

Summary
Denne protokol beskriver en ikke-invasiv metode til effektivt at identificere S-fase celler til downstream mikroskopi undersøgelser, såsom måling AF DNA reparation protein rekruttering ved laser mikro-bestråling.
Abstract
DNA-skade reparation opretholder den genetiske integritet af celler i et meget reaktivt miljø. Celler kan akkumulere forskellige typer af DNA-skader på grund af både endogene og udefrakommende kilder såsom metaboliske aktiviteter eller UV-stråling. Uden DNA-reparation bliver cellens genetiske kode kompromitteret, underminerer proteiners strukturer og funktioner og forårsager potentielt sygdom.
Forståelse af de forskellige DNA-reparationsvejes spatiotemporale dynamik i forskellige cellecyklusfaser er afgørende inden for dna-skadesreparation. Nuværende fluorescerende mikroskopi teknikker giver gode værktøjer til at måle rekruttering kinetik af forskellige reparation proteiner efter DNA-skader induktion. DNA-syntese i cellecyklussens S-fase er et ejendommeligt punkt i celle skæbne med hensyn til DNA-reparation. Det giver et unikt vindue til at screene hele genomet for fejl. Samtidig udgør DNA-syntesefejl også en trussel mod DNA-integriteten, som ikke opstår i ikke-dividerende celler. Derfor adskiller DNA-reparationsprocesser sig betydeligt i S-fasen sammenlignet med andre faser af cellecyklussen, og disse forskelle er dårligt forstået.
Følgende protokol beskriver forberedelsen af cellelinjer og måling af dynamikken i DNA-reparation proteiner i S fase på lokalt induceret DNA-skader steder, ved hjælp af en laser-scanning konfokal mikroskop udstyret med en 405 nm laser linje. Tagged PCNA (med mPlum) bruges som cellecyklusmarkør kombineret med et AcGFP-mærket reparationsprotein af interesse (dvs. EXO1b) til at måle rekrutteringen af DNA-skader i S-fasen.
Introduction
Flere DNA-reparationsveje har udviklet sig til at adressere de forskellige typer DNA-læsioner, der kan opstå i celler, som alle er stærkt reguleret i både rum og tid. En af de mest sårbare perioder i cellecyklussen er S-fase, når DNA-syntese opstår. Selv om spredning er grundlæggende for livet, er det også en stor udfordring. Celler skal sikre trofast replikation af deres genom for at undgå mutationer, der skal videregives til fremtidige generationer. Derfor er spredning et terapeutisk interventionspunkt, der er blevet anvendt til udvikling af terapeutiske tilgange inden for onkologi.
Alle de vigtigste teknikker, der anvendes til at studere protein rekruttering på DNA-læsioner har deres styrker og begrænsninger. Mikrobestråling har bedre rumlig og tidsmæssig opløsning1 end de fleste af de alternative metoder som immunfluorescent billeddannelse af imiterende strålingsfremkaldt foci (IRIF), kromatin-immunprecipitation (ChIP) eller biokemisk fraktionering. Mikrobestrålingen giver imidlertid robustheden af de ovennævnte teknikker, der kan prøve et stort antal celler på samme tid.
For at undersøge DNA-reparation i S-fase skal man kunne skelne S-faseceller i en asynkron cellekulturpopulation. Der er mange velkendte metoder til at løse dette, der involverer enten synkronisering af celler eller visualisering af de forskellige cellecyklusfaser. Begge tilgange giver dog betydelige udfordringer og mulige genstande. Kemiske synkroniseringsmetoder, der i vid udstrækning anvendes til at berige celler i den tidlige S-fase (f.eks. dobbelt thymidinblok, bladlusicolin og hydroxyurea-behandling) opnår synkronisering gennem induktion af replikationsstress og i sidste ende DNA-skader i sig selv. Dette begrænser brugen af disse metoder til at studere DNA-reparationsprocesser i S fase2. Synkronisering gennem serum sult og frigivelse gælder kun for et begrænset antal cellelinjer, stort set eksklusive kræft cellelinjer, der er mindre afhængige af vækstfaktorer for celle-cyklus progression i forhold til ikke-transformerede cellelinjer. Fluorescens Ubiquitin Cell Cycle Indicator (FUCCI) systemet er et særligt nyttigt værktøj til at studere cellecyklussen, men det har en grundlæggende begrænsning, når der skelnes mellem S- og G2-cellecyklusfaser3.
Her er det vist, at ved hjælp af fluorescerende mærkede PCNA som en ikke-invasiv markør for S fase begrænser ulemperne ved kemiske celle-cyklus synkroniseringsmetoder, samtidig med at der er mulighed for mere specificitet og fleksibilitet end FUCCI-systemet. Som en enkelt markør kan PCNA ikke kun fremhæve S-faseceller i en asynkron population, men det kan også vise den nøjagtige progression af celler inden for S-fase (dvs. tidlig, midt eller sen S-fase)4. Lave ekspressionsniveauer af eksogent, mærket PCNA sikrer minimal interferens med både cellecyklus progression og DNA-reparationsprocesser. Det er vigtigt, at PCNA også fungerer som en intern kontrol for korrekt induktion af DNA-skader, da det er involveret i reparation af flere DNA-læsioner og rekrutteres til lokalt inducerede DNA-skadessteder1,4.
De eksperimenter, der præsenteres her, viser, hvordan man måler rekrutteringsdynamikken i EXO1b i S-fasen, og hvordan dette påvirkes af den veletablerede PARP-hæmmer, olaparib. EXO1b nucleaseaktivitet er relevant for en lang række DNA-reparationsveje, herunder reparation af mismatch (MMR), nukleotid excision repair (NER) og dobbeltstrenget pause (DSB) reparation. I S-fasen spiller EXO1b en stor rolle i homolog rekombination (HR) gennem dannelsen af 3' sDNA-udhæng under DNA-resektion5. EXO1b har været yderligere impliceret i DNA-replikation med roller i checkpoint aktivering for at genstarte gået i stå DNA gafler samt primer fjernelse og Okazaki fragment modning på halter streng under streng forskydning i replikation5. EXO1b-rekruttering til beskadigede DNA-steder reguleres af den direkte interaktion med poly (ADP-ribose) (PAR)6,7. På grund af de mange cellecyklusspecifikke konsekvenser af EXO1b er det et glimrende valg for S-fase specifikke rekrutteringsstudier ved hjælp af PCNA.
Protocol
1. Dyrkning af humane osteosarcoma-afledte celler (U-2 OS)
BEMÆRK: U-2 OS celler er ideelle til disse undersøgelser, da de har en flad morfologi, stor kerne og stærkt knyttet til flere overflader, herunder glas. Andre cellelinjer med lignende egenskaber kan også anvendes.
