



Overview
Fuente: Alexander S. Gold1, Tonya M. Colpitts1
1 Departamento de Microbiología, Escuela de Medicina de la Universidad de Boston, Laboratorios Nacionales de Enfermedades de Infecciones Emergentes, Boston, MA
La transducción es una forma de intercambio genético entre bacterias que utiliza bacteriófagos, o fagos, una clase de virus que infecta exclusivamente organismos procaarios. Esta forma de transferencia de ADN, de una bacteria a otra a través de un fago, fue descubierta en 1951 por Norton Zinder y Joshua Ledererg, quienes denominaron el proceso "transducción" (1). Los bacteriófagos fueron descubiertos por primera vez en 1915 por el bacteriólogo británico Frederick Twort, luego descubiertos de nuevo de forma independiente en 1917 por el microbiólogo franco-canadiense Felix d'Herelle (2). Desde entonces, la estructura y función de estos fagos se han caracterizado ampliamente (3), dividiendo estos fagos en dos clases. La primera de estas clases son los fagos líticos que tras la infección se multiplican dentro de la bacteria huésped, alterando el metabolismo bacteriano, lysing la célula, y liberando fago progenie (4). Como resultado de esta actividad antibacteriana y la creciente prevalencia de bacterias resistentes a los antibióticos, estos fagos líticos han demostrado ser útiles recientemente como tratamiento sustituto de los antibióticos. La segunda de estas clases son los fagos lisogénicos que pueden multiplicarse dentro del huésped a través del ciclo lítico o entrar en un estado de reposo en el que su genoma se integra en el del huésped (Figura 1), un proceso conocido como lisógeno, con la capacidad de fago producción que se inducirá en varias generaciones posteriores (4).
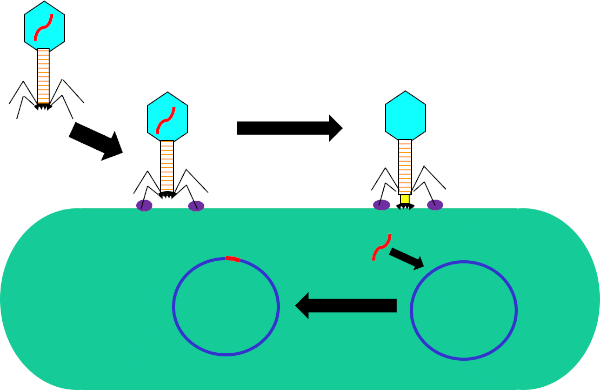
Figura 1: Infección de la célula huésped por bacteriófago. Adsorción por el fago a la pared celular bacteriana a través de interacciones entre las fibras de cola y el receptor (púrpura). Una vez en la superficie celular, el fago se une irreversiblemente a la célula bacteriana utilizando la placa base (negro) que se mueve a la pared celular por la vaina contráctea (amarillo). El genoma del fago (rojo) entra en la célula y se integra en el genoma de la célula huésped.
Mientras que la transducción bacteriana es un proceso natural, utilizando tecnología moderna ha sido manipulada para la transferencia de genes a bacterias en el entorno de laboratorio. Al insertar genes de interés en el genoma de un fago lisogénico, como el fago, uno es capaz de transferir estos genes a los genomas de las bacterias y, en consecuencia, expresarlos dentro de estas células. Mientras que otros métodos de transferencia de genes, como la transformación, utilizan un plásmido para la transferencia y expresión de genes, la inserción del genoma del fago en el de la bacteria receptora no sólo tiene el potencial de conferir nuevos rasgos a esta bacteria, sino que también permite mutaciones naturales y otros factores del entorno celular para alterar la función del gen transferido.
