

Summary
タイプBゼラチンベースの設計nanovectorsシステム(GENS)が膵臓癌の治療における全身遺伝子デリバリーとトランスフェクションのために開発されました。上皮成長因子受容体(EGFR)nanparticlesの表面上の特定のペプチドによる修飾により、彼らは、EGFR受容体をターゲットに可能性があり、そのような高い細胞内のグルタチオン濃度として、環境を還元条件下でプラスミド離します。
Abstract
32,000人以上の患者が年間米国で膵臓癌と診断され、この病気は非常に高い死亡率1に関連付けられています。緊急の必要性は、膵臓癌患者の悲惨な生存統計で改善できる新たな臨床的に翻訳可能な治療戦略を開発するために存在します。癌の遺伝子治療は、途方もない約束を示しているものの、主要な課題は、持続的な導入遺伝子の発現につながることができる安全で効果的なデリバリーシステムの開発中です。
ゼラチンは、広く食品や医薬品に使用される最も汎用性の高い天然の生体高分子の一つである。我々の研究室からのこれまでの研究では、タイプBゼラチンは物理的な細胞内送達時にプラスミドと改良されたトランスフェクション効率のスーパーコイル構造を維持したDNAを、カプセル化することができることが示されている。ゼラチンのチオール化により、スルフヒドリル基は、ポリマーとのとFに導入することができます全体の複雑度ジスルフィド結合を安定化ナノ粒子、内ORMのジスルフィド結合は、細胞質ゾルのグルタチオンの存在のために壊れている、ペイロードは2-5を放出される。ポリ(エチレングリコール)(PEG)修飾GENS、全身循環に投与、長期循環時間を提供し、優先的に強化された透過性と保持効果6により新生血管の超透過性に起因する腫瘍塊の対象とする。研究は7 PANC - 1ヒト膵臓腺癌細胞の上皮成長因子受容体(EGFR)の発現を介して示している。積極的に膵臓癌細胞株を標的とするためには、EGFR特異的ペプチドは、PEGのスペーサーを介して粒子表面上に結合させた8。
ほとんどの抗腫瘍遺伝子治療は、9セルのアポトーシス機能を復元するために、そのような野生型p53の腫瘍抑制遺伝子、(WT - P53)の管理に焦点を当てている10を制御する細胞増殖、の重要なシグナル伝達経路としてのp53機構機能。膵臓癌では、ほとんどの細胞がアポトーシス活性の損失を引き起こし、p53タンパク質に変異を持っている。 WT - p53の導入により、アポトーシスが修復される可能性があり、さらに癌細胞11に細胞死を誘発する。
上記の理論的根拠に基づいて、我々は、EGFRがWT - p53遺伝子のデリバリーとPANC - 1細胞で評価した配信の効率化とトランスフェクションのためにペプチドで修飾されたチオール化ゼラチンナノ粒子をターゲットに設計されている。
Protocol
1。プラスミドDNAをカプセル化されたEGFR標的ゼラチンナノ粒子の調製
- チオール化ゼラチンの合成
- チオール化ゼラチンは、タイプBゼラチンの第一級アミノ基の2 -イミノチオランと共有結合によって、従来の方法2-5のように合成した。ゼラチン1グラムを100mlの脱イオン水に溶解し、15時間室温で20 mgの2 - イミノチオラン塩酸塩とインキュベートした。
- 未反応の試薬は3時間ごとに、1mMのHCl溶液で5 mMの塩酸溶液、透析によって除去した。精製チオール化ゼラチンは、4℃でさらに使用するために乾燥し、保存されている凍結された。
- DNA含有ナノ粒子の調製
- 200mgをチオール化ゼラチンは水と溶液のpHで溶解し、0.2 M NaOH溶液の添加によって7に調節した。 1mgのDNAは、ゼラチン溶液を加え穏やかに混合した。
- しながら冷却したエタノールを混合物に徐々に添加した高速で攪拌しながらソリューション。溶媒組成が75%を水力発電 - アルコール溶液に変化したときナノ粒子が形成された。
- ナノ粒子は、さらに0.1ミリリットルの8%(v / v)のグリオキサール溶液をゆっくりと添加することによって架橋された。未反応試薬を0.5ml、0.2Mグリシン溶液でクエンチした。
- ナノ粒子は、30分、16,000 rpmで超遠心した。ペレット回脱イオン水で洗浄し、ナノ粒子を精製した凍結乾燥し、4℃で保存した。
- ナノ粒子の表面改質
- ナノ粒子を10 mg / mlの濃度で0.1 Mリン酸緩衝液(pH7.4)に懸濁させ、メトキシ- PEG -スクシンイミジルカルボキシメチルエステル(MPEG - SCM、MW 2000ダ)またはマレイミド- PEG -スクシンイミジルカルボキシの2倍の重量とインキュベートしたゆっくりと撹拌しながら室温で2時間メチルエステル(MAL - PEG - SCM、MW 2000ダ)。
- PEG化ナノ粒子を、30分間、16,000 rpmで超遠心分離で収集したS.ペレット回脱イオン水で洗浄し、ナノ粒子を精製した凍結乾燥し、4℃で保存した。
- MAL - PEG - SCM修飾されたナノ粒子は、10mg/mlの濃度で0.1Mリン酸緩衝液(pH6.5)に懸濁させ、ゆっくりと攪拌しながら室温で6時間EGFR特異的ペプチド(YHWYGYTPQNVI - GGGGC)の10%の重量でインキュベートした。