- Til dyrkning af U-2 OS-cellelinjer skal du bruge McCoys 5A-medium suppleret med 10% fosterkvægsserum (FBS) og antibiotika (100 U/mL penicillin og 100 μg/mL streptomycin). Inkuberes ved 37 °C i en befugtet atmosfære, der indeholder 5 % CO2. Til mikroskopiundersøgelser skal cellekulturen i en 10 cm skål for at give tilstrækkeligt celletal.
- Når celler nærmer sig 90% sammenløb (~7 x 106 celler/10 cm parabol), opdele cellerne.
- Skyl celler med PBS for at vaske trypsinhæmmere, der er indeholdt i serumet, væk.
- Tilsæt 1 mL Trypsin-EDTA og sørg for, at cellelaget er lige dækket.
- Inkuberes ved 37 °C, indtil cellelaget løftes af pladen (ca. 6 min).
- Brug de trypsiniserede celler i serum, der indeholder medier, til at inaktivere trypsin og tilføje 1/10th af volumen (~0,7 x 106 celler) i en ny 10 cm plade indeholdende 10 cm plade indeholdende 10 mL suppleret vækstmedium.
- Før forsøg, rutinemæssigt teste celler for mycoplasma forurening ved hjælp af Universal Mycoplasma Detection kit efter producentens anbefaling.
2. Retroviral infektion
BEMÆRK: For BSL-2 sikkerhedsforanstaltninger og mens du arbejder med rekombinante vira henvises til: NIH Retningslinjer, afsnit III-D-3: Rekombinant virus i vævskultur.
- Seed 4 x 106 HEK293T celler for at opnå ~ 60% sammenløb inden for 24 timer efter plating i en 10 cm kultur skål.
- For dyrkning hek293T følg dyrkning trin af U-2 OS beskrevet i 1.1-1.3 i denne protokol. For HEK293T erstatte McCoy's 5A medium for DMEM. Sørg for altid forsigtigt at vaske HEK293T-celler, når de fastgøres svagt til vævskulturplader.
- Transfect HEK293T celler ved hjælp af en lipid-baseret transfection reagens til viral emballage af plasmid.
- For retrovirale vektorer kombineres 1,5 μg VSV-G (Addgene #8454) og 1,5 μg pUMVC (Addgene #8449) emballagevektorer sammen med 3 μg af vektoren, der indeholder interessegen (i en retroviral vektor rygrad med puromycinresistens) i 250 μL Opti-MEM reducerede serummedier i et mikrocentrifugerør. Der tilsættes 1 μL P3000 reagens for hvert μg DNA, der tilsættes opti-MEM/DNA-blandingen (i dette tilfælde 6 μL), og blandes forsigtigt ved tapning. Hvirvle eller pipette op og ned.
- I et andet mikrocentrifugerør kombineres 2 μL pr. μg DNA (i dette tilfælde 12 μL) transfectionreagens med 250 μL Opti-MEM reducerede serummedier.
- Kombiner de to blandinger (500 μL kombineret, ikke vortex, kun blandes ved blid tapning) og lad det inkubere i 15 minutter ved stuetemperatur.
- Tilsæt forsigtigt blandingen dropwise til de seedede HEK293T-celler uden at løsne cellerne. Hvirvle pladerne forsigtigt.
- Virusinfektion for at generere stabile cellelinjer.
- Fjern viruset, der indeholder supernatant, fra HEK293T-cellerne 72 timer efter transfektion. Filtrer forsigtigt opløsningen med et 0,45 μm filter for at fjerne cellerester og løsrevne celler. Eventuelt tilsættes 8 μg/mL polybren til virus supernatant at lette virusinfektion.
- Tilføj virus, der indeholder supernatant til U-2 OS-celler ved ~ 50% sammenløb i en 10 cm skål (~ 3 x 106 celler). Seed U-2 OS celler dagen før.
- Infekt i 6-16 timer, før du fjerner og kasserer det virusholdige supernatant.
BEMÆRK: For at opnå den ønskede mængde overekspression for interessegen inkuberes en række virale fortyndinger i en bestemt mængde tid. Kontroller udtryksniveauerne for transgene i hver nyoprettet cellelinje med western blot, der sammenligner det med endogene niveauer. - Lad cellerne vælge i nærværelse af passende antibiotika (i 3-4 dage i tilfælde af puromycin ved 2 μg/mL endelig koncentration) og kontrollere ekspressionen af det fluorescerende proteinmærkede gen af interesse under et mikroskop.
- Gentag disse trin for at generere dobbeltmærkede cellelinjer. I de eksperimenter, der præsenteres her mPlum-PCNA blev udtrykt fra en retroviral vektor (pBABE) kombineret med EXO1B-AcGFP, også udtrykt fra en retroviral vektor (pRetroQ-AcGFP1-N1).
3. Fremstilling af celler til mikrobestråling
- Platingceller: 24 timer før forsøget, plade i alt 8,0 x 104 celler i et volumen mellem 500 μL-1 mL medier (for ca. 70% sammenløb) på en fire godt chambered coverglass med en No. 1.5 borosilicat glas bund, som leverer ideelle resultater for høj forstørrelse konfokal mikroskopi og laser mikro-bestråling. Et højere cellekonfluent giver mulighed for flere celler målt i et enkelt synsfelt (FOV); men fuldt sammenløbslidt dias vil indføre cellecyklus uregelmæssigheder.
- Billedbehandlingsmedier: En time før mikrobestråling udveksles regelmæssigt vækstmedium for FluoroBrite DMEM suppleret med 10% FBS, 100 U/mL penicillin og 100 μg/mL streptomycin, 15 mM HEPES (pH=7,4) og 1 mM natrium pyruvat. Dette billedmedie hjælper med at maksimere signal-støj-forholdet, hvilket gør det muligt at registrere meget svag fluorescens. Da den indeholder HEPES, stabiliserer den også pH i mangel af en CO2-atmosfære på 5%.
- Påfør yderligere behandling før billeddannelsen på dette trin. I de forsøg, der præsenteres her, blev cellerne forbehandlet en time før billeddannelse med enten olaparib (PARP-hæmmer, ved 1 μM endelig koncentration) eller en køretøjskontrol (DMSO)1,8,9.
4. Forberedelse af mikroskopet og valg af S-faseceller til billeddannelse.
- Brug et konfokalt system, der har de samme egenskaber som det system, der er skitseret her, for at opnå de bedste resultater. De eksperimenter, der præsenteres her, blev udført ved hjælp af et konfokalt mikroskop monteret på et omvendt mikroskopstativ (se Materialetabel).
BEMÆRK: Det mikroskop, der anvendes her var udstyret med en 50 mW 405 nm FRAP laser modul, og en 60x 1,4 NA olie plan-apochromat mål. Den konfokale scanhead havde to scanner muligheder: en galvano scanner (for høj opløsning) og resonant scanner (for high-speed imaging).- Indfør fluorescensgenopretning efter fotobleaching (FRAP) laser til prøven via en softwarestyret XY galvano-enhed. Brug en 488 nm laserlinje til at begejstre AcGFP og en 561 nm eller 594 nm laserlinje til at ophidse mPlum.
