

Summary
全細胞RNAは、短期的RNA合成と崩壊の変化と同様に、RNAプロセシングの動態を研究するための貧乏人のテンプレートが用意されています。ここでは、これらの制限を克服することができ、新たに転写されたRNAのチオール特有のビオチン化および精製し、4 - チオウリジンと新しく転写されたRNAの代謝標識を記述します。
Abstract
全トランスクリプトームマイクロアレイや次世代シーケンシングの開発は、細胞の遺伝子発現の複雑さの理解に革命をもたらしました。関与する分子機構の理解を深めるとともに、下地動態の正確な測定がますます重要になってきた。ここでは、これらの強力な方法論は、テンプレートサンプルそれら研究、 すなわち全細胞RNAの本質的な特性のための主要な制限に直面しています。多くの場合、全細胞RNAの変化が十分な解像度で、基礎となる分子事象とその動態を表現するためにいずれか遅すぎるか早すぎる起こる。また、RNA合成、加工、及び減衰の変化の寄与を容易に区別されていない。
最近では、これらの制限を克服するために、高解像度の遺伝子発現プロファイリングを開発した。我々のアプローチは、4 thiouriで新しく転写されたRNAの代謝標識に基づいていますチオール - 特異的ビオチンとストレプトアビジンでコートした磁気ビーズを使用して、新しく転写されたRNAの厳格な精製しダイン(ひいては4SUタグ付けとも呼ばれる)。それは、脊椎動物、 ショウジョウバエ 、および酵母を含む生物の広い範囲に適用可能である。我々は正常、転写因子活性のリアルタイム動態を研究するRNA半減期の正確な測定を提供し、RNAプロセシングの動態に新規な洞察を得るために4SUタギングを適用した。最後に、計算モデルは、基礎となる分子機構の統合された包括的分析を生成するために使用することができる。
Introduction
遺伝子発現プロファイリングは、細胞プロセス及び関連する複雑な相互作用ネットワークを研究するために使用される重要なツールである。 mRNAの存在量に関する研究は、通常、基礎となる分子メカニズムに基本的な洞察を得るための最適な方法となっている。全トランスクリプトームマイクロアレイ1と、RNA(RNA-seqの)2-4より最近では、次世代シーケンスの開発は、このアプローチを燃料とする。これらの技術は、細胞の遺伝子発現の複雑さの理解に革命をもたらしているが、彼 らは、テンプレートサンプル、 すなわち全細胞RNAの本質的な特性のための主要な制限に直面しています。トータルRNAレベルで最初の、短期的な変化は、転写率の変化と一致していませんが、それぞれの転写産物のRNAの半減期に本質的に依存している。転写因子のための短命のトランスクリプト、 例えばエンコードの五倍誘導は、トータルRNAで容易に検出される一方時間以内に、長命の転写産物の同じ誘導、代謝酵素のための例えばエンコードは、実質的には不可視のままです。加えて、たとえ完全なシャットダウン(> 1,000倍ダウンレギュレーション)五時間のRNAの半減期と平均遺伝子の転写率は、単にだけ二重減少するために、その全RNAレベルに5時間かかりますで。従って、トータルRNAの分析は、調節機能5と転写因子や遺伝子をコードする多くは短命転写産物のアップレギュレーションの検出に有利で ある。また、規制の真の運動カスケードが隠されている、プライマリシグナリングイベントは二次と区別することはできません。両方、順番に、下流のバイオインフォマティクス解析の大幅な偏りが生じる可能性があります。第二に、トータルRNAレベルの変化は、RNAの合成または減衰の変化に起因することはできません。後者の測定はtranscriptiをブロック細胞侵襲アプローチ、 などを必要とするアクチノマイシンD 6、そして時間をかけて継続的なRNA崩壊の拡張監視を使用してください。 5の哺乳類細胞における平均mRNAの半減期で- 10時間5,7、ほとんどの遺伝子のmRNAレベルは、二重転写逮捕の数時間後に未満減少しております。これらむしろ小さな違いは、基礎となる数学の方程式の指数関数的性質に起因する細胞遺伝子の大多数のmRNAの半減期の著しく不正確な測定値になる。我々の遺伝子の約半分が選択的スプライシングイベント8の対象であることを最後に、全細胞RNAのRNA-seqのが明らかになっている間には、基本となる動力学と同様、組織とRNAプロセシングのコンテキスト固有の規制を導くダイナミックメカニズムはよくわかっていないままです。また、特に非コーディングRNA用の差動遺伝子発現に対する処理RNAの寄与は、決定されていない。要するに、これらの制限のためには大きな障害を表す基盤となる分子機構のバイオインフォマティクス運動モデリング。
我々は、最近のアプローチを開発し、これらの問題を克服するために、5,7,9、高解像度の遺伝子発現プロファイリングと呼ばれる。なお、4 -チオウリジン(4SUタギング)、天然に存在するウリジン誘導体を使用して、新しく転写されたRNAの代謝標識に基づいており、細胞増殖および遺伝子発現の干渉を最小限( 図1参照)5と新たに転写された転写物への直接アクセスを提供する10-12。その急速な取り込み、4SU-三リン酸へのリン酸化、および新たに転写されたRNAへの取り込みにおける4SU結果に真核細胞の暴露。全細胞RNAの単離の後、4SU-標識されたRNA画分はチオール - 特異的ビオチンと新たに転写されたRNA間のジスルフィド結合を生成するビオチン化される。 '全細胞RNA'は、その後、定量的にラベル分け( '新たに転写')とラベル付けされていない( 'プレexistinすることができますストレプトアビジンでコーティングされた磁気ビーズを用いて高純度グラム ')RNA。最後に、標識されたRNAは、単にジスルフィド結合を切断する還元剤( 例えば、ジチオスレイトール)を添加し、ビーズから新たに転写されたRNAを解放することによってビーズから回収される。
新たに転写されたRNAは4SU暴露期間中にすべての遺伝子の転写活性を示しています。分のタイムスケールで4SUタギングは、このように、真核生物の遺伝子発現のスナップ写真とダウンストリームバイオインフォマティクス解析( 例えばプロモーター解析)のための理想的なテンプレートを提供しています。定常状態が想定される場合、新たに転写/合計の比率では、新たに転写/非標識及び非標識/全RNAは、正確なRNAの半減期7,13への非侵襲的なアクセスを提供します。また、4SUタギング(5分4SU-RNA)の、わずか5分で15歳未満と60分4SU-RNAである。後精製し、その新たに転写されたRNAを注意することが重要です超短と次第に長く4SUタギングRNA-seqのと組み合わせて、単一の実験設定で両方を行う場合は、RNAプロセシングの動態は、ヌクレオチド分解能9で明らかにされています。最後に、計算モデルと組み合わせて新たに転写され、総RNAの経時的分析は、RNA合成と崩壊14の統合的な分析を可能にする。
結論として、このアプローチは、真核細胞におけるRNA合成、加工、及び劣化のダイナミクス直接分析を可能にする。これは、哺乳類、昆虫( ショウジョウバエ )、両生類( アフリカツメガエル )、および酵母5,15,16を含むすべての主要なモデル生物で適用されます。これは、マイクロアレイ解析5,17、RNA-seqの9,13,14と直接互換性があり、 生体内で 12,15 に適用されます。ここでは、詳細な方法論を、ラベルを分離、培養した哺乳動物細胞で新たに転写されたRNAを精製する。また、ポテンショらの問題や落とし穴が議論されています。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1。 4チオウリジンで代謝標識
実験装置/スケジュールの詳細な計画を作成し、細胞培養に4SUを追加するときに、サンプルを収穫するときなど 。各条件の間には少なくとも5分間プラン。一度に一つの条件のセルを扱う。最大を扱う。 3 - 特定の時点で5皿。温度とCO 2濃度の変化を最小限に抑えるために、できるだけ早く、細胞を扱う。これは細胞タンパク質へ4SU標識RNAの架橋につながる可能性があるので4SUが追加された後に明るい光に細胞をさらすことは避けてください。
ラベリングの開始
- ただ滅菌ファルコンチューブに各条件のための使用と4SUのピペット必要量の前に解凍4チオウリジン(4SU)。
- 料理オフ細胞培養培地(10cmディッシュあたり5 ml)を必要量を取り、4SU含有ファルコンチューブに追加し、徹底的に混ぜる。食器から残りの培地を除去し、廃棄する。 <李>皿に戻し4SU含有培地を適用します。
ラベリングの終わり
