な方法を用いてさらなる分析のために選択することができる。
手順を開始する前に、ラボコートや手袋を含む適切な個人用保護具を着用してください。次に、70%のエタノールでワークスペースを殺菌し、表面を拭き取ります。
この後、LB塩溶液の3つの1ミリリットルのアリコートを調製する。さて、5ミリリットルのLB成長培地を5ミリリットルのLB成長培地を含む15ミリリットルの円錐バイアルに100マイクロリットルの大腸菌を500マイクログラムのアンピシリンで添加してドナー株培養を調製する。その後、通気と220 rpmで振って、摂氏37度で一晩培養を成長させます。翌日、70%のエタノールでベンチトップを拭き取ってから、振るインキュベーターから培養を取り除きます。次に、塩溶液を添加した新鮮なLBの990マイクロリットルにドナー株の10マイクロリットルを加えることによって100に一晩培養を希釈する。
細菌希釈を37°Cで2時間、通気と220rpmで振るようにします。細胞が早期ログ相に達したら、インキュベーターから培養物を取り出し、40マイクロリットルのP1ファージを培養に加え、再度インキュベートする。培養がリズされるまで1~3時間細胞を監視し続けます。次に、50~100マイクロリットルのクロロホルムをリサートに加え、渦で混ぜます。その後、ライサートを遠心分離して破片を除去し、上清を新鮮なチューブに移します。上清に数滴のクロロホルムを加え、1日以上摂氏4度に保存します。
経誘導手順を開始するには、レシピエント株の1ミリリットル培養物を得る。次に、100マイクロリットルのドナーファージリザートを1.5ミリリットルのマイクロ遠心分離管に移し、キャップを30分間開けて37°Cでインキュベートし、残りのクロロホルムを蒸発させます。ドナーファージがインキュベートする間、ペレットは穏やかな遠心分離を介して細胞を歪める。上清を廃棄し、硫酸100ミリモルマグネシウムと5ミリモルカルシウム塩化カルシウムを含む新鮮なLBの300マイクロリットルで細胞ペレットを再懸濁します。
次に、レシピエント株の100マイクロリットルとドナーファージリサートの100マイクロリットルをマイクロ遠心管に組み合わせて経誘導反応を設定する。次いで、レシピエント株の100マイクロリットルとLBの100マイクロリットルを硫酸マグネシウム及び塩化カルシウムと組み合わせて負の対照を設定する。インキュベーション後、クチン酸1モルナトリウム1ミリリットルとLB1ミリリットルを両管に加え、上下にゆっくりと配管して混ぜます。その後、チューブを1時間インキュベートした後、遠心分離を介して細胞を穏やかにペレットする。
遠心分離後、上清を廃棄し、100ミリモルクチン酸で100マイクロリットルのLBでペレット化した細胞を再懸濁する。溶液を渦にし、1Xアンピシリンを含むLB寒天プレートにトランスメ下サンプル全体をピペットします。最後に、アンピシリンなしでLB寒天プレートに負の対照細胞混合物の全容積をピペットする。37°Cで一晩プレートをインキュベートした後、無菌ピペットチップを使用して、トランスダクションプレートから3~4個のコロニーを選び、1Xアンピシリンと1モルクチン酸100マイクロリットルを含む新しいLB寒天プレートにストリークします。1つのモルクチン酸の100マイクロリットルのみを含む別のLB寒天プレート上の負の対照のためのこのめっき方法を繰り返します。その後、一晩37°Cでプレートをインキュベートし、ファージのないコロニーが成長できるようにします。
翌日、70%のエタノールでベンチトップを拭き取ってから、インキュベーターからプレートを取り出します。滅菌ピペットチップを使用して、トランスダクションプレートから3つのコロニーを選び、それぞれ5ミリリットルのLB培地を含む別々のチューブに追加します。次に、負の制御プレートから3つのコロニーを選択し、LB培地の5ミリリットルを含む別のチューブに追加します。220 rpmで通気と揺れで摂氏37度で一晩培養を成長させます。前に示したようにベンチトップを殺菌した後、DNAミニプレップキットを使用して、製造元の指示に従って各培養物の4.5ミリリットルからDNAを分離します。次いで、35マイクロリットルのヌクレアーゼフリー水でDNAを溶出し、ラボ分光光度計で得られた濃度を測定します。最後に、100%グリセロールの0.5ミリリットルに両方の細菌溶液の残りの0.5ミリリットルを追加することにより、グリセロールストックを調製します。
経電を確認するには、まず24個のqPCR反応に対して2つのqPCRマスターミックスを調製します。最初のマスターミックスでは、150マイクロリットルのqPCRバッファーミックスをマイクロ遠心管に加え、アンピシリン耐性遺伝子を増幅するように設計されたフォワードとリバースプライマーのそれぞれに12マイクロリットルを加えます。次に、マイクロ遠心管に150マイクロリットルのqPCRマスターミックスを加え、ハウスキーピング遺伝子を増幅するように設計されたフォワードプライマーとリバースプライマーのそれぞれ12マイクロリットルを追加して、第2のqPCRマスターミックスを準備します。