En comparación con otros métodos de transferencia de genes horizontales, como la conjugación, la transducción es bastante flexible en los criterios requeridos para las células donantes y receptoras. Cualquier elemento genético que pueda caber dentro del genoma del fago que se está utilizando puede transferirse de cualquier cepa de bacterias donantes a cualquier cepa de bacterias receptoras, siempre y cuando ambas sean permisivas para el fago, requiriendo la expresión de los receptores de fago necesarios en el superficies celulares. Una vez que este gen se mueve fuera del genoma del donante y se empaqueta en el fago, se puede transferir al receptor. Después de la transducción, es necesario seleccionar para las bacterias receptoras que contienen el gen de interés que deben seleccionarse. Esto podría hacerse mediante el uso de un marcador genético, como una etiqueta FLAG o una etiqueta de polihistidina, para marcar el gen de interés, o la función intrínseca del gen, en el caso de genes que codifican para la resistencia a los antibióticos. Además, la PCR podría utilizarse para confirmar aún más la transducción correcta. Mediante el uso de imprimaciones para una región dentro del gen de interés y la comparación de la señal con un control positivo, bacterias que tiene el gen de interés, y un control negativo, bacterias que se sometieron a los mismos pasos que la reacción de transducción sin fago. Si bien la transducción bacteriana es una herramienta útil en biología molecular, ha desarrollado y sigue desempeñando un papel importante en la evolución de las bacterias, particularmente en lo que respecta al reciente aumento de la resistencia a los antibióticos.
En este experimento, la transducción bacteriana se utilizó para transferir la codificación genética para la resistencia a la ampicilina antibiótica de la cepa W3110 de E.coli a la cepa J53 a través del bacteriófago P1 (5). Este experimento consistió en dos pasos principales. En primer lugar, la preparación de fago P1 que contiene el gen de resistencia a la ampicilina de la cepa del donante. En segundo lugar, la transferencia de este gen a la cepa receptora por transducción con el fago P1 (Figura 1). Una vez realizada, la transferencia exitosa del gen de resistencia a la ampicilina podría determinarse por qPCR (Figura 2). Si la transducción fuera exitosa, la cepa J53 de E. coli sería resistente a la ampicilina, y el gen que confiere esta resistencia es detectable por qPCR. De no tener éxito, no habría detección del gen de resistencia a la ampicilina y la ampicilina seguiría funcionando como un antibiótico eficaz contra la cepa J53.
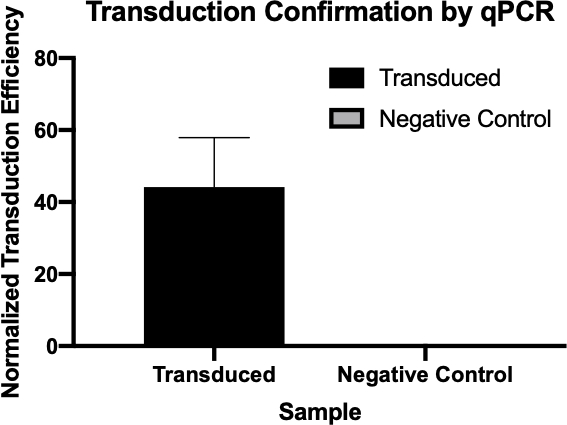
Figura 2: La confirmación de la transducción exitosa por qPCR. Al comparar los valores Cq detectados para el gen de interés de la reacción de transducción y la reacción de control negativa, y normalizar estos valores contra un gen de limpieza, se pudo confirmar que la transducción bacteriana fue exitosa.
Procedure
1. Configuración
- Antes de empezar cualquier trabajo que involucre microbios, esterilice el espacio de trabajo con 70% de etanol. Utilice siempre el EPP necesario (abrigo de laboratorio y guantes).
- Asegúrese de que las placas de agar LB con 1x ampicilina, solución de lysato de fago P1 disponible en el uso comercial, cloroformo, citrato de sodio 1 M, glicerol y una caja de puntas de pipeta de plástico preesterilizadas y esparcidores celulares están cerca.
- Preparar el LB estéril mediante autoclave y utilizarlo para hacer tres alícuotas de 1 ml de solución de sal LB.