- 細胞から細胞培養培地を除去する。各プレートにトリゾール5mlのを追加します。複数の時点または条件を含む複雑な実験では、このステップは最高の2人、培地を除去し1、他追加トリゾールによって行われ、ライセートを収穫されています。
- 完全な細胞溶解を室温で5分間インキュベートする。
- 追加トリゾールで慎重プレートをすすぎ、10ミリリットルピペットを使用してください。これは完全な細胞溶解と試料回収を助ける。皮膚や目に接触して取得するときトリゾールは極めて危険であるように取り扱いに注意して!手元フェノール火傷( 例えばポリエチレングリコール300または工業変性 アルコール(70:30)で400)のための解毒剤を持っている。ポリプロピレンチューブにサンプルを転送します。標準のファルコンチューブ)は、これらの高グラム力に抵抗はありませんのでご了承ください。サンプルは私が全RNAまで少なくとも1ヶ月、-20℃で保存することができますsが用意しました。
2。修正トリゾールプロトコルを使用したRNAの準備
- 1ミリリットルのクロロホルムを(MLトリゾール当たり0.2 ml)を加え、15秒間激しく振り。 3分 - 2室温でインキュベートする。
- 4℃で15分間、13,000×gで遠心℃、
- 新しい15ミリリットルのポリプロピレンチューブに水上相を(RNAを含む)を転送します。
- 追加½RNA降水バッファとイソプロパノールの双方の反応ボリューム(3ミリリットル上澄みに例えば 1.5ミリリットルRNA降水バッファー、1.5 mlのイソプロパノールを追加します)。
- よく混ぜる。 10分間室温でインキュベートする。
- 4℃で10分間、13,000×gで遠心℃、上清を捨てます。
- 簡単にスピンダウン(5,000×gで30秒間)と200μlのピペットで残留イソプロパノールを除去。
- 75%エタノールを等量を追加し、ペレットデタッチまでチューブを振る。これは残滓の除去を行うことができるように多くの小さな部分にそれを壊すことは避けてくださいリットルエタノール難しい。
- 4℃で10分間、13,000×gで遠心℃、上清を捨てます。
- 簡単にRNAをスピンダウンし、200μlのピペットで残りのエタノールを除去。ステップを繰り返して、20μlのピペットで残りのエタノールを除去。この2つの手順の後、ペレットのさらなる乾燥が行われるべきではない。
- 100μgの予想RNA収量当たりのH 2 Oを100μLを加え、5を上下にピペッティングでよく混ぜる- 6回RNAを溶解を助けるために。
- 溶解し、65に加熱したRNAを変性℃で10分間(シェーカー)のため、すぐに氷上に置きます。を
- 製造元の指示に従って、光度計分光光度計を用いて260nmのRNA濃度を測定します。このRNAは、少なくとも一ヶ月-80℃で保存することができる。
3。新たに転写されたRNAのチオール特有のビオチン化
- 60で始まる - 全細胞RNAの80μgのを。
- ラベリング反応を構成する。以下の中でピペットオーダー(μgのRNAあたり):
- 1μlの10倍のビオチン化バッファー
- 7μlのRNA(ヌクレアーゼフリーのH 2 Oで希釈した1μgのRNAを含有)
- 2μlのビオチンHPDP(1 mg / mlのDMF)
常に最後ビオチンHPDPを追加し、ピペッティングにより直ちに混ぜる。ビオチンが沈殿する場合には、DMF含有量は、40%の最終濃度まで増加させることができる。
- 回転に伴って1.5時間室温でインキュベートする。
- 等量のクロロホルムを加える。精力的に混ぜる。相を分離し始めると泡が消え始めるまで3分 - 2インキュベートする。
- 4℃で5分間、20,000×gで遠心慎重に新しいチューブに上部の水相を転送します。
- 一度ステップ3.4と3.5を繰り返します。あなたは、RNAの損失を減らすために2 mlの位相ロックジェルヘビーチューブに、この手順を実行することがあります。
- RNA沈殿物:追加1/10の5のNaClの量と等量の水相にイソプロパノール。
- 4℃で20分間、20,000×gで遠心上清を捨てます。
- 、75%エタノールを等量を追加4℃で10分間℃で20,000×gで遠心分離し、上清を捨てる。
- 簡単にスピンし、200μlのピペットで残留エタノールを除去。
- 簡単にスピンし、20μlのピペットで残留エタノールを除去。
- RNAは乾燥させないでください。 50でそれを再懸濁する- 100μlのH 2 O(1μgの入力RNA当たり約1μL)。 6回 - 5を上下にピペッティングしてよく混ぜる。
- RNAの分解を除外するelectrophoretical分析によってRNAの品質を確認してください。
4。 4SU-組み込み(オプション)のドットブロット分析
4SU取り込みを容易にビオチン化RNAのドットブロット分析によって決定することができる。これは、トラブルシューティングおよびビオチン化DNAオリゴコントロールに対する4SU取り込み率の推定を可能にするオプションの手順です。このアッセイのために我々Rステップ3.2 4SU標識RNAのビオチン化のために代わりにビオチンHPDPのヨードアセチル - ビオチン使っecommend。 4SU-RNAの不可逆的なビオチン化でこの結果。したがって、カラムベースの方法( 例えば 、RNイージー)ビオチン化RNAの非常に小さい量( 例えば、5μgの)の回収に使用することができる。 RNAはビオチンHPDPを用いてビオチン化しながらも、このアッセイに適しており、結果として得られる信号が弱いと不利な信号対雑音比( 図3)である。
- セクション1と2に記載されているように4SU標識及び全細胞RNAを単離するためのプロトコルに従ってください。
- ヨードアセチル-ビオチンとビオチンHPDPを交換するセクション3で説明し、完全に過剰なヨードアセチル-ビオチン残渣を除去するために2つのクロロホルム抽出を行うようにビオチン化RNA 4SU標識。
- 記載またはRNAの場合、少量(<10μgのでカラムベースのアプローチを( 例えばをRNeasy)使用としてイソプロパノール/エタノール沈殿によりビオチン化RNAを回復)が使用されます。
- 10分間ロッキングとヌクレアーゼフリー水でゼータメンブレンをインキュベートする。
- ヌクレアーゼフリー水の膜を取り出し、両者の間にきれいなペーパータオルで膜を配置し、しっかりと押して、過剰な水分を除去する。空気乾燥で5分間膜するとよりよいのドットになります。
- 各サンプルについて、氷冷ドットブロット結合バッファー(10 mMのNaOHを、1mMのEDTA)を使用して、200 ngの/μlのRNAを20μlを準備します。ピペッティングによりゼータ膜にこの希釈液5μl(RNA すなわち 1μgの)と同様に3以降の10倍希釈( すなわち 、それぞれ100、10、および1 ngのRNAを、)を適用します。ピペットチップの空のラックを介してピペットを均等に分散間隔を提供するために使用することができる。あるいは、製造元の指示に従ってドットブロット装置を用いる。
- 正CONTとして20〜ng /μLの20 PG /μL( すなわち 0.1〜100 ngのオリゴ)の範囲の濃度でビオチン標識DNAオリゴを5μlを適用ピペッティングにより膜へROL。ネガティブコントロールとしてビオチン化、4SU-ナイーブサンプルを使用してください。
- 空気乾燥5分間メンブレン。
- ロッキングと40ミリリットルブロッキング緩衝液中で30分間メンブレンをインキュベートする。
- 15分(5 mlのPBS + 5ミリリットルの20%SDS +10μlのストレプトアビジン - ホースラディッシュペルオキシダーゼ)が1:1000ストレプトアビジン - ホースラディッシュペルオキシダーゼ10mlのメンブレンをインキュベート
- 5分間40ミリリットルPBS + 10%SDS(20ミリリットルPBS + 20ミリリットルの20%SDS)で二回膜を洗浄してください。
- 5分間40ミリリットルPBS + 1%SDS(38 mlのPBS + 2ミリリットルの20%SDS)で二回膜を洗浄してください。
- 5分間40ミリリットルPBS + 0.1%SDS(40ミリリットルPBS + 200μlの20%SDS)で二回膜を洗浄してください。
- 2清潔なペーパータオルの間に膜を置き、しっかりとそれらを押すことによって、過度の流体を除去。
- メーカーの指示に従ってECLを用いた膜結合HRPを可視化する。
- プラスチックフォイル/袋にメンブレンを置き、気泡を除去し、暗所で2分間インキュベートする。
- 膜にさらさ1用フィルム - 5分。
5。ストレプトアビジンコート磁気ビーズを用いて標識し、非標識RNAの分離
- 熱は℃の水浴中で65に(サンプル当たり3ミリリットル)洗浄バッファー。