各qPCR反応について、各反応から100マイクログラムの実験DNAを14.5マイクロリットルのqPCRマスターミックスと組み合わせます。さて、前に示したように残りの反応を準備します。94°Cに予熱されたサーモサイクラーに反応を転送し、プログラムを開始します。最後に、qPCRによって生成されたサイクル定量(Cq)値を使用して、アンピシリン耐性遺伝子の正規化されたトランスダクション効率を計算します。
対象遺伝子のサイクル定量(Cq)値は、各陰性対照およびトランスネクテッドサンプルについて集計された。低いCq値は、通常29サイクル未満で、この例のトランス下サンプルのように、ターゲット配列の高量を示す。
ハウスキーピング遺伝子は、ここでも集計され、各反応におけるDNAの量を正規化するためのローディングコントロールとして、またqPCRが働いていることを確実にするための陽性対照として使用される。同じ量のハウスキーピング遺伝子がロードされている場合、各サンプルにおいて比較的同じ速度で見出される。
次に、各サンプルのデルタCq値を算出し、対応する標的遺伝子のCq値から各サンプルのハウスキーピング遺伝子のCq値を減算する。たとえば、最初の負のコントロールのデルタ Cq は 13.54 です。次に、この値を使用して、ここに示す式を使用して、各サンプルの正規化されたトランスダクション効率を計算します。最後に、各サンプル群の平均正規化トランスダクション効率を算出することができる。
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
バクテリオファージによる細菌との間の遺伝子の移動は、自然なプロセスながら、多くの研究目的のために非常に有用であることが証明されています。形質転換や結合などの遺伝子導入の他の方法は可能ですが、トランスダクションはバクテリオファージを一意に使用します。宿主ゲノムへの遺伝子統合を可能にするだけでなく、他の方法の影響を受けにくい複数の細菌への遺伝子送達も可能にする。このプロセスは、実験室で特に有用である一方で、最近出現している遺伝子治療分野においても使用されており、より具体的には代替遺伝子治療においては、細菌を利用して標的組織に治療薬を提供する治療戦略、その多くは他の送達方法の影響を受けにくく、多くの臨床的関連性を有する(8,9)。
Subscription Required. Please recommend JoVE to your librarian.
References
- Lederberg J, Lederberg E.M., Zinder, N.D., et al. Recombination analysis of bacterial heredity. Cold Spring Harbor symposia Quantitative Biol. 1951;16:413-43.
- Duckworth DH. "Who Discovered Bacteriophage?". Bacteriology Reviews. 1976;40:793-802.
- Yap ML, Rossman, M.G. Structure and Function of Bacteriophage T4. Future Microbiol. 2014;9:1319-27.
- Sulakvelidze A, Alavidze, Z., Morris, J. G. Bacteriophage Therapy Antimicrobial Agents and Chemotherapy 2001;45(3):649-59.
- Moore S. Sauer:P1vir phage transduction 2010 [Available from: https://openwetware.org/wiki/Sauer:P1vir_phage_transduction].
- Kobayashi A, et al. Growth Phase-Dependent Expression of Drug Exporters in
- Escherichia coli and Its Contribution to Drug Tolerance. Journal of Bacteriology. 2006;188(16):5693-703.
- Rocha D, Santos, CS, Pacheco LG. Bacterial reference genes for gene expression studies by RT-qPCR: survey and analysis. Antonie Van Leeuwenhoek. 2015;108:685-93.
- Pálffy R. et al. Bacteria in gene therapy: bactofection versus alternative gene therapy. Gene Ther. 2006 13:101-5.
- O'Neill JM, et al. Intestinal delivery of non-viral gene therapeutics: physiological barriers and preclinical models. Drug Discovery Today. 2011;16:203-2018.