- mM MgCl2 (952.11-2.3803 g), 5 mM CaCl2 (11.098 mg), 0.1-0.2% glucosa (100-200 g)
- mM MgSO4 (12.0366 mg), 5 mM CaCl2 (11.098 mg)
- citrato de sodio mM (25.806 mg)
- Una vez terminado, esterilizar todas las superficies, así como los guantes con 70% de etanol y lavarse las manos.
2. Protocolo
- Preparación del donante Phage Lysate
- Preparar un cultivo de 1 ml de cepa W3110 del donante E. coli en LB con 1x ampicilina cultivada durante la noche a 37oC con aireación y agitación a 220 rpm.
- Diluir este cultivo nocturno 1:100 en 1 mL de LB fresco complementado con 10-25 mM MgCl2, 5 mM CaCl2y 0.1-0.2% glucosa.
- Cultivar esta dilución bacteriana a 37oC durante 2 horas con aireación y agitación a 220 rpm.
- Una vez que estas células han alcanzado la fase de crecimiento logarítmico temprano (crecimiento notable y ligera turbidez), añadir 40 l de fago P1 disponible comercialmente y dejar a 37 oC con aireación y temblor a 220 rpm.
- Antes de la adición de fago, la densidad óptica medida a 600 nm de estas células debe ser aproximadamente 0,4 (6).
- Supervise las células durante 1-3 horas hasta que el cultivo se haya lisado.
- La lisis resultará en un aumento de los desechos celulares, así como una disminución notable en la turbidez (es decir, las células se considerarán lysed una vez que uno es capaz de ver a través del cultivo).
- Añadir varias gotas (50-100 l) de cloroformo al lisado y mezclar por vórtice.
- El cloroformo esteriliza el lisato del fago matando las células restantes del donante, dejando sólo fago y aumentando el titer de este lisato.
- El lisado de centrífuga a 14.000 rpm durante 2 minutos para eliminar los escombros y transferir el sobrenadante a un tubo nuevo.
- Añadir unas gotas de cloroformo y almacenar a 4 oC durante no más de un día.
- Transducción
- Preparar un cultivo de 1 ml de cepa Receptora J53 E. coli cultivada durante la noche en LB a 37oC con aireación y agitación a 220 rpm.
- Transfiera 100 ml de lisato de fago donante (2.1) a un tubo de microfúgelo de 1,5 ml e incubar con tapa abierta a 37 oC durante 30 minutos.
- Esta incubación permite que cualquier cloroformo restante en la solución de lisato P1 se evapore antes de ser añadido a las células receptoras.
- Células de deformación de cepa receptoras de pellets suavemente por centrifugación a 6.000 rpm durante 5 minutos.
- Resuspenda estas células en 300 ml de LB fresco que contenga 100 mM MgSO4 y 5 mM CaCl2. (P1 fago requiere que el calcio sea infeccioso).
- Configurar dos reacciones utilizando las células bacterianas receptoras y el lisato de fago de donante preparado: 1) reacción de transducción combinando cepa J53 receptor a 100 l E53 y lisato de fago donante de 100 ol, y 2) control negativo que combina cepa J53 receptor de 100 l E. coli y 100 l de LB que contienen 100 mM MgSO4 y 5 mM CaCl2.
- Incubar a 37oC durante 30 minutos con agitación a 220 rpm.
- Añadir 1 ml de LB y citrato de sodio de 200 ml y 1 M (pH-5,5) e incubar durante 1 hora a 37 oC con agitación a 220 rpm.
- El citrato se utiliza para reducir la infectividad de P1 quelándose con calcio, evitando la lisis de las bacterias receptoras.
- La incubación de esta solución permite la expresión del marcador de resistencia a la ampicilina.
- Células de pelet por centrifugación a 6.000 rpm durante 5 minutos.
- Resuspender los gránulos celulares en 100 ml de LB con citrato de sodio de 100 mM (pH 5.5). Vórtice y solución entera de placas para ambas reacciones en dos placas de agar LB.
- La placa LB debe tener 1 x amperio para la muestra transducida y ningún amplificador para el control negativo.