- ヌクレアーゼフリーH 2 Oの中で新鮮な100mMのジチオスレイトール(DTT)を準備超微細スケールの上に置かれ、クリーン50ミリリットルファルコンチューブにDTTパウダー30mgの - 15デカントによってそう。ヌクレアーゼフリーのH 2 Oの必要量を計量して加える
- 熱は10分変性し、直ちに氷上に場所にするために、65℃にRNAサンプルをビオチン化。
- 磁気スタンドにμMACS列を置きます。 ( - 8サンプル最適である6)我々は一度に12以上のサンプルを処理しないことをお勧めします。
- 1ミリリットルの室温洗浄バッファーでミルテニー列を事前平衡。これは、約15分かかります。
- 一方、50にストレプトアビジンビーズを100μlを加える - ビオチン化RNAを100μl。回転で15分間室温でインキュベートする。 <リチウム>列のいずれかが、今では排出開始していない場合、これは穏やかに手袋をはめた指でカラムの頂部を押すことによって容易にすることができる。いったん流れが列が容易に排水を開始しました。
- 列にRNA /ビーズを適用します。フロースルーは未標識RNA画分を回収したい場合を除きます(セクション7を参照)捨てる。
- 65°Cの洗浄緩衝液(65℃のバッファをペッティングするとき1ミリリットルピペットチップが縮小℃)0.9ミリリットルで3回洗浄する。
- 0.9ミリリットルの室温洗浄バッファーで3回洗浄する。
- ピペット新しい2 mlチューブに700μlのBuffer RLT(をRNeasyのMinEluteクリーンアップキット、キアゲン)と列の下に配置します。
- 列にの100mM DTTを100μlを加えることによりRLTバッファに新たに転写されたRNAを溶出。
- の100mM DTTの別の100μlのを追加することで、同じチューブに3分後に2回目の溶出ラウンドを行う。
6。新たに転写されたRNAの回収
ontent ">製造者の指示に従ってのRNeasyのMinEluteクリーンアップ(キアゲン)プロトコルを続行します。光度計分光光度計を用いて25μlのヌクレアーゼフリーH 2 Oを測定RNA濃度の溶出。提出する前に、RNAを解凍し、再凍結する必要性を回避するためにそれは、ハイスループットアッセイに、我々は新たに転写されたRNAを精製された直後にcDNAを準備をお勧めします。製造元の指示に従ってcDNA合成のために、20μlのcDNA合成ミックスで新たに転写されたRNAの2.5μlを使用する1を用いて定量RT-PCRのコントロールを実行:cDNAのミックスの10希釈-80で保存RNA℃まで。7。ラベルなし、バインドされていないRNAの回収(オプション)
結合していない場合には、RNAを回収する必要があり、フロースルー(列にRNA-ストレプトアビジンビーズ溶液を添加した後)およびその後の沈殿のための最初の洗浄を収集し、組み合わせる。通常、それはTHIとして結合していないRNAの50%だけ沈殿するのに十分であるsが出発物質の> 80%が含まれています。
- 等体積のイソプロパノールを追加する(まだ塩が洗浄緩衝液は、すでに1MのNaClを含有するように添加する必要はない)。
- 4℃で20分間、20,000×gで遠心上清を捨てます。
- 、75%エタノールを等量を追加4℃で10分間℃で20,000×gで遠心分離し、上清を捨てる。
- 簡単にスピンし、200μlのピペットで残留エタノールを除去。
- 簡単にスピンし、20μlのピペットで残留エタノールを除去。
- RNAは乾燥させないでください。 100μlのH 2 Oに再懸濁し6回 - 5を上下にピペッティングしてよく混ぜる。振とうしながら10分間65℃でインキュベートし、氷に直接転送します。
- RNAの分解を除外するelectrophoretical分析によってRNAの品質を確認してください。
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
1。出発物質と期待利回り
4SU暴露の1時間(時間)に続いて新たに転写されたRNAは、約1を表す - 全細胞RNAの4%。彼らはもはや細胞増殖/複製を考慮してRNAを合成しないので、これは成長逮捕細胞で低くなります。全RNAの80μgの - 1時間ラベリングするとき、我々は60でアッセイを開始をお勧めします。ビオチン化工程の後に参照するのは難しいです小さなRNAペレットの総RNAの結果未満の30μgの始まるので、簡単に失われる可能性があります。入力RNAレベルは、ラベリングの非常に短い期間(10 - 10分間、 例えば 、5)のような多くの150のような目安に増加させることができる。 RNAラベリングの持続時間は、1時間に5 分〜60%〜80%、9〜新たに転写されたRNAが増加に短命のイントロン配列の寄与から短縮された場合。イントロンは、実質的に長いコード配列、ならびに5'-および3'-UTRを、新しく転写の量と比較される短期あるいは超短4SUタグ付け後に精製することができるRNAは、直線的に低下しない。このように、我々は、非接着性ヒトB細胞株9 4SUタギングの5分後の総RNAの> 0.5%を得た。しかしながら、標識4SUと若干長い期間、より高い濃度の付着細胞において同様4SU取り込み率を達成するために必要とされてもよいことに留意すべきである。たとえ低4SU-取り込み率が低いウリジン含有量の大きい、ウリジン豊富な転写産物の効率的な回収と精製、非常に短い転写を可能にしますが( 例えば miRNA)は高い4SU濃度(> 1 mM)を使用している場合であっても精製を脱出する可能性があります。 5 100ヌクレオチド(NT) - NIH-3T3マウス線維芽細胞では、200μM4SU暴露の1時間は50あたり4SU約一残基で新たに転写されたRNAのラベルが付いた。長さが1000 NT - これは転写産物> 500の高効率回収を許可する必要があります。したがって、我々は唯一のマイナーなトランスクリプトのサイズを観察したバイアスマウス線維芽細胞およびヒトB細胞の7の両方で4SU 200μMを使用して1時間ラベリング。 200μMの4SUの1時間、マウス線維芽細胞における細胞の転写産物レベルの任意の有意な変化をもたらさなかったが、≥200μMの4SUへの細胞の長期暴露は、24時間以内に測定可能な成長の赤字(未発表データ)に結果を行います。したがって、標識および採用4SU濃度の持続時間の両方が異所性又は毒性を回避するために最小にすべきである。新たに転写されたRNAを効率的に回収するために必要な最小限の4SU濃度を決定するための簡単な方法は4SU濃度の増加( - 1600μM 例えば 、50)で4SUラベリング後に新たに転写されたRNAを精製することである。として図2Aおよび図2Bに示すように、第一のヒト線維芽細胞で1時間ラベル新たに転写されたRNAの回収率は50〜200μM4SUから大幅に増加したが、その後高原に始めました。
2。ドット4SU定款ブロットの定量化(オプション)
場合によっては、トータルRNAの4SU配合量を測定するために重要であり得る。これは、最高のストレプトアビジンコンジュゲートを用いてビオチン化されたRNA上のドットブロット分析によって行われる。その化学的性質のためにヨードアセチルビオチンは、新たに転写されたRNAにおける実質的にすべての4SU残基のビオチン化で得られたビオチン-HPDPよりチオール基と反応性が高い。ビオチンHDPDように、ヨードアセチルビオチンが水溶性ではなく、ビオチン-HPDPを行って効率よくクロロホルム抽出により除去されることに注意することが重要である。したがって、同一の反応条件および濃度は、ビオチン-HPDPを使用する場合などに用いることができる。しかし、ヨードアセチル-ビオチンは可逆的ではありません。従って、列ベースのアプローチで新たに転写されたRNAの精製に使用することができない。ヨードアセチルビオチンの使用は4SU-取り込みを定量化することを可能にするが、ビオチン-HPDPに基づく測定の両方を検討4SU·定款及びビオチン化効率。同じサンプルに2ビオチン化試薬を採用するRNA-法人4SUのビオチン化効率の測定が可能になります。 4SU 標識RNA用ビオチンHPDPのビオチン化効率は、新たに転写されたRNAの唯一の約1 4SU 3の残基が実際にビオチンHPDP( 図3)により、ビオチン化されていることを示すヨードアセチル-ビオチンのそれよりも約3倍未満であると思われる。ビオチン化対照DNAオリゴサンプル信号強度を比較することにより、ビオチン化密度を測定することができる。ほとんどの哺乳動物細胞株の場合は正の信号がまだ200μM4SUラベルの1時間後にビオチン化RNA 10ngの中で検出する必要があります。弱いバックグラウンド信号は未標識RNAの最高濃度(1μgの)通常検出可能である。