BEMÆRK: Følgende filterkombination giver optimale resultater: Ved hjælp af et 560 nm langt gennemløbsfilter blev emissionslyset med en bølgelængde på under 560 nm passeret gennem et emissionsfilter på 525/50 nm til AcGFP, mens emissionslys med en bølgelængde på over 560 nm blev passeret gennem et emissionsfilter på 595/50 nm for mPlum. Alle relevante filtersæt (f.eks. FITC/TRITC, GFP/mCherry, FITC/TxRed), der sikrer, at der kan anvendes minimal fluorescensudblødning.
- Indfør fluorescensgenopretning efter fotobleaching (FRAP) laser til prøven via en softwarestyret XY galvano-enhed. Brug en 488 nm laserlinje til at begejstre AcGFP og en 561 nm eller 594 nm laserlinje til at ophidse mPlum.
- Tænd for miljøkammeret og mikroskopkomponenterne.
- Tænd for opvarmningen (fase, mål og miljøkammer, når det er muligt), CO2-forsyning og fugtighedsregulatoren mindst 4 timer før forsøgets start for at sikre termisk ligevægt for stabil billedopsamling.
- Initialiser lyskilder sammen med laserlinjerne mindst 1 time før overførsel af cellerne til mikroskopet.
- Vælg S-fase celler i en asynkron population ved hjælp af fluorescerende mærket PCNA som en markør. Gør dette ved at følge nedenstående trin.
- Se efter det unikke lokaliseringsmønster for den mPlum-taggede PCNA i S-fase, hvilket gør det muligt at identificere denne cellecyklusfase. PCNA har en fuldstændig homogen fordeling i kernen i G1- og G2-faserne i cellecyklussen, samtidig med at den udelukkes fra nucleolien. I S-fase danner PCNA foci på placeringen af repliomer i kernen. Figur 1 viser de forskellige mønstre af PCNA foci i hele S-fase, hvilket gør det muligt at differentiere tidligt, midt og sent S-fase.
- Kig gennem okulæren for at vælge en FOV, der har nok S-fase celler til mikro-bestråling. Asynkrone U-2 OS celler har normalt 30-40% af deres befolkning i S fase.
- Prøv at undgå ekstremer i udtryksniveauer (både lyse og svage celler) for både PCNA og proteinet af interesse (POI), i dette tilfælde EXO1b-AcGFP, hvilket kan føre til eksperimentelle artefakter.
- Når du finder en passende FOV, så prøv at undgå at scanne feltet i lang tid for at minimere fotografering og uønsket DNA-skade.
- Indstil den ønskede interesseregion (ROI) til mikrobestråling. Brug den tilknyttede software (se Materialetabel) skal du angive det ønskede investeringsafkast ved først at indsætte binære linjer (angiv det ønskede antal linjer og afstand). Klik på Binær, og klik derefter på Indsæt linje | Cirkel | Ellipse til at tegne det ønskede antal linjer.
- Konverter disse binære linjer til investeringsafkast, og konverter til sidst disse investeringsafkast til stimulerings-INVESTERINGSAFKAST. Det kan du gøre ved først at klikke på INVESTERINGSAFKAST, derefter klikke på Flyt binær til investeringsafkast, derefter højreklikke på et af investeringsafkastene og vælge Brug som stimulerings-INVESTERINGSAFKAST: S1. Placer disse linjer i FOV at passere gennem kernen af cellerne. INVESTERINGSAFKAST med en længde på 1024 pixel, der strakte sig over hele FOV, blev brugt i hele protokollen.
5. Mikrobestråling til immunfluorescencefarvning eller tidsforladt billeddannelse.
- Bestemmelse af optimale mikrobestrålingsindstillinger.
- Før mikrobestråling af cellerne skal du tage et billede af FOV'en med højere opløsning for at identificere PCNA foci til senere analyse. I stedet for sekventiel scanning skal du samtidig registrere begge anvendte optiske kanaler (grøn og rød) for at undgå cellebevægelse mellem scanning ved de to bølgelængder. For korrekt opløsning af foci bruge mindst 1024 x 1024 pixels / felt opløsning med 1x zoom (0,29 μm pixel størrelse på billedsystemet, der anvendes her), med 1/8 ramme / s scanningshastighed (4,85 μs /pixel) med 2x gennemsnit. Når disse parametre er angivet i A1 LFOV Compact GUI og vinduerne A1 LFOV Scan Area, skal du trykke på knappen Hent for at optage FOV.
BEMÆRK: Det er vigtigt at bevare den samme pixelstørrelse gennem eksperimenter for at sikre sammenlignelige resultater. - Hvis du vil konfigurere mikrobestrålingen, skal du åbne fanen ND Stimulation i billedsoftwaren for at få adgang til tidsskemaet (A1 LFOV / Galvano Device). Dette bruger galvano scannere til at erhverve en række pre-stimulation billeder, stimulere (ved hjælp af LUN-F 50 mW 405 nm FRAP laser), og derefter erhverve en række post-stimulation billeder igen ved hjælp af galvano scannere. Konfigurer først tre faser i vinduet Tidsplan. Vælg Anskaffelse | i kolonnen Acq/Stim Blegning | Erhvervelse for henholdsvis de tre faser. For blegning fase, sæt S1 som ROI.
BEMÆRK: I eksperimentet, der præsenteres her, blev der ikke erhvervet billeder i stimuleringsfasen. - I Galvano XY-vinduetskal du konfigurere nøglefaktorerne for mikrobestråling: 405 nm lasereffekt, dvæletid (gentagelse er som standard 1 på dette system). I de eksperimenter, der præsenteres her, celler blev bestrålet med 405 nm FRAP laser (50 mW ved fiberspids) ved 100% effekt med en 1000-3000 μs ophold tid.
BEMÆRK: Da laserbo tiden er på en per pixel basis, så længe pixel størrelse forbliver den samme, forholdet mellem dvæle tid og effekttæthed vil være sammenlignelig mellem forskellige FOVs. Figur 2A viser brugen af DNA-skadesrespons (DDR) pathway specifikke proteiner (FBXL10 for DSBs og NTHL1 for oxidativ base skader) for at optimere laser effektindstillinger for specifikke skader induktion. Disse stabile cellelinjer blev genereret med virusinfektion efter afsnit 2 i protokollen.
- Før mikrobestråling af cellerne skal du tage et billede af FOV'en med højere opløsning for at identificere PCNA foci til senere analyse. I stedet for sekventiel scanning skal du samtidig registrere begge anvendte optiske kanaler (grøn og rød) for at undgå cellebevægelse mellem scanning ved de to bølgelængder. For korrekt opløsning af foci bruge mindst 1024 x 1024 pixels / felt opløsning med 1x zoom (0,29 μm pixel størrelse på billedsystemet, der anvendes her), med 1/8 ramme / s scanningshastighed (4,85 μs /pixel) med 2x gennemsnit. Når disse parametre er angivet i A1 LFOV Compact GUI og vinduerne A1 LFOV Scan Area, skal du trykke på knappen Hent for at optage FOV.