- La contaminación por fago P1 en esta placa requiere volver a rayar antes de que se puedan preparar las existencias del congelador.
- Si no se elimina el fago, los cultivos cultivados de estas colonias no crecerán a menos que en presencia de un quelante de calcio.
- Escoja alrededor de 3-4 colonias de ambas placas y vuelva a rayar en dos placas de agar LB esparcidas con 100 ml de citrato de sodio de 1 M (pH-5,5).
- La placa LB debe tener 1 x amperio para la muestra transducida y ningún amplificador para el control negativo.
- Incubar las placas a 37oC durante la noche para permitir que crezcan colonias libres de fagos.
- Recoger las colonias de ambas placas y utilizarlas para cultivar cultivos nocturnos en 5 mL de LB a 37 oC con aireación y agitación a 220 rpm.
- Aísle el ADN de estos cultivos mediante el miniajuste de ADN utilizando 4,5 ml del volumen total de cultivo.
- Eluda el ADN con 35 ml de agua libre de nucleasas.
- Mida la concentración resultante por Nanodrop. El ADN puro generará una relación de absorbancia (A260/280)de aproximadamente 1,8.
- Utilice los 0,5 ml restantes de cada cultivo para preparar 1 ml de reservas de glicerol haciendo una mezcla de 1:1 de 100% glicerol y cultivo bacteriano.
- Conservar las existencias de glicerol bacteriano a -80 oC.
3. Análisis de datos y resultados
- Confirmación de transducción por qPCR
- Preparar dos mezclas maestras qPCR para seis reacciones qPCR, tres usando imprimaciones qPCR para el gen de resistencia a la ampicilina, y las otras tres usando imprimaciones qPCR para un gen de limpieza (14,5 l por reacción): mezcla tampón qPCR de 12,5 l + imprimación directa de 1 L + imprimación inversa de 1 L.
- En este experimento, usamos SYBR Green master mix.
- Las imprimaciones genéticas de limpieza se diseñaron para amplificar un segmento de ADN dentro de la codificación del gen bacteriano para la gengensa b (7) del ADN.
- Para cada reacción qPCR, combine 100 g de ADN de cada reacción (10,5 l) con 14,5 ml de mezcla maestra qPCR.
- Utilizando una máquina qPCR y el protocolo de termociclismo enumerado en la Tabla 1, la amplificación se midió para los genes de resistencia a la ampicilina y limpieza para las seis reacciones.
- Los valores Cq generados por qPCR se utilizaron para calcular la eficiencia normalizada de transducción del gen de resistencia a la ampicilina (Figura 3), confirmando la transducción exitosa del gen de resistencia a la ampicilina.
- El valor Cq, o valor de cuantificación de ciclo, de una muestra es el número de ciclo PCR más antiguo en el que se detecta una señal excedida el umbral de fondo. Los valores Bajos de Cq corresponden a más secuencia de destino, viceversa.
- La eficiencia normalizada de transducción de un gen dentro de una muestra se puede calcular utilizando estos valores Cq restando el valor del gen de limpieza del gen de destino, generando un valor de "Cq", que puede utilizarse para calcular la transducción normalizada eficiencia en 2(-Cq).
- Preparar dos mezclas maestras qPCR para seis reacciones qPCR, tres usando imprimaciones qPCR para el gen de resistencia a la ampicilina, y las otras tres usando imprimaciones qPCR para un gen de limpieza (14,5 l por reacción): mezcla tampón qPCR de 12,5 l + imprimación directa de 1 L + imprimación inversa de 1 L.