- ペプチド修飾されたナノ粒子は、30分で16,000 rpmで超遠心分離で収集した。ペレット回脱イオン水で洗浄し、ナノ粒子を精製した凍結乾燥し、4℃で保存した。
2。 EGFR標的ナノ粒子のキャラクタリゼーション
- 粒子径とゼータ電位測定
ナノ粒子は、1mg/mlの濃度で水に懸濁した。懸濁液をゼータサイザーナノ(マルバーン社)を用いて分析した。粒度分析は、25で90度の散乱角で実施させたC.ゼータ電位は25℃の水の誘電率、屈折率と粘度のデフォルトパラメータ℃で測定した - 走査型電子顕微鏡
凍結乾燥ナノ粒子はアルミニウム試料のマウントにマウントし、スパッタコーティングされたパラジウムと導電性を向上させ、電荷の蓄積を最小限にする。サンプルは、3 kVで走査型電子顕微鏡日立4800電界放出で表面形態を観察した。 - 化学分析用電子分光(ESCA)
コントロールの凍結乾燥製剤、PEG化およびペプチド改変ナノ粒子は、ESCAで分析した。それは、国立ESCAおよび生物医学問題の表面分析センター(NESAC / BIO)、ワシントン大学(シアトル、ワシントン州)で行った。- サンプルは、超高真空中に入れ、表面から二次光電子の放出を誘導低エネルギーX線ビームにさらされた。
- ビンの関数として検出される電子数をプロットして鼎エネルギー、観測スペクトルのピークは、それぞれの化学成分に割り当てられていた。
- C1sのスペクトルの高分解能の分析)は286.4mVでエーテル(CO)、炭化水素(285mVでCCやCH)からの正確な化学組成を決定するために行われ、カルボニル(288.1 mVでC = O)、および各機能の相対組成された曲線下の面積により決定された。
- カプセル化されたプラスミドの安定性
カプセル化されたプラスミドDNAの安定性は、プレキャストゲルで抽出されたDNAを実行することによって確認された。ナノ粒子は、同時にまたは連続して、別々にPBS(37℃で30分)、0.2 U / mlのDNaseを(室温で10分)を含む0.2 mg / mlのプロテアーゼで消化した。次に、試料をウェルあたり18μLのボリュームで100ng/wellの濃度で1.2%アガロースゲル(GP)(E -ゲル、Invitrogen社、CA)上に負荷された。プラスミド裸をコントロールとしてロードされており、ゲルを30分間75 Vで実行されました。コダックデジタルX線検体(DXS)システムは、bを視覚化するために使用されたUV transluminescenceで扱う。 - プラスミド荷重の決定
プラスミドカプセル化されたナノ粒子は、℃で30分間、1mg/mlに懸濁させ、37℃で0.2mg/mlプロテアーゼで消化した。溶液を10分間、13,000 rpmで遠心分離し、上清をPicoGreenアッセイ(Invitrogen社)を持つプラスミド濃度のために収集し、テストされています。カプセル化率は、初期荷重0.5%(w / w)のでカプセル化されたプラスミド濃度を除して算出した。
3 PANC - 1膵臓癌細胞の in vitro トランスフェクション研究で
- 細胞の培養条件
PANC - 1およびCapan - 1膵臓の腺癌細胞株、SKOV3卵巣腺癌細胞株およびNIH - 3T3マウス繊維芽細胞株は、ATCCから入手した。 PANC - 1、NIH - 3T3は、37 L -グルタミン、ペン- strepおよび10%ウシ胎児血清に付属のDMEMで成長させた℃、5%CO 2、Capan - 1必須のDMEM中にはウシ胎児血清20%を供給。SKOV3、10%ウシ胎児血清に付属のRPMI - 1640で増殖させた。 - EGFR発現のウェスタンブロット解析
- 細胞ライセートを2万個の細胞から採取し、BCAアッセイ(Pierce)を用いて、総タンパク質濃度について分析した。 NIH - 3T3は、ネガティブコントロールとSKOV3はEGFRの発現のポジティブコントロールとして使用されたとしても使用されました。
- 総タンパク質抽出物10μgを90分間135Vでプレキャストドデシル硫酸ナトリウム - ポリアクリルアミドゲル電気泳動(SDS - PAGE)システム上で実行されました。
- その後、ゲルをiBlotドライブロッティングシステム(インビトロジェン社)により、PVDF膜に転写した。
- 膜は、室温で1時間のTween含有トリス緩衝生理食塩水(TBS - T)の5%脱脂乳でブロッキングした。
- 膜は4℃で別々に一晩一次ウサギβ-アクチン抗体の1:1000希釈と一次ウサギのEGFR抗体の1:1000希釈で切断し、インキュベートした℃に
- 膜は、その後で2回洗浄し、TBS - Tと室温で1時間、TBS - Tで二次抗ウサギ西洋ワサビペルオキシダーゼ標識IgGの1:2000希釈液とインキュベート。
- TBS - Tと水で余分な抗体を洗浄した後、4ミリリットルECL基質(Pierce、ロックフォード、イリノイ州、米国)が5分間メンブレンを加えてインキュベートした。
- 化学発光バンドはその後、コダックのデジタルX線検体(DXS)システムを用いて可視化した。
- 異なる製剤で細胞の生存率の研究
- PANC - 1細胞を一晩補充したDMEMの200μLでウェルあたり10,000細胞で96ウェルプレートで増殖させた。