- Tid bortfalder billeddannelse.
- Konfigurer tidsforkortningsafbildning for det ønskede tidsvindue og intervaller ved hjælp af tidsskemaet, A1 LFOV Compact GUI og vinduerne A1 LFOV Scan Area. I de eksperimenter, der præsenteres her, blev rekrutteringen af EXO1b og PCNA afbildet i 12 min, scanning af FOV hvert 5. sekund, ved 1024 x 1024 pixels / felt, ved hjælp af 1x zoom (hvilket resulterer i 0,29 μm pixelstørrelse på billedsystemet, der bruges her) med 0,35 ramme / s scanningshastighed (1,45 μs / pixel) uden gennemsnit at reducere fotoblegning.
- Optimer lasereffekten %, gain og offset-indstillingerne for at reducere fotoblegning under billeddannelsen i A1 LFOV Compact GUI-vinduet. Hvis man har til formål at måle både POI og PCNA, skal du bruge samtidig scanning i stedet for sekventiel scanning for at undgå cellebevægelse mellem scanning af feltet for de to separate fluorophorer.
- Billedsystemet blev brugt sammen med følgende indstillinger. For 488 nm laser linje (20 mW): 7% laser effekt, gain: 45 (GaAsP detektor) med og offset af 2, for 561 nm laser linje (20 mW): 4% laser magt, få 40 (GaAsP detektor) med og offset af 2.
- Afhængigt af proteinets kinetika skal intervallet mellem billederne eller varigheden af den samlede tid bortfalder. Angiv det ønskede interval og den ønskede varighed for rækken anskaffelse i tredje fase i vinduet Tidsplan.
- Tryk på Kør nu for at udføre mikrobestrålingen og den efterfølgende tid bortfalder billeddannelse.
- Ved afslutningen af tiden bortfalder billeddannelse, gemme stimulation ROIs som separate billeder, som vil være en nyttig hjælp til at identificere koordinaterne for mikro-bestråling i enhver downstream software, der anvendes til analyse.
- Immunofluorescence farvning.
BEMÆRK: Trin 5.1.3 og figur 2A viser brugen af kendte DNA-reparationsproteiner til at vurdere de typer DNA-læsioner, der indføres ved mikrobestråling. Visse DNA-læsioner kan også påvises ved hjælp af specifikke antistoffer efter fastsættelse af cellerne. Det er også muligt at opdage rekrutteringen af POI ved antistofdetektering af det endogene protein. Visualiseringen af γH2A.X for at kontrollere, om DSBs er vist nedenfor (Figur 2B). Figur 3 viser konsistensen af PCNA-lokalisering og rekruttering i hele cellecyklussen for både endogene og eksogene pcna-mærkede PCNA.- Efter trin 5.1.3 skal du kun tage et billede efter mikrobestråling for at sikre en korrekt FRAP-begivenhed baseret på rekruttering af mPlum-PCNA. Vær opmærksom på de nøjagtige koordinater for FOV for at finde feltet senere efter immunfluorescent mærkning.
- Tag cellekulturkammeret ud af mikroskopet, og inkuber cellerne ved 37 °C i en befugtet atmosfære, der indeholder 5 % CO2 i 5-10 min.
BEMÆRK: Paraformaldehyd (PFA) er giftigt, og arbejdet skal udføres i et godt ventileret område eller en røghætte. Al efterfølgende vask og inkubation vil ske med 0,5 mL volumener i 4 brøndkammer dias. Efter inkubationstiden vaskes cellerne med 0,5 mL PBS (137 mM NaCl, 2,7 mM KCl, 8 mM Na2HPO4og 2 mM KH2PO4) og fastgør med 0,5 mL på 4% PFA i PBS i 10 min ved stuetemperatur (RT). - Vask cellerne en gang med PBS, vask dem derefter med 50 mM NH4Cl for at slukke resterende PFA.
- Permeabilize cellerne i 15 min på RT med 0,1% Triton X-100 i PBS.
- Bloker prøverne i 1 time med blokerende buffer (5% FBS, 3% BSA, 0,05% Triton X-100 i PBS).
- Blokeringsopløsningen fjernes, og det fortyndede primære antistof (anti-γH2A.X, 1:2000) tilsættes i blokeringsbufferen i 1 time ved RT.
- Vask brøndene med blokerende buffer 3 x 10 min.
- Tilføj fortyndet sekundært antistof (anti-mus Alexa 488 Plus konjugat, 1:2000) i blokering buffer for 1 time på RT.
- Vask brøndene med blokerende buffer 3 x 10 min.
- Kernen skal modvirkes med 1 μg/mL DAPI-opløsning i PBS i 15 min.
- Vask cellerne én gang med PBS. Billeddannelsen kan udføres direkte i PBS eller en PBS-løsning med antifade reagenser (f.eks. AFR3) for at reducere fotografering.
6. Rekrutteringsanalyse
BEMÆRK: Figur 4A viser repræsentative billeder af Exo1b- og PCNA-rekruttering i nærværelse af DMSO eller olaparib. Figur 4B viser et repræsentativt billede til dataanalyse. Gennemsnitlige fluorescensværdier blev beregnet ved at måle gennemsnitlige AcGFP intensiteter ved hjælp af et rektangel langs lasersporet fremhævet af mPlum-PCNA (A, gule rektangler) på tværs af forskellige tidspunkter ved hjælp af Fiji. PCNA kan fungere som en intern kontrol for at fremhæve vellykket bestråling langs ROI-koordinaterne. Tilsvarende blev der også beregnet gennemsnitlige acGFP-fluorescensværdier for ubeskadigede områder af kernen (B, blå rektangler). Baggrundssignalintensiteten blev målt i ubefolkede områder (C, røde rektangler) og blev trukket fra de gennemsnitlige fluorescerende værdier (Figur A og B). Således blev den relative gennemsnitlige fluorescerende enhed (RFU) for hvert dataindsamlingspunkt beregnet ved ligningen RFU = (A − C)/ (B − C)8,9. De resulterende RFU-værdier i det mikrobestrålede område normaliseres til RFU-værdierne før mikrobestråling.
- Ved fastlæggelsen af region A på det mikrobestrålede område skal du udelukke nukleolære regioner, replikations-foci og uregelmæssige nukleare områder af cellen fra måling. Hold skifttasten mellem tegning af to investeringsafkast i Fiji for at gruppere to separate områder som ét.