| Temperatura | hora | |
| Desnaturalización | 94 oC | 2 min |
| 40 ciclos: | ||
| Desnaturalización | 94 oC | 15 seg |
| Lectura de recocido, extensión y fluorescencia | 60 oC o 5 oC por debajo de la imprimación más baja Tm | 1 min |
Tabla 1: protocolo de termociclismo qPCR
Las bacterias pueden adaptarse rápidamente a un entorno que cambia rápidamente mediante el intercambio de material genético y una manera de hacerlo es a través de la transducción, el intercambio de material genético mediado por virus bacterianos. Un bacteriófago, a menudo abreviado como fago, es un tipo de virus que infecta las bacterias al adherirse primero a la superficie del huésped y luego inyectar su ADN en la célula bacteriana. A continuación, degrada el propio ADN de la célula huésped y replica su genoma viral, mientras secuestra la maquinaria de la célula para sintetizar muchas copias de sus proteínas. Estas proteínas de fago se autoensamblan y empaquetan los genomas del fago para formar progenie múltiple. Sin embargo, debido a la baja fidelidad del mecanismo de empaquetado de ADN, ocasionalmente, el fago empaqueta fragmentos de ADN bacteriano en la cápside del fago. Después de inducir la lisis del huésped, la progenie del fago se libera y, una vez que un fago infecta otra célula huésped, transfiere el fragmento de ADN de su huésped anterior. Esto puede recombinarse y incorporarse permanentemente al cromosoma del nuevo huésped, mediando así la transferencia de genes entre las dos bacterias.
Para llevar a cabo la transducción de fagos en el laboratorio se requiere una cepa de donante que contenga un gen de interés, una cepa receptora que carece de ella, un fago que pueda infectar ambas cepas y un método para seleccionar las bacterias transducidas. En la mayoría de los casos, este será un medio de crecimiento sólido selectivo que apoya el crecimiento de bacterias transducidas pero inhibe el crecimiento de las no transducidas. Para empezar, la cepa del donante que contiene el gen del interés se cultiva en un medio de crecimiento líquido. Cuando todas las bacterias se dividen activamente en la fase de registro de su crecimiento, el cultivo se inocula con el fago objetivo. Después de tres a cuatro horas de incubación, cuando casi todas las bacterias han liado y liberado las partículas de fago, el lisato de fago donante se inocula en un cultivo recién cultivado de la cepa bacteriana receptora. Después de una breve incubación de una hora, el cultivo ahora debe contener una mezcla de células bacterianas transducidas y no transducidas y esto se examinan para las células transducidas mediante la propagación de una fracción de la suspensión en un medio de crecimiento sólido selectivo apropiado. Tras una mayor incubación, las células transducidas deben crecer y multiplicarse para producir colonias visibles. Estas colonias se pueden seleccionar para un análisis posterior utilizando una variedad de métodos para confirmar aún más la transducción exitosa, como PCR de colonias, secuenciación de ADN o PCR cuantitativa.
Antes de iniciar el procedimiento, ponte cualquier equipo de protección personal adecuado, incluyendo una capa de laboratorio y guantes. A continuación, esterilizar el espacio de trabajo con 70% de etanol y limpiar la superficie.
Después de esto, prepare tres alícuotas de un mililitro de solución de sal de LB. Ahora, preparar un cultivo de cepa de donante mediante la adición de 100 microlitros de E. coli a un vial cónico de 15 mililitros que contiene cinco mililitros de medio de crecimiento LB con 500 microgramos de ampicilina. Luego, cultivar el cultivo durante la noche a 37 grados centígrados con aireación y temblor a 220 rpm. Al día siguiente, limpie la parte superior del banco con 70% de etanol antes de retirar el cultivo de la incubadora de temblores. A continuación, diluir el cultivo nocturno de uno a 100 mediante la adición de 10 microlitros de cepa de donante a 990 microlitros de LB fresco complementado con solución de sal.
Permita que la dilución bacteriana crezca a 37 grados centígrados durante dos horas con aireación y agitación a 220 rpm. Una vez que las células han alcanzado la fase de registro temprano, eliminar el cultivo de la incubadora, añadir 40 microlitros de fago P1 al cultivo e incubar de nuevo. Continúe monitoreando las células durante una o tres horas hasta que el cultivo se haya lisado. A continuación, añadir 50 a 100 microlitros de cloroformo al lisado y mezclar por vórtice. Luego, centrifugar el liso para eliminar los escombros y transferir el sobrenadante a un tubo nuevo. Añadir unas gotas de cloroformo al sobrenadante y almacenarlo a cuatro grados Celsius por no más de un día.