- 増殖培地は、0、0.5ナノ粒子、1、2、4、6 mg / mlの異なる濃度で含有する無血清培地と交換した。 1mg/ml PEI、既知の細胞傷害性カチオン性ポリマーは、陽性対照として使用した。
- 細胞は6時間200μLナノ粒子で処理し、20μLMTS試薬と完全な100μLに置き換えられた培地。
- 37℃で3時間後のインキュベーションの後、℃、5%のCO 2が 、ホルマザン産物の吸光度をBioTekのSynergyHTプレートリーダー(ウィヌースキ川、VT)で490nmの吸測定した。
- 細胞のパーセント生存率は100倍したネガティブコントロール(0mg/ml)からの相対ポリマー処理した細胞の吸光度の比として表され、ポリマーの濃度の関数としてプロットした。
- 細胞の人身売買の研究
- ローダミンBイソチオシアネート(RBITC)はアミン基との反応によりチオール化ゼラチンに結合するために使用されていました。透析および凍結乾燥後、RBITCはチオール化ゼラチンは、ナノ粒子の調製に使用したラベルが付いて。
- 脱溶媒する前に、25μlのPicoGreenは1分間プラスミド1mgのと混合し、標識したプラスミドをゼラチン溶液に加えた。別の製剤は、従来の方法に以下の行われた。
- PANC - 1細胞は20万セルのpとガラスカバースリップを含む6ウェルプレートで増殖させたよくER。一晩増殖させた後、標識されたナノ粒子の2ミリリットルを無血清培地に1mg/mlの濃度で各ウェルに処理した。
- 異なる時点の後、15分から6時間に、培地を室温で15分間のインキュベーションのためHoest 33342(Invitrogen社)の1μg/mlのを含む培地と交換した。 4%パラホルムアルデヒド溶液の2ミリリットルを、細胞を固定するために各ウェルに置き換えられました。次いで、細胞をPBSで2回洗浄した。
- Coversilpsをガラススライドにマウントした。レーザー走査型共焦点蛍光顕微鏡は、固定された細胞の画像を撮影するために使用されていました。
- pEGFPを- N1カプセル化されたナノ粒子と蛍光顕微鏡によるトランスフェクション効率の定性定量
- pEGFPを- N1プラスミドは、ナノ粒子に封入し、さらに治療のためのml当たり10μgのプラスミドと同等の濃度で無血清培地に懸濁した。
- PANC - 1細胞を6ウェルプレートcontaiで一晩増殖させたウェルあたり20万セルとガラスカバースリップを寧。 pEGFPを- N1プラスミドカプセル化されたナノ粒子の2ミリリットルを各ウェルに処理した。プラスミドの20μgを20μlのリポフェクチン、陽イオン性脂質トランスフェクション試薬と混合し、および未処理細胞をネガティブコントロールとして使用されていた間、それは、陽性対照として使用した。
- 細胞を6時間ごとに異なる処方でインキュベートした。
- 培地は、24、48、72および96時間後にトランスフェクションした培養液と細胞に置き換えられた。
- ポストトランスフェクション後、培地をHoest 33342(Invitrogen社)の1μg/mlのを含む培地に交換したし、室温で15分間細胞とインキュベート。
- カバーガラスは、ガラススライド上にマウントされ、細胞内のGFPの発現を蛍光顕微鏡で観察した。微分干渉コントラスト(DIC)と蛍光画像は、画像Jソフトウェアで処理されたオリンパスBX61顕微鏡とデジタル画像を使用して取得した。
- pEGFPを- N1プラスミドは、ナノ粒子に封入し、さらに治療のためのml当たり10μgのプラスミドと同等の濃度で無血清培地に懸濁した。
- PANC - 1細胞を、ウェル当たり20万セルで6ウェルプレートで一晩増殖させた。 pEGFPを- N1プラスミドカプセル化されたナノ粒子の2ミリリットルを各ウェルに処理した。プラスミドの20μgは、ネガティブコントロールとして使用した陽性コントロールおよび未処理細胞として使用された20μlのリポフェクチン、陽イオン性脂質トランスフェクション試薬と混合した。
- 細胞を6時間ごとに異なる処方でインキュベートした。
- 培地は、24、48、72および96時間後にインキュベートした培養液と細胞に置き換えられた。
- ポストトランスフェクション後、細胞溶解物を各ウェルから回収し、BCAアッセイ(Pierce)を用いて、総タンパク質濃度について分析した。
- ウェルプレートは、COAだ各ウェルにモノクローナル抗GFP抗体の1:1000希釈液100μlとテッド。 2時間のインキュベーション後、プレートをPBS - 0.5%(w / v)の4回のTween - 80で洗浄した。
- 300μlTBSブロッキングバッファーを各ウェルに添加し、2時間インキュベートした。その後、プレートをPBS - 0.5%(w / v)の4回のTween - 80で洗浄した。
- 各群のタンパク質の30μgをプレートに添加し、4℃で一晩インキュベートした。その後、プレートをPBS - 0.5%(w / v)の4回のTween - 80で洗浄した。
- アルカリホスファターゼの相対的な二次抗GFP抗体の1:2400希釈液100μlを各ウェルに添加し、1時間インキュベートした。その後、プレートをPBS - 0.5%(w / v)の4回のTween - 80で洗浄した。