BEMÆRK: Proteinrekruttering vil variere mellem forskellige gener og bestrålingsbetingelser; derfor skal størrelsen af region A bestemmes individuelt. Når pixelbredden af område A er bestemt, skal den forblive konstant for eventuelle sammenlignende rekrutteringer. I de eksperimenter, der præsenteres her, blev der brugt 7 pixelbredderektangler. - Udelad celler, der er flyttet under varigheden af de optagede videoer, fra analyse. For at medtage meget mobile celler skal den beskrevne analyse udføres ramme for ramme.
- For at visualisere rekrutteringsprofilen skal du plotte de normaliserede RFU-værdier mod tiden ved hjælp af en statistisk software.
- Forskellen beregnes på et angivet tidspunkt mellem DMSO og olaparib (n=31) behandling ved hjælp af en Mann-Whitney-test.
Representative Results
Celler adresserer hver type DNA-læsion på en bestemt måde, der også afhænger af, hvilken cellecyklusfase de er i. For eksempel vil dobbeltstrengede pauser (DSB) efter mikrobestråling blive behandlet enten ved ikke-homolog ende sammenføjning (NHEJ) eller HR afhængigt af cellecyklusfasen. Nukleaser, der virker mest omfattende i S- og G2-faserne i cellecyklussen, skaber DNA-udhæng, der er afgørende for korrekt HR. For at fremme evalueringen af celler i S-fasen blev PCNA anvendt som en enkeltfarvet cellecyklusmarkør. Figur 1A viser lokaliseringsprofilen for mPlum-PCNA under cellecyklusforløb. PCNA har en fuldstændig homogen fordeling i kernen i G1- og G2-fasen (samtidig med at det for det meste er udelukket fra nucleoli). I S fase lokaliserer PCNA til steder med DNA-replikation, som kan visualiseres som lyspunkter i kernen. I tidlige S fase celler, pletterne er relativt små og ligeligt fordelt i hele kernen af cellen. Fremad i midten af S fase, pletterne bliver sløret og lokalisere mere mod omkredsen af kernen og nucleoli. I sen S fase, pletter reducere i antal, men bliver stadig større som PCNA koncentrater på sene replikeringssteder (Figur 1B). Vigtigere, udefrakommende PCNA udtryk fra pBABE vektor rygraden var mindre end de endogene niveauer, men var nok til påvisning ved mikroskopi, som minimerer potentielle artefakter i cellecyklus progression og DDR. Figur 1C viser omfanget af PCNA-overekspression sammenlignet med endogene niveauer. Bemærk, at båndet svarende til mPlum-PCNA migrerer langsommere på grund af dets større størrelse.
Vi havde til formål at indføre DSBs under mikrobestråling for at undersøge den PARP1/2-afhængige rekruttering af EXO1b til disse læsioner i S-fasen. Figur 2A viser, at lave doser energi (1000 μs opholdstid) ikke fremkalder rekruttering af EGFP-FBXL10, en DSB-responder (komponent i FRUCC-komplekset 8),mens det var tilstrækkeligt at fremkalde rekruttering af NTHL1-mCherry, en base excision reparation (BER) pathway protein, rekruttering til steder med oxidativ DNA-skade10,11,12. Ved 3000 μs dvæle tid, både EGFP-FBXL10 og NTHL1-mCherry rekruttere, der viser en laser output, der genererer både oxidative læsioner og DSBs. Styrkelse af disse resultater, Figur 2B viser immunofluorescence farvning mod γH2A.X (DSB markør), som er klart mere tydeligt, når du bruger højere energi doser. PCNA fungerer både som en celle cyklus markør og en markør for vellykket mikro-bestråling, da det tilstrækkeligt rekrutter med både laser ophold tidsindstillinger. Det er vigtigt, at både eksogene og/eller endogene fluorescerende proteinmærkede PCNA kan bruges til denne reporterfunktion, da de opfører sig på samme måde (Figur 3). Endogent mærkede PCNA blev manipuleret ved at indsætte mRuby i ramme med den første exon i en allel af PCNA locus13 (cellelinjen var en slags gave af Jörg Mansfeld).
Figur 4A og figur 4C viser rekrutteringen af ACGFP-mærkede EXO1b i S-faseceller. EXO1b når det maksimale akkumuleringsniveau på mikrobestrålingssteder omkring 1 minut og begynder derefter langsomt at frigøre sig fra DNA-læsionerne bagefter. Berigelser på mikrobestrålingssteder er betegnet med en > 1 relativ fluorescensenhed på grafen. I nærværelse af olaparib er akkumuleringen af EXO1b ved laserstriben ved 1 minut betydeligt mindre sammenlignet med køretøjets kontrol. Disse resultater er i overensstemmelse med litteraturen6,7. Figur 4B viser repræsentative regioner for kvantificering (område A, B og C) som beskrevet i punkt 6 i protokollen. Figur 4D viser de sammenlignelige ekspressionsniveauer for endogene EXO1b og eksogene EXO1b-AcGFP i celler, der anvendes til mikrobestråling.
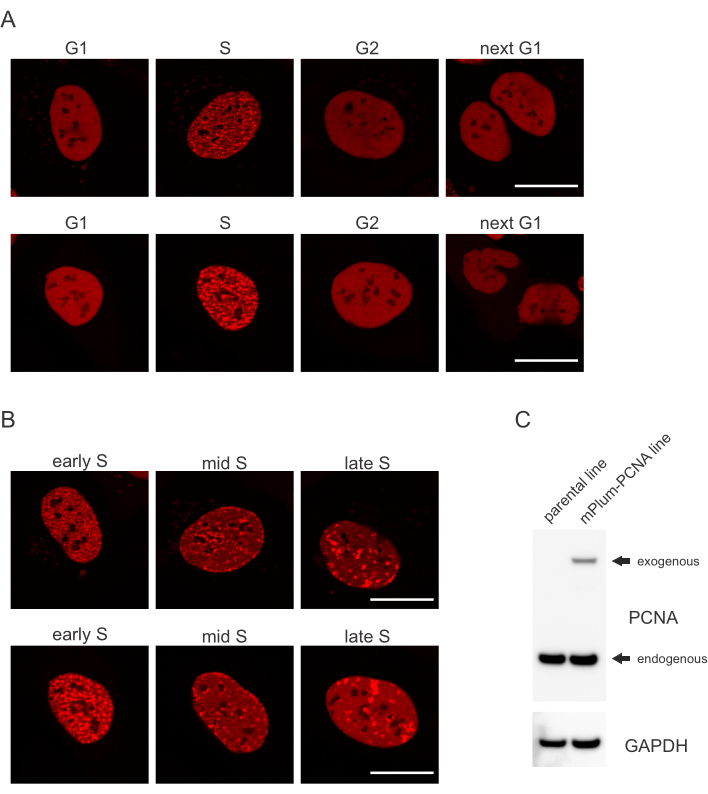
Figur 1: Lokaliseringsmønster for PCNA. (A) Billeder viser lokaliseringsmønster af stabilt integreret, eksogent PCNA i hele cellecyklussen i U-2 OS-celler. (B) Billeder viser PCNA foci mønstre i forskellige stadier af S fase (tidligt, midten og sent) i U-2 OS celler. (C) Western blot viser endogene og eksogene niveauer af PCNA i U-2 OS celler, der anvendes til billeddannelse. Skalalinjen repræsenterer 20 μm. Klik her for at se en større version af dette tal.