3。新たに転写されたRNAの精製
新たに転写されたRNAの回収率は非常に泉ありtitative。同じRNA濃度で開始した場合は、すべてのサンプルのために新たに転写されたRNAの同じ量を得ることが期待できます。 OD 260の測定を妨げる可能性がある260 nmの(洗濯バッファ由来界面活性剤の存在) -多数のカラムベースアッセイと同様に、をRNeasyのMinEluteキットを使用して新たに転写されたRNAのコレクションは230で、追加の吸収になることがあります。各遠心分離工程のための新たな2 mlのコレクションチューブを使用する場合には、より少ない程度に見られる。それにもかかわらず、任意の不当に高いODの測定は、(>他の試料に比べ2倍以上)OD 280分の260比が<1.7アール場合は特に、慎重に考慮されるべきである。ダウンストリーム解析のためには、すべてのサンプルについて鋳型RNA量の同量を使用するのが最もことである。標識RNAの収量はelectrophoretical解析によるRNA分解の兆候期待チェックよりも低いケースでは。新たに転写されたRNAは、かなり大きな金額が含まれています典型的なrRNAのバンドが( 図4)あまり目立つことを持つ大規模な、スプライス転写産物の。
4。新たに転写されたRNAの定量
最後に、新たに転写されたRNAで4SUの混入率は、直接330 nmおよびOD 260分の330比率5,18で4SUの吸収極大に基づく分光分析により定量することができる。イソプロパノール/エタノール沈殿によって - これは、少量(20μlの10)に集中して標識RNAの> 3μgのを必要とします。ヌクレアーゼフリーグリコーゲン30μgの(フェルメンタス、#R0551)と小さなRNAペレット共沈を失うことを避けるために行われるべきである。追加のピークは新しく転写されたRNA( 図5)に4SUの取り込み率を反映した330 nmで表示されている。
/ files/ftp_upload/50195/50195fig1highres.jpg "/>
図1。 。全細胞RNAの準備が続く時間- 4 -チオウリジン(4SU)で代謝標識の原理 4SUは、必要(120分5)のために細胞に添加されています。チオール - 特異的ビオチン化の後、全細胞RNAを、ストレプトアビジンでコーティングされた磁気ビーズを用い4SU標識し、新たに転写されたRNA、および非標識、既存のRNAに分離される。新たに転写されたRNAは、ビーズに新たに転写されたRNAをリンクジスルフィド結合を切断する還元剤を用いてビーズから回収される。 より大きい数字を表示するには、ここをクリックしてください 。
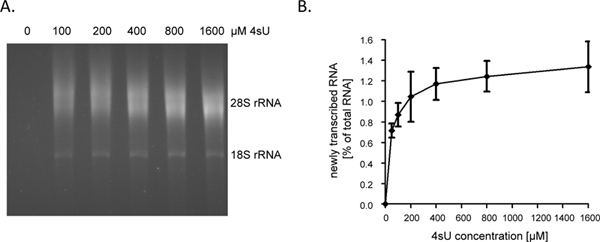
図2。続いて新たに転写されたRNAの回収 4SU の濃度を増加させ、(A)主要なヒト包皮線維芽細胞(HFF)は100と共にインキュベートし、4SU 200、400、800または1,600μM。新たに転写されたRNAは、50μgの全細胞RNAから精製し、electrophoretical分析を行った。予想どおり、回収新たに転写されたRNA濃度依存的増加が高濃度でプラトーに開始した観察された。(B)新たに転写されたRNAの精製の 量は1.45s ImageJのソフトウェアを用いて定量した。新たに転写されたRNAの量には4つの独立した実験の結合されたデータは、50に至るまで4SU標識、異なる濃度の後に回復した- 800μM4SU(N = 2)または100 - 1,600μM4SU(N = 2)が表示され、ここをクリックしてください大きい数字を表示する 。
upload/50195/50195fig3.jpg "ALT ="図3 "のfo:コンテンツ幅=" 4.5in "のfo:SRC =" / files/ftp_upload/50195/50195fig3highres.jpg "/>
図3。ドットブロット分析を用いて4SU標識したトータルRNAで4SU取り込みを推定する。総RNAは、一時間4SU 200μMと共にインキュベートNIH-3T3マウス線維芽細胞またはヒト包皮線維芽細胞(HFF)から単離した。まだ4SUは陰性対照として一皿に添加しなかった。 HFFのために両方の接点は(N =非増殖する細胞)を阻害し、増殖する細胞は、(y)は含まれていた。 RNAをトリゾール試薬およびビオチンHPDPまたはヨードアセチル-ビオチンにその後共役を用いて単離した。各サンプルの濃度はすべてあったが、200 ngの/μlのは、この希釈液5μlの(RNA すなわち 1μgの)、ならびに以降3回の10倍希釈( すなわち 100、10、および1 ngのRNA、それぞれ)に調整したゼータ膜の部分にスポット。ビオチン標識DNAオリゴ5μlの希釈をconcentratで陽性対照として膜上に置かれたng /μLのダウン20から20 PG /μL( すなわち 、それぞれ0.1〜100 ngの、)までイオン。ビオチン密度は、ストレプトアビジン - ホースラディッシュペルオキシダーゼコンジュゲートを用いてプローブした。
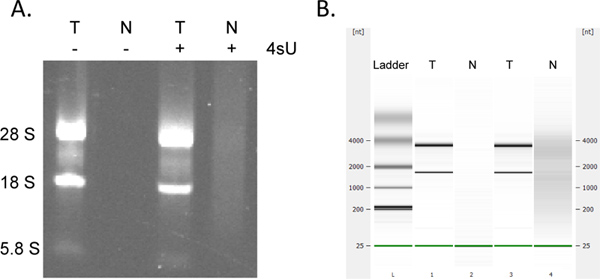
図4。新たに転写され、総RNAのElectrophoretical分析。総RNA(T)と新たに転写されたRNA(N)は 1時間をアガロースゲル電気泳動により分析したところ、500μM4SUの存在下および非存在下の両方で培養したマウスNIH-3T3線維芽細胞()から調製と(同じ順序で)アジレントBioanalyser(B)を使用。いいえRNAは細胞の4SU治療せずに回収されなかった。新たに転写されたRNAを精製して、高分子量のmRNAの量が大きいと合計よりも大幅成熟rRNAsを含んでいる28S、18S、および5.8S rRNAのバンドの間に顕著なようにRNA。 より大きい数字を表示するには、ここをクリックしてください 。
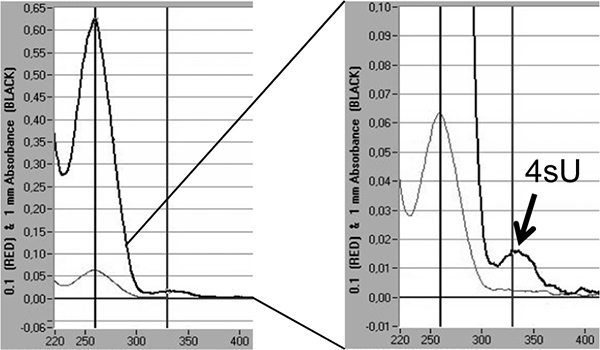
図5。分光分析によって新たに転写されたRNAで4SU取り込みの定量。マウスNIH-3T3線維芽細胞における4SU200μmの1時間後に2×100μgの全RNAから精製された新規転写されたRNA。新たに転写されたRNAは、ヌクレアーゼフリーグリコーゲン30μgのを追加した後イソプロパノール/エタノールで沈殿させた。ナノドロップ1000分光光度計で得られた新たに転写されたRNAの吸光光度分析が示されています。厚い、濃い灰色の線が1mm流体カラムで測定を表して薄い灰色線は0.1ミリメートルで測定を表す。 Tを表す絶滅のピークの右側に、倍率彼組み込ま4SU-残基が示されています。 4SU 18の共同効率的な消光をもとにして4SUの混入率を推定することができる。
| ラベル[分]の持続時間 | 推奨4SU濃度[μM] |
| 120 | 100から200 |
| 60 | 200から500 |
| 15から30 | 500 - 1000 |
| <10 | 500 - 2000 |
表1。 4SU 濃度を推奨。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
新たに転写されたRNAの代謝標識は、実質的に興味のある生物学的な問題に対処するためのより適切なテンプレートを提供することで、マイクロアレイとRNA-seqのようなハイスループット技術の能力を強化しています。現在のプロトコルでは、広範な最適化を行った。これは、新たに転写されたRNAの> 1,000倍濃縮を可能にし、再現性の高い結果を提供します。
新たに転写されたRNAは4SUへの細胞の曝露時間の間だけリアルタイム転写活性を描写するよう4SUタギング実験の実験計画は非常に重要である。刺激後に転写率の実際の変更は、すでに沈静化している場合は、合計RNAレベルでの変更がまだ検出可能であっても新たに転写されたRNAを解析する際に、これらは惜しまれることでしょう。したがって、基本的な生物学をよく理解は実験と同様に最適な期間Oを定義することが重要です4SU露出のためのF時間。以下では、お薦めなどの最も重要な手順については、一般的な落とし穴を避けるための方法を提供します。
原液とプラスチックの構造の作製