- 100μlのアルカリホスファターゼの基質を各ウェルに添加し、30分〜1時間インキュベートした。プレートは、405nmの吸光度をBioTekのシナジーHTプレートリーダーで測定した。
- 発現したGFP濃度はマイルあたりナノグラムとして報告された総タンパク質のlligrams。
- WT - p53のプラスミドをナノ粒子に封入し、さらに治療用10μgのプラスミド1ml当たりの濃度と同等と無血清培地に懸濁した。
- PANC - 1細胞を、ウェル当たり20万セルで6ウェルプレートで一晩増殖させた。 WT - p53のプラスミドカプセル化されたナノ粒子の2ミリリットルを各ウェルに処理した。プラスミドの20μgは、ネガティブコントロールとして使用した陽性コントロールおよび未処理細胞として使用された20μlのリポフェクチン、陽イオン性脂質トランスフェクション試薬と混合した。
- 細胞を6時間ごとに異なる処方でインキュベートした。
- 培地は48時間後にインキュベートした培養液と細胞に置き換えられた。
- mRNAは、T(高純度RNAアイソレーションキット(ロシュ、インディアナポリスを、)を使用して各ウェルから抽出され、光度計2000で測定したhermo科学、Wilminton、DE)。
- RT - PCRは、QIAGENを使用して行われたワンステップRT - PCRキット(QIAGEN、バレンシア、カリフォルニア州)。 p53のためのプライマー、バックス、Bcl - 2を、β-アクチン、DR5、Apaf - 1の、プーマ、サバイビンはEurofins MWGオペロン(ハンツヴィル、アラバマ州)により合成した。
- PCR産物をゲル電気泳動で評価し、cDNAのバンドのピクセルは、ImageJのソフトウェアを使用して分析した。
- WT - p53のプラスミドナノ粒子に封入し、さらに治療のためのml当たり10μgのプラスミドと同等の濃度で無血清培地に懸濁した。
- PANC - 1細胞を、ウェル当たり20万セルで6ウェルプレートで一晩増殖させた。 WT - p53のプラスミドカプセル化されたナノ粒子の2ミリリットルを各ウェルに処理した。プラスミドの20μgは、陽性対照および未処理細胞であったとして使用された20μlのリポフェクチン、陽イオン性脂質トランスフェクション試薬と混合し、ネガティブコントロールとして使用。
- 細胞を6時間ごとに異なる処方でインキュベートした。
- 培地は、24、48、72および96時間後にインキュベートした培養液と細胞に置き換えられた。
- クロマチン凝縮/膜透過性/死細胞アポトーシスキット(Invitrogen社、カールスバッド、カリフォルニア州)は、異なる色素でアポトーシス細胞、ネクローシス細胞と生細胞を標識するために使用されていました。
- CompuCyteからICYS研究イメージングサイトメーター(ウエストウッド、MA)は、治療後のアポトーシスのレベルを分析し、比較するために使用された。蛍光顕微鏡像に基づいて、すべての色の強度を記録し、異なる集団を算出したのカウントと割合に対してプロット。
- 細胞に対する治療法がなかったことを意味ネガティブコントロールと比較し、倍の変化アポトーシス細胞は外に計算し、グラフに記載されていた。
- 3.8.8アポ- ONE均質なカスパーゼ-3 / 7アッセイキット(プロメガ社、マディソン、WI)は、pを調べるために使用されたWT - p53のプラスミドのトランスフェクション後のROアポトーシス活性。 1mg/ml PEIは、すべてのアポトーシス活性を排除するネガティブコントロールとして使用した。 、カスパーゼ3 /の基質である、ポストトランスフェクション後、細胞はローダミン110、ビス - (Z - DEVD - R110 N - CBZL -アスパルチル- L -グルタミル- L -バリル- L -アスパラギン酸のアミド)で処理した7、最大18時間。
- プレートは、520分の490 nmの蛍光用BioTekのシナジーHTプレートリーダーで測定した。緑色の蛍光の強度に基づいて、プロアポトーシス活性を評価することができます。
4。代表的な結果
1。 EGFR標的ナノ粒子の合成とChatacterization
スキームは図1に示したようにEGFR標的ペプチドで修飾されたナノ粒子を合成した。脱溶媒和が作成したナノ粒子は、粒子径とゼータ電位のために特徴づけられた。チオール化ゼラチンから製造された粒子の平均粒径と表面電荷チオール化の程度が異なると、表1に記載されています。異なるナノ粒子の平均粒径は150から250 nmの間であった。チオール化ナノ粒子は、粒子内部のジスルフィド架橋の形成のために、ゼラチンナノ粒子に小さいサイズを可能性と比較している。さまざまな表面改質と、ナノ粒子のサイズが増加している。異なる製剤のゼータ電位は-20 mVの程度であった。 SEMの分析で、大きさ、表面形態とナノ粒子の球状の形状を観察したとサイザーの結果に対応する。ゼラチンナノ粒子とチオール化ゼラチン粒子にDNAローディングの効率は95%(表1)よりも高かった。
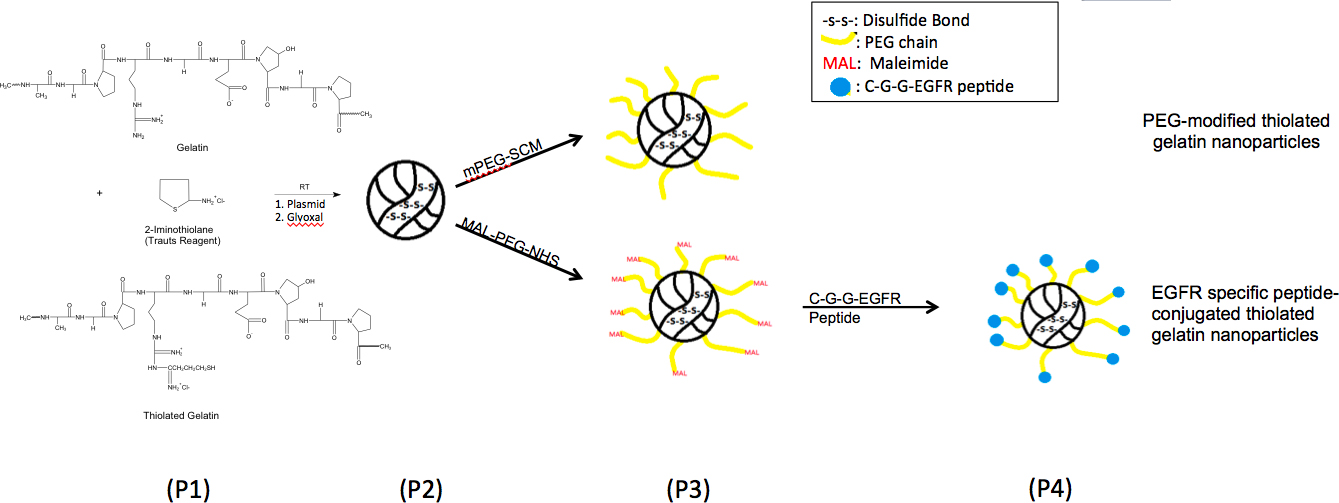
上皮成長ファクトとチオール化ゼラチンナノ粒子の表面の変形例を示す図1。化学反応スキーム、R受容体(EGFR)ポリ(エチレングリコール)(PEG)スペーサーを介して結合ペプチド。