Figur 2: Induktion af DSBs gennem optimeret lasereffekt. (A) Laserindstillinger kan optimeres til at fremkalde forskellige former for DNA-skader. U-2 OS-celler, der stabilt udtrykte både EGFP-FBXL10 og NTHL1-mCherry, blev brugt til at identificere henholdsvis DSBs og steder med oxidative læsioner. Mikrobestråling med en 405 nm laserlinje blev udført på asynkrone U-2 OS-celler med enten 1000 μs eller 3000 μs opholdstid. Skalastang repræsenterer 20 μm. (B) Immunofluorescent farvning mod γH2A.X blev udført på human retinale pigment epitelceller (hTERT RPE-1) med mRuby-mærket endogen PCNA. Cellerne blev fastgjort og behandlet 5 minutter efter mikrobestråling med enten 1000 μs eller 3000 μs opholdstid. Skalalinjen repræsenterer 20 μm. Klik her for at se en større version af dette tal.

Figur 3: Sammenlignelig rekruttering af endogene mRuby-PCNA og eksogene mPlum-PCNA til mikrobestrålingssteder ved 1000 μs eller 3000 μs laser opholdstid. Både endogene og udefrakommende tagget PCNA form replikering foci i S fase. Klik her for at se en større version af dette tal.

Figur 4: PARP1/2-afhængig rekruttering af EXO1b i S-fasen. U-2 OS-celler, der stabilt udtrykte EXO1b-AcGFP og mPlum-PCNA, blev mikrobestrålet med 405 nm FRAP-laserlinje ved hjælp af 3000 μs opholdstid. (A) Repræsentative billeder af mikrobestrålede celler på det angivne tidspunkt efter forbehandling med enten køretøjskontrol (DMSO) eller olaparib (1 μM). Skalalinjen repræsenterer 20 μm. (B) Repræsentative billeder af definerede områder I A, B og C til rekrutteringsanalysen. Skalalinjen repræsenterer 20 μm. (C) DNA-skade rekruttering dynamik blev fanget af levende celle billeddannelse. Relative gennemsnitlige fluorescensværdier og billeder blev erhvervet hver 5 s i 12 min. For hver tilstand blev ≥30 celler evalueret. Gennemsnitlige relative fluorescensværdier (faste sorte linjer) og standardfejl (område visualiseret af et skraveret område) blev afbildet mod tiden. Stiplet linje viser rekrutteringsværdier på 1 min efter mikrobestråling. Forskellen mellem DMSO (n=32) og olaparib (n=31) behandling blev beregnet ved hjælp af en Mann-Whitney-test. Asterix betegner p<0.0001. (D) Western blot sammenligner udtryksniveauerne for endogene EXO1b og eksogene EXO1b-AcGFP i celler, der anvendes til mikrobestråling. Klik her for at se en større version af dette tal.
Discussion
Kritiske trin og potentiel fejlfinding/-ændringer af protokoller
Korrekt vævskulturbeholder til mikrobestråling er afgørende for succes. De fleste billedsystemer med høj opløsning er optimeret til 0,17 mm dækglastykkelse. Brug af højere eller lavere tykkelse billeddannelse kamre eller dem fremstillet af plast polymerer (ikke optimeret til 405 nm billeddannelse), kan reducere billedkvaliteten betydeligt. Når du bruger glasoverflader, skal du sørge for, at de er vævskulturbehandlet for at forbedre celle vedhæftning. Hvis de ikke er vævskulturbehandlet, skal disse kamre f.eks. Ved plating af celler i det chambered coverglass er ideel celletæthed altafgørende for at undgå uregelmæssigheder i cellecyklussen og yderligere stress til cellerne. Korrekt termisk ekvilibrering af mikroskopkomponenterne før forsøg for at opretholde en stabil temperatur er afgørende for både at opretholde fokus gennem hele tiden bortfalder billeddannelse og er også nødvendig for at sikre en homogen DDR på tværs af tid og prøver.
Det er afgørende, at cellerne er i en sund tilstand forud for mikrobestråling for at reducere artefaktiske data. Hvis celler har uregelmæssig morfologi efter infektion/udvælgelse, skal cellerne udvikle sig gennem flere passager, indtil morfologien vender tilbage til normal. Sørg altid for, at de anvendte cellelinjer er fri for mycoplasmaforurening. Blandt de mange bivirkninger af mycoplasma infektion, det forårsager også DNA-skader på værtscellerne og kan påvirke deres DDR veje14,15. Den mest følsomme måde at opdage mycoplasma i cellekulturen er gennem PCR (versus detektion med DAPI eller Hoechst).
Optimal overekspression af reparationsproteinet af interesse bør kunne sammenlignes med endogene niveauer, men høje nok til påvisning. Promotoren, der anvendes på de virale vektorer, den virale titer under infektion, og længden af infektionen tid kan alle justeres for ideelle udtryk niveauer. For at opnå ensartede resultater skal du isolere individuelle cellekloner for at sikre homogene udtryksniveauer og normal cellemorfologi. Det anbefales at bruge vektorkonstruktioner, der ikke overekspresserer mærket PCNA på højere end endogene niveauer for korrekt cellecyklus og DNA-reparationsmarkørfunktion. Selv lave niveauer af PCNA overekspression er tilstrækkelige til at diskriminere S-fase celler. Retroviral pBABE vektorer er blevet anvendt med succes til dette formål (Addgene # 1764, # 1765, # 1766, # 1767). PCNA kan mærkes med monomerisk rød(f.eks mPlum, mCherry, mRuby, etc.) eller monomeriske grønne fluorescerende proteiner (f.eks mEGFP, AcGFP, mWasabi, mNeonGreen, mEmerald osv.), som derefter kunne kombineres med en skiftevis mærket POI. Overekspressering af et fluorescerende mærket POI har nogle begrænsninger og overvejelser. Fluorescerende tags kan forstyrre normal proteinfunktion og lokalisering. Således skal placeringen af mærket (N eller C-terminal) overvejes. Brug altid monomeriske fluorescerende proteiner, da oligomerisering af ikke-monomeriske varianter kan påvirke POI'ens funktion.