すべての原液をヌクレアーゼフリー水を用いて調製しなければならない。水は還元剤が含まれている場合、脱イオン水、精製、社内で使用すると問題が発生する可能性があります。あるケースでは、これはすべての標識されたRNAの完全な損失をもたらした。そこで、我々は強く既製ヌクレアーゼフリーのNaCl、トリス-CL、EDTA、クエン酸ナトリウムと水を買ってお勧めします。常時ヌクレアーゼフリーの条件を確認してください。ジメチルホルムアミド(DMF)は、いくつかのプラスチック材料を溶解する。我々は、ビオチン-HPDPストック溶液を調製し50ミリリットルファルコンチューブに株式のガラス瓶からDMFを転送することを25mlの細胞培養プラスチックピペットを使用して実質的に全部のアッセイから新しく転写されたRNAの収量を減少させるのに十分であることがわかった。興味深いことに、これは負のビオチニルに影響を及ぼさなかったATION効率(としてドットブロットによってテスト)が、ビーズから回収することができ、新たに転写されたRNAの75〜> 90%の損失をもたらした。標識の持続時間は60〜30分以下に低下したときに損失が最も顕著であった。ほとんどの場合、DMFによりプラスチックピペットから溶出した物質は、部分的にストレプトアビジンビーズのコーティングを破壊した。したがって、DMFと互換性があることが知られていないプラスチック材料の使用は、すべての手段によって回避されるべきである。同じ理由から、セルスクレイパーを細胞培養プレートからトリゾールサンプルの回収を向上させるために使用されるべきではない。それはDMFまたはトリゾールによってプラスチックから溶出推定物質が明らかにもクロロホルム抽出もイソプロパノール/エタノール沈殿で除去されなかったことに注意することは興味深い。
細胞培養
プレート上のセル密度が非常に重要である。 - 細胞が少しあまりにコンフルエントであるように思われた一つの実験(90100%)、我々はインターフェロンの100 U / mLで30分間NIH-3T3マウス線維芽細胞を治療(IFN)αまたはγ。少ないコンフルエントの細胞においてIFN治療のさえ15分は、すでに5の結果- IRF1またはSOCS3 5のような遺伝子の8倍の誘導に。細胞が少しあることとあまりにコンフルエントマイクロアレイ解析は、最も急速にIRF1またはSOCS3のような遺伝子を誘導するためであってもIFN-誘導性遺伝子の任意の誘導を示さなかった。従って、セル密度は4SU-標識実験のための重要な因子であり、すべての細胞培養プレートを慎重にラベリングを開始する前に検討すべきである。
4SUを効率的に365nmの光源への暴露後のタンパク質に架橋されている光活性リボと4SU含有RNAである。 4SU処理細胞は避けるべきで暗い部分と明るい光にさらされる中で培養されるべきである。トリゾールRNAの単離による細胞タンパク質を除去した後、このリスクを実質的に低減される。
<Pクラス= "jove_content"> 4SUは、細胞のDNAに組み込まれていません。しかしながら、トータルRNAは依然として細胞のDNAを少量含むことに留意すべきである。 4SU タギングを使用し、Q-RT-PCR解析サイトメガロウイルス感染でウイルス遺伝子発現を研究するときに我々はコンカテマーウイルスゲノム19を除去するためのプロトコルでDNアーゼIダイジェスト工程を含むことが必要と認められる。 DNAの存在に敏感ではない下流のプロトコルを使用している場合、これはおそらく必要ありません。4SU混入率と最適な4SU濃度
4SU は容易に最も可能性が高い分9,16未満内平衡内および細胞外レベルで細胞に取り込まれている。 4SUの取り込みと混入率は濃度に依存します。したがって、4SU濃度は便利標識採用期間に応じて調整することができます。 表1は、相対で4SU濃度にアドバイスを提供しています私たちの最高の個人的な経験に基づいてラベリングの期間にATION。哺乳動物細胞における4SU標識1時間、200μMの4SUは、線維芽細胞における新規転写されたRNAは50〜100ヌクレオチド当たり約一4SU渣の結果、ほとんどのアプリケーションで十分であろう。
ここ数年では、線維芽細胞、内皮細胞、上皮細胞、骨髄間質細胞、マクロファージやT細胞を含むヒトおよびマウス由来の細胞型の広い範囲に4SUタギングを適用している。また、 ショウジョウバエ及びアフリカツメガエル由来の細胞が正常に使用した。これらの実験の全てにおいて、4SU取り込みは異なる細胞型用4SU濃度の最小限の調整を必要とする非常に効率的であることが判明した。新しいセルの種類の方法を設定する場合、我々は4SU-濃度( 例えば 、50〜1600μMの範囲)の増加に伴って、ラベル細胞にお勧め、新たに転写された精製されたとの関係を分析するであろうR適用4SU-濃度NA( 図2A / Bを参照)。精製された新たに転写されたRNAの量がプラトーに入射する4SU濃度が選択されるべきである。
高度に合流、接触阻害細胞が使用されているケースでは、我々は効率的4SU取り込みを確実にするためにわずかに高い4SU濃度( 例えば 500の代わりに200μM)を使用することをお勧めします。また、非常に短い新たに転写された転写産物のキャプチャ(<200ヌクレオチド)が特に重要である場合には、4SU濃度も増加する必要があるかもしれません。これは、異所性の効果や毒性を避けるために、長時間のラベリング倍( 例:> 1時間)と組み合わせることはありません。最後に、細胞培養培地の体積が小さすぎるを使用する4SU取り込み効率を減少させることができることを見出した。したがって、我々はそれぞれ、5ミリリットルまたは10 cm以上を15cmディッシュあたり培地10mlを使用することをお勧めします。
全細胞RNAの調製
このプロトコルの成功のためには、クリーン、RNaseフリーの全細胞RNAを得ることが重要です。を15cmディッシュあたり5ミリリットルトリゾールを使用するヌクレアーゼの自由きれいなRNAを生成します。私たちは、Chomczynski らによって修正されたトリゾールプロトコルを使用することをお勧めします。20。まず、良好な洗浄工程の間に取り扱いが容易である硬くペレット中の強化された遠心力の結果として、多量のRNA(> 100μgの)を単離するのに適している。正規の15ミリリットル実験ファルコンチューブは6,000×gでより多くを生きていけないようにしかし、これは特殊なポリプロピレンチューブやアダプタを使用する必要があります。第二に、DNAおよび糖タンパク質の除去を改善する。器官または組織からのRNAを調製する場合に特に明らかになる。第三に、単離することができるトータルRNAの最大量を限定するものではない。我々はまた、適切な品質のRNAを提供するために、列ベースのRNA単離方法( 例えばをRNeasy)、標準列ARを見つけましたが、電子のみにより出発物質の量を制限する全RNA100μgのまでキャプチャすることができ。最後に、残留エタノールを除去するために、ピペットで二回、残りのエタノールを除去したRNAの乾燥によって、もはや必要ありません。これは、その後再び溶解することが困難であるRNAを、過乾燥の危険性を排除する。原則として、4SUタグ付けは、マウスの静脈内注射によって例えば 、 生体内で適用されます。しかし、RNAの純度が新たに転写されたRNA(未発表データ)の精製の前にポリA転写産物の精製を必要とする主要な問題を表していることに留意。
ビオチンと結合していないビオチンの除去
ビオチン-HPDPは、100%チオール固有であり、ビオチン残基とチオール標識されたRNA転写さ新規分子間ジスルフィド結合を形成する。 4SU 標識されたRNAのビオチン化効率は、ドットブロット分析によって決定された5〜約30%である。ビオチン-HPDPは水溶性ではないのでそれは効率的にクロロホルム抽出により除去することができる。シングルクロロホルム抽出ステップは、非結合ビオチンの大半を除去するのに十分ですが、我々は定期的に完全な除去を確実にするためにこの手順を繰り返します。クロロホルム抽出ステップ2 mlのフェーズロックゲルヘビーチューブ(エッペンドルフ)中にRNAの損失を減らすために、製造業者の指示に従って使用することができる。最初のステップのテンプレートボリュームはしばしばこれらのチューブと直接互換性があることが高すぎるとして、通常、我々は唯一の第二クロロホルム抽出工程に対する位相ロックチューブを使用しています。非結合ビオチン-HPDPを除去した後、RNAをイソプロパノール/エタノール沈殿によって回収される。それは、彼らが提供バッファにどの裂ジスルフィド結合を還元剤を含んでおり、新たに転写されたRNAからビオチンを除去するように使用すべきではないビオチン化RNAを(QIAGEN などからのRNeasy)を回復するために、その商用カラムベースのキットを注意することが重要です。
新しいの精製LY転写されたRNA