ナノ粒子のキャラクタリゼーション
| 製剤 | ナノ粒子の直径(nm)の | ゼータ電位(mV) | プラスミドDNAの積載効率(%) |
| ゲルNP | 151.4 ± 23.5 | -17.1 ± 5.23 | 95.6 ± 2.2 |
| SH -ジェルNP | 132.6 ± 17.9 | -24.6 ± 5.16 | 97.0 ± 3.8 |
| SH -ゲル- PEG | 179.0 ± 30.9 | -22.3 ± 9.50 | 95.8 ± 6.5 |
| SH -ジェルPEGペプチド | 230.8 ± 41.5 | -18.1 ± 4.02 | 94.8 ± 5.1 |
TABLE(1)制御とEGFR標的ゼラチンとチオール化ゼラチンナノ粒子の粒径、表面電荷、およびプラスミドDNAのカプセル化効率。
高解像度C 1S化学分析用電子分光法(ESCA)のスキャンは、ペプチドで修飾されたチオール化ゼラチンターゲティングチオール化ゼラチン(SH -ゲルNP)、PEG修飾チオール化ゼラチン(SH -ゲルPEG)とEGFRの表面の成分を分析するために使用されたナノ粒子(SH -ジェルPEGペプチド)。表2の結果は、ピークの285.0で、CH(炭化水素)、CO(エーテル)、およびC = O(カルボニル)グループの強度、286.3、および288.1 eVの、それぞれを示した。エーテルのCOの信号は、PEG修飾の後に増加し、ペプチドコンジュゲーションの後に減少している。窒素組成は、PEG修飾後に減少し、ナノ粒子にEGFRを標的とするペプチドの存在を確認したペプチドの変更、後に増加しているが。 ESCA分析では、さらにPEGとペプチド表面改質を認識しています。
ナノ粒子の表面組成の化学分析用電子分光
| 製剤 | C 1S(%) | O 1S(%) | N 1S(%) |
| SH -ジェルNP | 59.3 ± 0.8 | 22.9 ± 0.5 | 12.9 ± 0.1 |
| SH -ゲル- PEG | 58.2 ± 0.6 | 28.0 ± 1.2 | 9.5 ± 0.7 |
| SH -ジェルPEGペプチド | 56.7 ± 0.8 | 25.9 ± 0.7 | 12.3 ± 0.6 |
| 製剤 | CC(%) | CO、N(%) | C = O(% ) |
| SH -ジェルNP | 51.5 | 26.6 | 21.9 |
| SH -ゲル- PEG | 17.1 | 63.1 | 19.8 |
| SH -ジェルPEGペプチド | 33.1 | 42.8 | 24.1 |
化学分析用電子分光法の表2。C 1S高解像度スキャン(ESCA)
カプセル化されたプラスミドの安定性を調べるために、ナノ粒子はsimuntaneouslyまたは順次に、プロテアーゼまたはDNase別々に処理した。電気泳動後、図2の結果は、すべてのナノ粒子内にカプセル化されたプラスミドDNAは裸のプラスミドDNAに匹敵するナノ粒子と安定、で保護されていることが示されている。調査これらはすべて、これらのナノ粒子がカプセル化とカプセル化の後にプラスミドの構造を保つことができることが示されている。
/ files/ftp_upload/3612/3612fig2.jpg"/>
図2。チオール化ゼラチンでカプセル化されたプラスミドDNAの安定性、PEG修飾はゼラチンチオール化、およびEGFRは、アガロースゲル電気泳動によってチオール化ゼラチンナノ粒子をペプチドで修飾された。ナノ粒子はナノ粒子のマトリックス内にプラスミドDNAのカプセル化を証明するためにプロテアーゼ0.2 mg / mlの処理した
2。膵臓癌細胞におけるベースラインのEGFR発現
二つのヒト膵臓腺癌細胞株(PANC - 1およびCapan - 1)はEGFRの発現のためのウェスタンブロットにより分析した。ヒト卵巣腺癌(SKOV3)とマウス線維芽細胞((NIH - 3T3)細胞をそれぞれ、正および負の対照として選ばれた。β-アクチンは、タンパク質のローディングコントロールとして解析した。PANC - 1細胞は、Capan - 1に比べて高いEGFRの発現を示しているそしてこの細胞株は、in vitro試験は、次のために使用された
3。コントロールとSurfaの細胞毒性CE - Modifiedのチオールゼラチンナノ粒子
ナノ粒子の細胞間相互作用を評価するために、細胞毒性のアッセイは、ナノ粒子による治療後に実施された。図3の結果に基づいて、両方の制御と表面修飾ナノ粒子は、PEIの比較でも高濃度でPANC - 1細胞で比較的安全と生体適合性であった。以下の研究は1mg/mlナノ粒子を用いて行った。

としてテトラゾリウム色素(MTS)アッセイにより評価したPANC - 1細胞におけるナノ粒子製剤の濃度の関数として図3。パーセント細胞生存率
4。 PANC - 1細胞における受容体介在性細胞取り込み