Laserindstillingerne skal bestemmes for hvert billedsystem, da mange komponenter i den optiske sti vil påvirke den faktiske effekt, der leveres i cellerne. Laser mikro-bestråling kan forårsage flere typer af DNA-læsioner afhængigt af excitation bølgelængde, effekt af FRAP laser og hvis nogen præ-sensibiliserende midler (som Bromodeoxyuridin eller Hoechst) blev brugt. 405 nm lasere kan forårsage oxidativ DNA-skade, enkelt og dobbelt strandede pauser16,17. Ved at bruge højere indstillinger for laserudgang øges mængden af DSBs. I denne protokol pre-sensibilisering metoder blev ikke udnyttet, men disse teknikker er i høj grad dækket i litteraturen og re-capped i diskussionen nedenfor. Efter vores mening er den bedste måde at teste, om den ønskede læsion genereres ved at teste for rekruttering af kendte DNA-skader vej specifikke gener. Rekruttering af NTHL1 eller OGG1, komponenter i BER-stien, tyder på induktion af oxiderede DNA-baser10,11,17,18,19, mens FBXL10 eller XRCC5 angiver tilstedeværelsen af DSBs8,20,21. Rekruttering af XRCC1 kan indikere både tilstedeværelsen af oxiderede DNA-baser og enkelte strandede pauser (SSB)22,23. XPC (dvs. RAD4) er en god indikator for NER, der fjerner de voluminøse DNA-adducts genereret af ultraviolet lys (UV)17,24. Fordi rekruttering af udefrakommende proteiner kan indføre visse uregelmæssigheder, kan immunofluorescent farvning af endogene DNA-reparationsproteiner eller markører (som γH2A.X for dobbeltstrengede pauser) bekræfte tilstedeværelsen af specifikke DNA-læsioner. Alternativt kan antistoffer rejst mod specifikke typer dna-læsioner også anvendes. For at justere den leverede lasereffekt kan både opholdstiden og lasereffekten ændres.
Ved hjælp af matematisk modellering kunne der udføres en detaljeret kinetisk analyse, der kan give værdifuld indsigt i POI's rekrutteringsegenskaber (f.eks. bidrag fra flere DNA-bindende domæner, følsomhed over for forskellige signalhændelser osv.). Automatiseret rekrutteringsevaluering og cellesporing kan kombineres for at skabe robuste arbejdsgange 1,25.
Fordele og begrænsninger ved DNA pre-sensibilisering
Præfølsomhed af DNA før mikrobestråling er et almindeligt anvendt værktøj til rekruttering af DNA-reparation af protein16,17. Sensibiliserende DNA før mikrobestråling gør det mere modtageligt for DSBs. De to mest almindelige metoder til DNA-præfølsomhed er forbehandling af celler med enten Bromodeoxyuridin (BrdU) eller Hoechst farvestof. For systemer, der ikke er i stand til mikrobestråling ved høje laserkræfter, kan disse metoder være nødvendige for at fremkalde DNA-læsioner som DSBs. Derudover fungerer Hoechst som både et præfølsomt signal, der fremhæver cellekernen (f.eks. når man studerer rekrutteringen af ukodede endogene DNA-reparationsproteiner), som både et præfølsomt værktøj og en fluorescerende atomplet. Dna pre-sensibilisering kan dog introducere betydelige komplikationer. BrdU (anvendes ved en endelig koncentration på 10 μM) skal tilsættes cellerne 24 timer (eller tid svarende til en fuld cellecyklus i den anvendte cellelinje) for korrekt at indarbejde i DNA og kan forårsage cellecyklusinterferens26. Hoechst 33342 (anvendes i en endelig koncentration på 1 μg/mL) er cytotoksisk efter lange inkubationsperioder, men kræver tilstrækkelig tid til at mætte kernen med farvestoffet. Derfor bør det kun anvendes 15-20 minutter før mikrobestråling; Ellers vil rekrutteringsdataene ikke være konsistente. Cellerne farves på denne måde kan ikke holdes i kultur i mere end et par timer27,28. Sørg for ikke at bruge Hoechst 33358, som ikke er så celle permeable som Hoechst 33342 farvestof. Præsensibilisering kan også indføre unødvendig varians blandt eksperimenter og gør eksperimentet endnu mere følsomt over for forskelle i celletæthed (da dette vil påvirke mængden af indarbejdet farvestof / celle).
Fordele og begrænsninger ved konfokal mikroskopi
Billedhastighed af konfokal mikroskopi kan være begrænsende i forhold til widefield mikrokopi. Men et konfokalt mikroskop udstyret med en resonant scanner kan enormt forbedre billedhastigheden (på bekostning af opløsning) kommer tæt på hastigheder på spinning-disk mikroskopi. Tre funktioner gør A1R HD25 confocal-systemet til et glimrende valg til den protokol, der præsenteres her. For det første gør systemets 25 mm FOV det muligt at afbilde mellem 15-20 celler i et enkelt scannet felt (vs. 5-10 celler i regelmæssige opsætninger), hvilket begrænser antallet af erhvervelser, der er nødvendige for at få nok celler til statistisk analyse. For det andet gør FRAP-modulet og to scanheads det muligt at afbilde og mikroforbedre cellerne samtidigt, ikke kun sekventielt. Endelig giver fleksibiliteten ved at have både resonant og galvano scannere mulighed for nemt at skifte mellem høj tidsmæssig opløsning billeddannelse med enestående hastighed, som minimerer slukning af fluorophores, og høj-rumlig opløsning billeddannelse, der udnytter langsommere scanningshastigheder til at producere billeder med et højere signal til støj ratio. Mens det anvendte system gjorde det muligt for ovennævnte fleksibilitet, at ligne mere bredt tilgængelige konfokale mikroskop konfigurationer, kun galvano scanneren blev brugt i de præsenterede eksperimenter (for både mikro-bestråling og efterfølgende billeddannelse).
Fordele og begrænsninger ved mikrobestråling
Mens mikrobestråling giver uovertruffen rumlig og tidsmæssig opløsning, er det ikke uden begrænsninger. DNA-skader ved laser mikro-bestråling er stærkt grupperet til bestemte dele af kernen i forhold til naturligt forekommende skadelige stoffer. Kromatinrespons på grund af mikrobestråling kan således variere i forhold til homogent fordelt skade. Derudover er mikrobestråling tidskrævende og kan kun udføres på et par dusin celler, mens store befolkningsbaserede biokemiske metoder (kromatinfraktionering, immunprecipitation, ChIP) kan give øget robusthed ved at studere tusindvis af celler ad gangen. Kontrol af observationer foretaget af mikrobestråling med traditionelle biokemiske teknikker er en effektiv strategi for pålidelige konklusioner. Selvom samtidig mikrobestråling af mange celler i en bestemt FOV er mulig, vil billedsystemet have brug for mere tid til at udføre opgaven. Derfor begrænser målingen af dynamikken i proteiner, der rekrutterer meget hurtigt til DNA-læsioner, antallet af mulige ROI'er for mikrobestråling, der anvendes samtidigt. På det billedsystem, der anvendes til denne protokol, tager mikrobestrålingen af et enkelt 1024 pixel langt ROI 1032 ms ved hjælp af 1000 μs opholdstid og 3088 ms ved hjælp af 3000 μs dvæle tid til at fuldføre. Brug af flere linjer af ROI vil øge den tid, der er nødvendig for at afslutte mikrobestråling (f.eks 7 x 1024 pixel lang ROI tager 14402 ms ved hjælp af 1000 μs dwell tid og 21598 ms ved hjælp af 3000 μs opholdstid). Denne gang er tabt fra billederhvervelse og skal tages i betragtning. Når du billeddannelse hurtige rekruttering begivenheder, bruge den kortest ROI muligt og kun mikro-bestråle en celle ad gangen.