100μlのストレプトアビジンビーズに100以上μlのビオチン化RNAを追加しないでください。より少ない量を添加することが好ましい。しかしながら、RNA同量の全てのサンプルのために添加されるべきである。あなたは、単にビーズに1×TEの必要量を追加することで、ストレプトアビジンビーズに追加RNAの入力音量を(サンプル間)を調整します。新鮮なヌクレアーゼフリー100mMのDTTを作るための簡単な方法は、超高感度スケール上に配置さファルコンチューブにDTT粉末の十分な量をデカントし、その後の100mM DTTを生成するためにヌクレアーゼフリーのH 2 Oの必要量を添加することである(1mgのDTTあたり64.8μlの水)。
4SUタギングの開発中に様々なサプライヤーからストレプトアビジンビーズをテストしました。それらの数は、バックグラウンドを大量に生成された。そこで、我々は強く、これまでのところ、我々は組織Cから未標識RNAのキャリーオーバーの問題を経験したことのない、ミルテニーストレプトアビジンビーズとして使用することをお勧めしますulture由来RNAサンプル。このように、標識RNAのわずか150 ngのは、特にストレプトアビジンビーズを100μlを用いて150μgのビオチン化RNA(水100μl中)から精製することができる。ビーズを供給平衡緩衝液で平衡化ビーズを行ってもよいし、わずか13捕獲率を向上させることができる。
品質管理
我々は、高スループット分析のためにそれをかける前に、新たに転写されたRNA上のQ-RT-PCRコントロールを行うお勧めします。これにより、差動的に与えられた実験的な設定で調節されることが知られているいくつかのリファレンス遺伝子の定量を含んでもよい。 4SU タグ付けがRNAの減衰率を研究するために採用されているケースでは、我々は短命トランスクリプト( 例えば MYC、FOS)と合計し、新たに転写された両方のRNA中の長寿命1( 例えば GAPDH)を定量化することをお勧めします。新たに転写された/全RNAの比( - 〜10倍〜5)は、実質的に高くする必要があります短命の転写産物のために。参照遺伝子のRNA半減期をもとに、RNAの半減期を決定することができる。 3つすべてのRNA画分(トータルRNA、新たに転写されたRNA及び非標識既存RNA)が四つ以上の遺伝子が分析されている場合、異なるRNAサブセットの正規化は、線形回帰分析によって行うことができる、図7に記載されるような品質管理のスコアを決定することができる、21。
Q-RT-PCR解析のために、我々は20μlのcDNA合成ミックスの標識RNA2.5μlの使用をお勧めします。 Q-RT-PCRの結果の最適な比較のために最初に使用する前に、5μLのアリコート中のcDNAを凍結。使用直前に解凍管は、H 2 Oの45μlのとq-RT-PCR解析に希釈の対象5μLを加える。これは、大幅に異なるPCRラン間の比較可能性を向上させます。
新たに転写されたRNAサンプルは、それらを施す前に、AgilentのBioanalyserを使用してRNA分解の兆候をチェックする必要がありますハイスループット分析(マイクロアレイまたはRNA-seqの)。しかしながら、追加の帯域が時々アジレントBioanalyserによって観察されることに留意すべきである。この生物学的意義は不明である。新たに転写されたRNAは、大幅に少ないリボソームRNAが含まれているように、これらのサンプルは、時折アジレントBioanalyser品質コントロールを失敗する。これは許容可能な品質の可視RNA分解サンプルによるものでない場合は、通常、ハイスループット分析に供される微細である。
ダウンストリーム分析と新たに転写されたRNAの互換性
新たに転写されたRNAは、トータルRNAよりも実質的mRNAが含まれています。これは4SUタグ付けの期間が短縮されたときに増加新しく転写されたRNAにおけるイントロン配列を多量によるものである。これは出発物質の多量を必要とするため、我々は定期的に新たに転写されたRNAサンプルからrRNAsの枯渇を負わないものとしながら、提供します:非rRNAのでりむしろ少し(〜倍)ゲインが読み込まれます。最後に、次世代シーケンスのためのcDNAライブラリーを調製する際に、新たに転写されたRNA中に存在するスプライス、高分子量転写物の大きな割合が、追加断片化を必要とするかもしれないことに留意されていない。サイズの断片化のステップの結果は、したがって、品質を慎重に制御する必要があります。
RNA半のライブの測定のためのデータの正規化
RNA半減期の測定のための実験データを正規化するための標準的なアプローチは、以前の実験で決定された特定の細胞型で十分に特徴付けられたハウスキーピング遺伝子又はメジアンRNAの半減期のRNAの半減期にすべてのデータを正規化することである。哺乳動物細胞において、後者は、5〜10時間6,7の範囲にある。このアプローチはまた4SUベースの測定に非常にうまく動作しますが中央のRNAの半減期は、MAを知られているか、そればされていない場合は、正規化のための他の手段が必要とされるyがさえノックアウトRNA崩壊経路のことで、たとえば 、研究対象のセルラーシステムの変化に影響される。 4SU タグ付けは、3つすべてのRNA画分、 すなわち全細胞RNA、新たに転写されたRNA、およびラベルのない既存のRNAの分析に基づいて、中央値RNAの半減期を推定するユニークな方法を提供しています。全細胞RNAのような単純な線形回帰モデルを互いに、3つのRNA画分を正規化し、メディアンRNA半減期7,16を決定するために使用することができる後者の二つのRNA画分に分離される。ソフトウェアパッケージには、これらの分析22を実行するために、オンラインで入手可能です。
低ウリジン含有量の転写産物の非効率的なキャプチャが人為的に低く、新たに転写された/全RNA比と延長されたRNAの半減期で得られたRNAの半減期の測定値に影響を与える可能性があります。この問題の程度はuridiに対してRNAの半減期またはログ(新規転写/全RNA比)をプロットすることによって評価することができるすべての転写産物7,15のね内容。これはまた、異なるサンプルや条件間4SU-混入率の差を評価するために、良好な品質管理を提供します。ウリジンコンテンツへの実質的な相関が観察される場合、これは、バイオインフォマティクス手段15によって補正することができる。しかし、新たに転写されたRNA転写物で成熟の寄与を容易にはるかに大きく、したがって、はるかウリジンリッチ前駆体から区別することができないことに留意すべきである。与えられたトランスクリプトの処理速度論は、単に低ウリジン含有量(非効率的なキャプチャ)を補正すると、肉眼的RNAの半減期を歪めるかもしれない(彼らは通常ではありませんが)が知られていない限り。このように、我々は最近9非常に非効率的であることがほとんどの人間snoRNAsの処理を発見した。我々はかなり小さい(70から300 NT)の低いウリジンコンテンツに新たに転写された/全RNA比を修正していた場合snoRNAs、これは非常に短いsnoRNA半リヴをもたらしたであろう100%を超える多数の新しく転写された/トータルRNA比がエス(<5分)。したがって、我々は一般的にRNAの半減期を測定するとき、低ウリジン含有量を補正することはお勧めしません。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
著者は、彼らが競合する経済的利益を持っていないことを宣言します。
Acknowledgments
私たちは、原稿を注意深く読むためアミエリーガンに感謝したいと思います。この作品は、CCFにLDとDFG助成FR2938/1-1にNGFNプラス助成金#01GS0801、MRCフェローシップ助成G1002523とNHSBT助成WP11-05でサポートされていました
Materials
| Name | Company | Catalog Number | Comments |
| 4-thiouridine | Carbosynth | T4509 | Prepare 50 mM stock in sterile H2O, store at -20 °C in aliquots of 50-500 μl, discard unused reagent, do not refreeze. |
| Trizol | Invitrogen | 15596026 (100 ml), 15596018 (200 ml) | WARNING - CORROSIVE and HAZARDOUS TO HEALTH! Ensure immediate access to Phenol antidote (PEG-Methanol); Store at 4 °C. |
| Chloroform | Sigma | 372978 | WARNING - HAZARDOUS TO HEALTH |
| Isopropanol | Sigma | 650447 | |