EGFR標的化ペプチドとナノ粒子の受容体介在性エンドサイトーシス取り込みの表面のアクセシビリティを確認するには、システムは、各共同標識することにより設計されました細胞内のナノ粒子の取り込みと人身売買の可視化のための異なる蛍光でmponent。このラベリングシステムで、プラスミドDNA、ナノ粒子と細胞核を同定することができた。レーザー走査型共焦点蛍光顕微鏡は、15分から6時間に、異なる時点で画像を撮影するために使用されていました。異なる製剤の画像を比較することにより、ペプチド結合したゼラチンナノ粒子は、30分以内に高速取り込みとプラスミド放出を示した。この結果はさらにEGFRペプチドコンジュゲートナノ粒子は非特異的なエンドサイトーシスを受けた他のナノ粒子に比べて、はるかに高速だった細胞表面上のEGFR特異的ペプチドとEGFR受容体、間の迅速な相互作用を容易にエンドサイトーシスを受けたことを証明した。
細胞の人身売買に関する研究

図4共焦点蛍光顕微鏡PANC - 1細胞におけるDNA -カプセル化されたナノ粒子の取り込みと人身売買のalysis。 (赤=ローダミン標識ナノ粒子、緑=プラスミドDNA PicoGreen標識、および青= DAPI標識核)。レーザパワーは、下のパネルの最後の4つの図で7回以下であった。
5。強化緑色蛍光タンパク質との定性的および in vitroトランスフェクションの定量
図5と図6の蛍光顕微鏡分析におけるELISAは変更されずに、PEG修飾およびEGFRペプチドで修飾されたチオール化ゼラチンナノ粒子の投与の際PANC - 1細胞において定性的および定量的なGFPのトランスフェクション効率を測定するために使用されていました。 EGFR標的ナノ粒子が提供するプラスミドはリポフェクチン - 複合DNAを含む他のコントロールに対して48時間、後にGFPの発現の最高レベルとなりました。

図5。GFPの発現ELISAによるnalyzedは、時間制御とEGFR標的ナノ粒子におけるプラスミドDNAの投与後の関数としてプロット。
GFPのトランスフェクションのためのFluoresence顕微鏡分析

図6。EGFP - N1の24、48、72および96時間後にトランスフェクション後のepifluoresence顕微鏡によるPANC - 1細胞に緑色蛍光タンパク質の発現の定性分析。リポフェクチン- DNA複合体は、ポジティブコントロールとして使用した。
PANC - 1細胞の6。野生型p53 を用いたin vitroトランスフェクションのプラスミド
EF -1α/ HTLVハイブリッドプロモーターと野生型p53プラスミドPORF - hp53は、E.から抽出された大腸菌とアポトーシス治療効果を研究するナノ粒子に封入。 PANC - 1細胞を6時間粒子、さらに24のためのポストトランスフェクション、48、72、および96で処理した時間。
p53は細胞内で、この機能を達成するためにアポトーシスを誘導する可能性があるため、多くの下流の転写因子が関与し、直接重量- p53の発現により調節される。それらの、バックス、カスパーゼ-3、カスパーゼ-9、DR5の中で、PUMAとApaf - 1のは、p53の発現がアップレギュレートされるとBcl - 2の間に、サバイビンがダウンレギュレートされるでしょう。これらの転写因子のレベルを調べるために、mRNAは48時間後にトランスフェクション後のPANC - 1細胞から抽出し、RT - PCRに使用した。製品は、ゲル電気泳動で評価し、バンドはImageJを用いて分析した。その結果に基づいて、図7に示した、サバイビンが他の治療法に比べてチオール化ゼラチンナノ粒子を対象とEGFRの治療で有意に減少し、明らかな変化は、カスパーゼ-3、カスパーゼ9、DR5、のBcl - 2、Bax及び表現は見られませんでしたPUMAとApaf - 1increased標的ナノ粒子治療を。
les/ftp_upload/3612/3612fig7.jpg"/>
図7。WT - p53発現の下流の因子のmRNAレベルは、48時間後にトランスフェクション後、RT - PCRで比較した。
WT - p53のトランスフェクションの後、クロマチン凝縮/膜透過性/死細胞アポトーシスキットは異なる色素でアポトーシス細胞、ネクローシス細胞と生細胞を区別するために使用されていました。 CompuCyteからICYS研究イメージングサイトメーター(ウエストウッド、MA)は、治療後のアポトーシスのレベルを分析し、比較するために使用された。陰性対照と比較して、アポトーシス細胞倍の変化が出算出し、図8に示します。 EGFR標的チオール化ゼラチンnanopaticlesは、トランスフェクション後の後の最も高いアポトーシス細胞集団を示している。カスパーゼ-3 / 7活性の分析はまたEGFR標的ナノ粒子が急速に内部化し、PANC - 1細胞のアポトーシス活性の最も高いレベルを持っていたことが明らかになった。
 図8。ICYSを使用してコントロールWT - p53のトランスフェクションPANC - 1細胞·イメージングサイトメーターのプロアポトーシス活性のサイトメトリー分析
図8。ICYSを使用してコントロールWT - p53のトランスフェクションPANC - 1細胞·イメージングサイトメーターのプロアポトーシス活性のサイトメトリー分析
Discussion
制御とEGFR標的とチオール化ゼラチンナノ粒子は、効率的なDNAのカプセル化と安定性を用いて調製した。これらのシステムのすべての粒子サイズは直径150〜250 nmの範囲であった。ゼータ電位は、このシステムはわずかに負のシステムであることを証明しています。 SEMの分析では、ナノ粒子のサイズはゼータサイザーの結果と同じであった。 ESCA分析では、PEGとペプチド表面改質を確認することができた。