Para comenzar el procedimiento de transducción, obtenga un cultivo de un mililitro de cepa receptora. A continuación, transferir 100 microlitros de lisato de fago donante en un tubo de microcentrífuga de 1,5 mililitros e incubarlo a 37 grados centígrados con la tapa abierta durante 30 minutos para permitir que cualquier cloroformo restante se evapore. Mientras que el fago donante lisato incuba, pellet las células de cepa receptoras a través de una centrifugación suave. Deseche el sobrenadante y resuspenda el pellet celular en 300 microlitros de LB fresco que contenga 100 milimolares de sulfato de magnesio y cinco milimolares de cloruro de calcio.
A continuación, configure la reacción de transducción combinando 100 microlitros de la cepa receptora y 100 microlitros del lisato de fago donante en un tubo de microcentrífuga. A continuación, configurar el control negativo mediante la combinación de 100 microlitros de la cepa receptora y 100 microlitros de la LB con sulfato de magnesio y cloruro de calcio. Después de la incubación, agregue 200 microlitros de un citrato de sodio molar y un mililitro de LB a ambos tubos, y mezcle pipeteando suavemente hacia arriba y hacia abajo. Luego, después de que los tubos hayan sido incubados durante una hora, pelete suavemente las células a través de la centrifugación.
Después de la centrifugación, deseche el sobrenadante y resuspenda las células peletadas en 100 microlitros de LB con citrato de sodio de 100 milimolares. Vortex las soluciones y pipetear toda la muestra transducida en una placa de agar LB con 1X ampicilina. Por último, pipetee todo el volumen de la mezcla de células de control negativas en una placa de agar LB sin ampicilina. Después de incubar las placas durante la noche a 37 grados centígrados, utilice una punta de pipeta estéril para recoger de tres a cuatro colonias de la placa de transducción y fíjelas en una nueva placa de agar LB que contenga 1X ampicilina y 100 microlitros de un citrato de sodio molar. Repita este método de chapado para el control negativo en otra placa de agar LB que contenga sólo 100 microlitros de un citrato de sodio molar. Luego, incubar las placas a 37 grados centígrados durante la noche para permitir que las colonias libres de fago crezcan.
Al día siguiente, limpie la parte superior del banco con 70% de etanol antes de retirar las placas de la incubadora. Usando una punta de pipeta estéril, escoja tres colonias de la placa de transducción y agréguelas cada una a un tubo separado que contenga cinco mililitros de medios LB. A continuación, seleccione tres colonias de la placa de control negativa y agréguelas a otro tubo que contenga cinco mililitros de medios LB. Cultivar los cultivos durante la noche a 37 grados centígrados con aireación y agitación a 220 rpm. Después de esterilizar la mesa de trabajo como se demostró anteriormente, utilice un kit de minipresión de ADN para aislar el ADN de 4,5 mililitros de cada cultivo de acuerdo con las instrucciones del fabricante. Luego, eluye el ADN con 35 microlitros de agua libre de nucleasas y mida la concentración resultante por espectrofotómetro de laboratorio. Finalmente, preparar las existencias de glicerol añadiendo los 0,5 mililitros restantes de ambas soluciones bacterianas a 0,5 mililitros de 100% de glicerol.
Para confirmar la transducción, primero prepare dos mezclas maestras qPCR para 24 reacciones qPCR. Para la primera mezcla maestra, agregue 150 microlitros de mezcla tampón qPCR a un tubo de microcentrífuga y 12 microlitros cada uno de una imprimación delantera y inversa diseñada para amplificar el gen de resistencia a la ampicilina. A continuación, prepare una segunda mezcla maestra qPCR añadiendo 150 microlitros de mezcla maestra qPCR a un tubo de microcentrífuga y luego agregando 12 microlitros cada uno de una imprimación hacia adelante y una imprimación inversa diseñada para amplificar un gen de limpieza.