| Sodium citrate, nuclease-free | Sigma | C8532 | Prepare 1.6 M stock solution using nuclease-free water. |
| 5M nuclease-free NaCl | Sigma | 71386 | Stock solution |
| Nuclease-free H2O | Sigma | W4502 | Make 1 ml aliquots in nuclease-free tubes. |
| RNA precipitation buffer | 1.2 M NaCl, 0.8 M sodium citrate in nuclease free water. Prepare in advance under strictly nuclease-free conditions. Store at room temperature in 50 ml falcon tubes. | ||
| Ethanol | Sigma | 459844 | Use with nuclease-free water to prepare 80% ethanol, store at -20 °C. |
| 1 M nuclease-free Tris Cl, pH 7.5 | Lonza | 51237 | Stock solution |
| 500 mM nuclease-free EDTA, pH 8.0 | Invitrogen | 15575-020 | Stock solution |
| 10x Biotinylation Buffer (BB) | 100 mM Tris pH 7.4, 10 mM EDTA in nuclease-free water, make aliquots of 1 ml. | ||
| Dimethylformamide (DMF) | Sigma | D4551 | |
| EZ-Link biotin-HPDP | Pierce | 21341 | Prepare 1 mg/ml stock solution by dissolving 50 mg biotin-HPDP in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. |
| Phase Lock Gel Heavy tubes 2.0 ml | Eppendorf | 0032 005.152 | Optional for the chloroform extraction step. |
| Zeta membrane | BIORAD | 162-0153 | |
| 10x Dot blot binding buffer | 100 mM NaOH, 10 mM EDTA | ||
| Biotin-oligo | 5'-biotin, 25 nucleotides, any sequence | ||
| Sodium dodecyl sulphate | Fisher | BPE9738 | For 100 ml 20% stock solution, add 20 g SDS to 80 ml PBS pH 7-8 and adjust volume to 100 ml. Keep all high-percentage SDS solutions above 20 °C. Warm the solutions slightly should SDS precipitate. |
| EZ-Link Iodoacetyl-LC-Biotin | Pierce | 21333 | Prepare 1 mg/ml stock solution by dissolving 50 mg iodoacetyl-biotin in 50 ml DMF. Gentle warming enhances solubilisation. Store at 4 °C in aliquots of 1 ml. Generates irreversible, thiol-specific biotinylation. |
| Phosphate buffer saline | Gibco | 10010-015 | |
| Dot blot blocking buffer | Mix 20 ml 20% SDS with 20 ml 1 x PBS pH 7-8 and add EDTA to the final concentration of 1 mM. | ||
| Streptavidin-horseradish peroxidase | Vector Laboratories | SA5004 | Store at -20 °C. Mix 10 ml 20% SDS with 10 ml 1 x PBS. Add 20 μl Streptavidin-HRP before use. |
| ECL reagent | GE Healthcare | RNP2109 | Use following the manufacturer's instructions. |
| Super RX, X-RA Film, 18x24 cm | Fujifilm | 47410 19236 | |
| μMacs Streptavidin Kit | Miltenyi | 130-074-101 | Store the beads at 4 °C. |
| Tween 20 | Sigma | P1379 | |
| Washing buffer | 100 mM Tris pH 7.4, 10 mM EDTA, 1 M NaCl, 0.1% Tween 20 in nuclease-free H2O. | ||
| Dithiothreitol (DTT) | Sigma | 43817 | Prepare as 100 mM DTT in nuclease-free H2O, always prepare fresh before use. |
| RNeasy MinElute Kit | Qiagen | 74204 | Store columns at 4 °C, remaining components of the kit at room temperature. |
| 1.5 ml screw-top polypropylene tubes | Sarstedt | 72.692.005 | Compatible with Dimethylformamide |
| 2.0 ml screw-top polypropylene tubes | Sarstedt | 72.694.005 | Compatible with Dimethylformamide |
| 15 ml tubes | BD Falcon | 352096 | Compatible with Dimethylformamide |
| 50 ml tubes | BD Falcon | 352070 | Compatible with Dimethylformamide |
| All solutions/reagents should be stored at room temperature unless otherwise specified. | |||
| Equipment | |||
| UV/VIS spectrophotometer | Thermo Scientific | NanoDrop 1000 | Or equivalent. Use low volume (1-2 μl) for measurements of low RNA concentrations to avoid excessive sample loss. |
| Polypropylene 15 ml centrifuge tubes | VWR International | 525-0153 | In contrast to standard 15 ml tubes, these tolerate up to 15,000 × g |
| High-speed centrifuge | Beckman Coulter | Avanti J-25 | Or equivalent equipment capable of reaching 13,000×g |
| High-speed rotor | Beckman Coulter | JLA-16250 | Or equivalent equipment capable of reaching 13,000×g |