ウェスタンブロット分析は、PANC - 1細胞は、高いEGFRの発現のレベルを持っていたし、この細胞株はin vitro試験のために使用されることを示した。 PEIと比較して、コントロールと表面修飾ナノ粒子の両方がPANC - 1細胞では比較的少ない細胞毒性であった。
細胞の人身売買の研究は急速な取り込みとPANC - 1細胞におけるEGFR標的ナノ粒子のプラスミド放出を示した。 EGFR標的ナノ粒子で表現するレポータープラスミドDNAの配信は、最高レベルの結果リポフェクチン-複合DNAを含む他のコントロールに対する相対的なGFP発現。同じシステムで、WT - p53のプラスミドでトランスフェクションは、下流のアポトーシス経路およびPANC - 1細胞において誘導急速にアポトーシスを誘発した。
これらの予備的な結果は、EGFRを標的とチオール化ゼラチンナノ粒子は、膵臓癌の治療として遺伝子治療のための安全かつ効率的なDNAの配信システムとして提供できることを示唆している。
Disclosures
利害の衝突は宣言されません。
Acknowledgments
この研究はがんナノエクセレンス(CCNE)助成U54 - CA151881のためのがんのセンターのナノテクノロジーの国立がん研究所のアライアンスによってサポートされていました。
Materials
| Name | Company | Catalog Number | Comments |
| Type B gelatin, Bloom 225 | Sigma-Aldrich | G9391 | |
| 2-iminothiolane hydrochloride | Sigma-Aldrich | I6256 | |
| pEGFP-N1 plasmid | Elim Biopharm | N/A | |
| pORF-hp53 E. coli | Invitrogen | porf-hp53 | |
| Glyoxal solution (40wt. % in H2O) | Sigma-Aldrich | 128465 | |
| Glycine | Sigma-Aldrich | 410225-250G | |
| QIA filter Plasmid Mega kit | Qiagen | 12281 | |
| Beckman LE 80K Ultracentrifuge | Beckman Coulter Inc. | N/A | |
| FreeZone 6 Liter Console Freeze Dry Systems | Labconco Corp. | 7753020 | |
| mPEG-SCM, MW 2,000 Da | Laysan Bio Inc. | mPEG-SCM-2K-1g | |
| MAL-PEG-SCM, MW 2,000 Da | Jenkem Technology | A5001-1 | |
| Zetasizer Nano | Malvern Instruments | Zetasizer Nano ZS | |
| Hitachi 4800 field emission scanning electron microscope | Hitachi | S-4800 UHR FE-SEM | |
| Quant-iT PicoGreen dsDNA Reagent and Kits | Invitrogen | P7589 | |
| Lipofectin Transfection Reagent | Invitrogen | 18292011 | |
| DMEM | Mediatech, Inc. | 10 013 CM | |
| RPMI | Mediatech, Inc. | 50 020 PB | |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific, Inc. | 23225 | |
| iBlot Dry Blotting System | Invitrogen | IB1001 | |
| XCellSureLock Mini-Cell and XCell II Blot Module Kit CE Mark | Invitrogen | EI0002 | |
| Pierce ECL Western Blotting Substrate | Thermo Fisher Scientific, Inc. | 32109 | |
| Kodak Digital X-ray Specimen (DXS) System | Kodak | N/A | |
| CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS) | Promega Corp. | G3580 | |
| BioTek SynergyHT plate reader | BioTek | N/A | |
| Nanodrop 2000 | Thermo Fisher Scientific, Inc. | N/A | |
| One-step RT-PCR kit | Qiagen | 210212 | |