Para cada reacción qPCR, combine 100 microgramos de ADN experimental de cada reacción con 14,5 microlitros de mezcla maestra qPCR. Ahora, prepare las reacciones restantes como se demostró anteriormente. Transfiera las reacciones a un termociclador precalentado a 94 grados Celsius y luego inicie el programa. Por último, utilice los valores de cuantificación del ciclo, o Cq, generados por qPCR para calcular la eficiencia de transducción normalizada del gen de resistencia a la ampicilina.
Los valores de cuantificación de ciclo, o Cq, para los genes de interés se tabularon para cada uno de los controles negativos y muestras transducidas. Los valores Bajos de Cq, normalmente por debajo de 29 ciclos, como las muestras transducidas en este ejemplo, indican cantidades altas de la secuencia de destino.
Un gen de limpieza, también tabulado aquí, se utiliza como un control de carga para normalizar la cantidad de ADN en cada reacción y como un control positivo para asegurar que el qPCR está funcionando. Siempre que se carguen las mismas cantidades del gen de limpieza, se encuentra a una velocidad relativamente igual en cada muestra.
A continuación, para calcular el valor delta Cq para cada muestra, reste el valor Cq del gen de limpieza para cada muestra del valor Cq de su gen de destino correspondiente. Por ejemplo, el delta Cq del primer control negativo es 13.54. A continuación, utilice este valor para calcular la eficiencia de transducción normalizada de cada muestra utilizando la fórmula que se muestra aquí. Por último, se puede calcular la eficiencia media de transducción normalizada para cada grupo de muestra.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
La transferencia de genes hacia y desde bacterias por bacteriófago, mientras que un proceso natural, ha demostrado ser extremadamente útil para una multitud de propósitos de investigación. Mientras que otros métodos de transferencia de genes como la transformación y la conjugación son posibles, la transducción utiliza de manera única los bacteriófagos; no sólo permite la integración génica en el genoma del huésped, sino también la entrega de genes a múltiples bacterias que no son susceptibles a otros métodos. Este proceso, aunque especialmente útil en el laboratorio, también se ha utilizado en el campo recientemente emergente de la terapia génica, más específicamente en la terapia génica alternativa, una estrategia terapéutica que utiliza bacterias para entregar terapias a los tejidos diana, muchos de los cuales no son susceptibles a otros métodos de administración y tienen mucha relevancia clínica (8,9).
Subscription Required. Please recommend JoVE to your librarian.
References
- Lederberg J, Lederberg E.M., Zinder, N.D., et al. Recombination analysis of bacterial heredity. Cold Spring Harbor symposia Quantitative Biol. 1951;16:413-43.
- Duckworth DH. "Who Discovered Bacteriophage?". Bacteriology Reviews. 1976;40:793-802.
- Yap ML, Rossman, M.G. Structure and Function of Bacteriophage T4. Future Microbiol. 2014;9:1319-27.
- Sulakvelidze A, Alavidze, Z., Morris, J. G. Bacteriophage Therapy Antimicrobial Agents and Chemotherapy 2001;45(3):649-59.
- Moore S. Sauer:P1vir phage transduction 2010 [Available from: https://openwetware.org/wiki/Sauer:P1vir_phage_transduction].
- Kobayashi A, et al. Growth Phase-Dependent Expression of Drug Exporters in
- Escherichia coli and Its Contribution to Drug Tolerance. Journal of Bacteriology. 2006;188(16):5693-703.
- Rocha D, Santos, CS, Pacheco LG. Bacterial reference genes for gene expression studies by RT-qPCR: survey and analysis. Antonie Van Leeuwenhoek. 2015;108:685-93.
- Pálffy R. et al. Bacteria in gene therapy: bactofection versus alternative gene therapy. Gene Ther. 2006 13:101-5.
- O'Neill JM, et al. Intestinal delivery of non-viral gene therapeutics: physiological barriers and preclinical models. Drug Discovery Today. 2011;16:203-2018.