| Adaptors for 15 ml tubes | Laborgeräte Beranek | 356964 | Or equivalent equipment capable of reaching 13,000×g |
| Refrigerated table-top centrifuge | Eppendorf | 5430 R | Or equivalent. |
| Thermomixer | Eppendorf | Thermomixer compact | Or equivalent. |
| Magnetic stand | Miltenyi Biotec | 130-042-109 | One stand holds 8 μMacs columns. |
| Waterbath | Grant | SUB Aqua 5 | Or equivalent. |
| Ultra-fine scale | A&D | GR-202 | Or equivalent. |
| E-Gel iBase Power System | Invitrogen | G6400UK | For RNA gels; or equivalent. |
| E-Gel EX 1% agarose precast gels | Invitrogen | G4020-01 | For RNA gels; or equivalent. |
References
- Brown, P. O., Botstein, D. Exploring the new world of the genome with DNA microarrays. Nature Genetics. 21, 33-37 (1999).
- Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods. 5, 621-628 (2008).
- Nagalakshmi, U., Wang, Z., et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 320, 1344-1349 (2008).
- Wilhelm, B. T., Marguerat, S., et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 453, 1239-U1239 (2008).
- Dölken, L., Ruzsics, Z., et al. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 14, 1959-1972 (2008).
- Yang, E., van Nimwegen, E., et al. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res. 13, 1863-1872 (2003).
- Friedel, C. C., Dölken, L., Ruzsics, Z., Koszinowski, U. H., Zimmer, R. Conserved principles of mammalian transcriptional regulation revealed by RNA half-life. Nucleic Acids Res. 37, e115 (2009).
- Nilsen, T. W., Graveley, B. R. Expansion of the eukaryotic proteome by alternative splicing. Nature. 463, 457-463 (2010).
- Windhager, L., Bonfert, T., et al. Ultra short and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res. , (2012).
- Melvin, W. T., Milne, H. B., Slater, A. A., Allen, H. J., Keir, H. M. Incorporation of 6-thioguanosine and 4-thiouridine into RNA. Application to isolation of newly synthesised RNA by affinity chromatography. Eur. J Biochem. 92, 373-379 (1978).
- Cleary, M. D., Meiering, C. D., Jan, E., Guymon, R., Boothroyd, J. C. Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nature Biotechnology. 23, 232-237 (2005).
- Kenzelmann, M., Maertens, S., et al. Microarray analysis of newly synthesized RNA in cells and animals. Proc. Natl. Acad. Sci. U.S.A. 104, 6164-6169 (2007).
- Schwanhäusser, B., Busse, D., et al. Global quantification of mammalian gene expression control. Nature. 473, 337-342 (2011).
- Rabani, M., Levin, J. Z., et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat. Biotechnol. , (2011).
- Miller, M. R., Robinson, K. J., Cleary, M. D., Doe, C. Q. TU-tagging: cell type-specific RNA isolation from intact complex tissues. Nat. Methods. 6, 439-441 (2009).
- Miller, C., Schwalb, B., et al. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol. Syst. Biol. 7, 458 (2011).
- Weintz, G., Olsen, J. V., et al. The phosphoproteome of toll-like receptor-activated macrophages. Mol. Syst. Biol. 6, 371 (2010).
- Lipsett, M. N. The isolation of 4-thiouridylic acid from the soluble ribonucleic acid of Escherichia coli. Journal of Biological Chemistry. 240, 3975-3978 (1965).
- Marcinowski, L., Liedschreiber, M., et al. Real-time Transcriptional Profiling of Cellular and Viral Gene Expression during Lytic Cytomegalovirus Infection. PLoS Pathog. 8, e1002908 (2012).
- Chomczynski, P., Mackey, K. Short technical reports. Modification of the TRI reagent procedure for isolation of RNA from polysaccharide- and proteoglycan-rich sources. Biotechniques. 19, 942-945 (1995).
- Friedel, C. C., Dölken, L. Metabolic tagging and purification of nascent RNA: implications for transcriptomics. Mol. Biosyst. 5, 1271-1278 (2009).
- Friedel, C. C., Kaufmann, S., Dölken, L., Zimmer, R. HALO - A Java framework for precise transcript half-life determination. Bioinformatics. , (2010).