| Chromatin Condensation/Membrane Permeability/Dead Cell Apoptosis Kit | Invitrogen | V23201 | |
| Apo-ONE Homogeneous Caspase-3/7 Assay kit | Promega Corp. | G7790 | |
| Hybaid PCR Sprint Thermal Cycler | Thermo Fisher Scientific, Inc. | N/A | |
| EGF Receptor Antibody | Cell Signaling Technology | 2232 | |
| β-Actin Antibody | Cell Signaling Technology | 4967 | |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling Technology | 7074 | |
| Mouse Monoclonal GFP Antibody | Novus Biologicals | NB600-597 | |
| Goat Polyclonal GFP antibody (Alkaline Phosphatase) | Novus Biologicals | NB600-1502 | |
| Phosphatase Substrate Kit | Thermo Fisher Scientific, Inc. | 37620 |
References
- Vimalachandran, D. Genetics and prevention of pancreatic cancer. Cancer. Control. 11, 6-14 (2004).
- Kommareddy, S., Amiji, M. Preparation and evaluation of thiol-modified gelatin nanoparticles for intracellular DNA delivery in response to glutathione. Bioconjug. Chem. 16, 1423-1432 (2005).
- Kommareddy, S., Amiji, M. Poly(ethylene glycol)-modified thiolated gelatin nanoparticles for glutathione-responsive intracellular DNA delivery. Nanomedicine. 3, 32-42 (2007).
- Kommareddy, S., Amiji, M. Antiangiogenic gene therapy with systemically administered sFlt-1 plasmid DNA in engineered gelatin-based nanovectors. Cancer. Gene. Ther. 14, 488-498 (2007).
- Kommareddy, S., Amiji, M. Biodistribution and pharmacokinetic analysis of long-circulating thiolated gelatin nanoparticles following systemic administration in breast cancer-bearing mice. J. Pharm. Sci. 96, 397-407 (2007).
- Kaul, G., Amiji, M. Tumor-targeted gene delivery using poly(ethylene glycol)-modified gelatin nanoparticles: in vitro and in vivo studies. Pharm. Res. 22, 951-961 (2005).
- Bardeesy, N., DePinho, R. A. Pancreatic cancer biology and genetics. Nat. Rev. Cancer. 2, 897-909 (2002).
- Li, Z. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB. J. 19, 1978-1985 (2005).
- McCormick, F. Cancer gene therapy: fringe or cutting edge. Nat. Rev. Cancer. 1, 130-141 (2001).
- Green, D. R., Kroemer, G. Cytoplasmic functions of the tumour suppressor p53. Nature. 458, 1127-1130 (2009).
- Barton, C. M. Abnormalities of the p53 tumour suppressor gene in human pancreatic cancer. Br. J. Cancer. 64, 1076-1082 (1991).