Fordele og begrænsninger i forhold til synkroniseringsmetoder
I forbindelse med cellecyklusspecifikke undersøgelser omfatter de eksisterende metoder enten synkronisering af celler i specifikke cellecyklusfaser eller ved hjælp af fluorescerende reportere til at identificere cellens specifikke cellecyklusfase. Men hver af disse metoder giver deres egne udfordringer og begrænsninger.
FUCCI system3 (afhængig af fluorescerende protein mærkede afkortede former for CDT1 og Geminin) er et særligt nyttigt værktøj til celle cyklus undersøgelser, men har begrænsninger, når det kommer til at skelne mellem S og G2 faser af celle cyklus. Geminin niveauer er allerede høje fra midten af S fase og forblive høj indtil M fase, hvilket gør disse faser vanskeligt at adskille. Brug af FUCCI-systemet betyder også, at to optiske kanaler i mikroskopet ikke kan bruges til billeddannelse af POI.
Ikke-kræft cellelinjer kunne synkroniseres i G0 ved fjernelse af vækstfaktorer findes i serum (serum sult) forårsager ringe eller ingen DNA-skader på cellerne. Men, de fleste kræft cellelinjer vil delvist fortsætte med at udvikle sig gennem celle cyklus selv uden tilstrækkelige mængder af serum i deres medier. Derudover begynder cellerne delvist at miste synkroniseringen i den sene G1-fase, tidlige S-fase. Ud over serum sult, der er mange kemiske metoder til at opnå celle cyklus synkronisering. Hydroxyurea, bladlusicolin og thymidinblokke er metoder til at stoppe DNA-replikation for at synkronisere celler i den tidlige S-fase. Mens disse metoder er billige og enkle, introducerer de replikationsstress, hvilket resulterer i DNA-skade. Disse DNA-replikationshæmmere har vist sig at fremkalde fosforylering af H2A. X, en velkendt markør for DSBs2,29. Metoden til at bruge tagged-PCNA som en markør for S-fase celler reducerer potentialet for artefakter forårsaget af kemisk synkronisering og kan anvendes på en bred vifte af cellelinjer i forhold til serum sult.
Konklusion
DNA-skader er en drivkraft for genetiske sygdomme, hvor mutagene læsioner kan føre til ondartet transformation af celler. Målretning af DNA syntese maskiner er en grundlæggende terapeutisk strategi til behandling af hyperproliferative sygdomme som kræft. For at behandle disse sygdomme på en mere målrettet måde har vi brug for en bedre forståelse af de proteiner, der reparerer DNA-læsioner. Den protokol, der er beskrevet her, hjælper mikrobestrålingsbaserede undersøgelser i S-fasen ved at minimere de udfordringer, der præsenteres af traditionelle synkroniseringsmetoder for at reducere mulige genstande og øge reproducerbarheden af forsøgene.
Disclosures
Forfatterne anfører, at offentliggørelsen af det præsenterede arbejde var sponsoreret af Nikon Corporation. Forfatterne erklærer, at der ikke findes konkurrerende interesser.
Acknowledgments
Forfatterne takker M. Pagano for hans fortsatte støtte samt D. Simoneschi, A. Marzio og G. Tang for deres kritiske gennemgang af manuskriptet. B. Miwatani-Minter takker R. Miwatani og B. Minter for deres fortsatte støtte. G. Rona takker K. Ronane Jurasz og G. Rona for deres fortsatte støtte.
Materials
| Name | Company | Catalog Number | Comments |
| Ammonium chloride | Sigma-Aldrich | A9434-500G | For quenching formaldehyde |
| Anti-EXO1 Rabbit Polyclonal Antibody | Proteintech | 16253-1-AP | primary antibody |
| Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301 | Millipore | 05-636 | primary antibody |
| Bovine Serum Albumin | Sigma-Aldrich | 3117332001 | BSA for blocking |
| BrdU (5-Bromo-2'-deoxyuridine) | Merck | 19-160 | pre-sensitizing agent |
| Citifluor™ Mountant Solution AFR3 | Electron Microscopy Sciences | 17973-10 | antifade containing PBS solution for imaging |
| DAPI | Sigma-Aldrich | D9542-1MG | nucleic acid stain |
| DMEM Medium | Thermo Fisher Scientific | 10569010 | Cell culture medium for HEK293T cells |
| DMSO | Sigma-Aldrich | D2650-100ML | Vehichle control and dissolution solvent |
| EGFP-FBXL10 | Addgene | #126542 | viral expression vector for EGFP-FBXL10 |
| EXO1b-AcGFP (in pRetroQ) | custom cloning | na | EXO1b cDNA was cloned in the NheI, BamHI sites of pRetroQ-AcGFP1-N1 vector. |
| Fetal Bovine Serum | Gibco | 16140071 | Media supplement |
| FluoroBrite DMEM | Thermo Fisher Scientific | A1896701 | Phenol red free medium for microscopy |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | A32723 | secondary antibody |
| HEK293T cells | ATCC | ATCC CRL-3216 | Cell line for viral packaging |
| HEPES | Sigma-Aldrich | H0887-100ML | Buffering agent to supplement live cell imaging medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | pre-sensitizing agent |
| Lipofectamine 3000 | Thermo Fisher Scientific | L3000015 | Transfection reagent |
| McCoy’s 5A (Modified) Medium | Life Technologies | 16600-108 | Cell culture medium for U-2 OS cells |
| mCherry-PCNA | Addgene | #55117 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA | Addgene | #55994 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA (in pBABE) | custom cloning | na | mPlum-PCNA cDNA was cloned from Addgene #55994 in the BamHI, SalI sites of pBABE (puro) |
| Nikon A1R-HD25 Confocal Scanhead and Controller | Nikon | na | confocal imaging system |
| Nikon LUN4 laser unit | Nikon | na | excitation system |
| Nikon LUN-F 50 mW 405 nm FRAP laser unit | Nikon | na | FRAP laser unit |
| Nikon NIS Elements Confocal Controller Software | Nikon | na | Confocal controlling software |
| Nikon Ti2-E Inverted Microscope | Nikon | na | inverted epifluorescent microscope base |
| Nikon Ti2-LAPP Modular Illumination System | Nikon | na | illumination system |
| NTHL1-mCherry (in pRetroQ) | custom cloning | na | NTHL1 cDNA was cloned in the NheI, SalI sites of pRetroQ-mCherry-N1 vector. |
| Nunc Lab-Tek II Chambered Coverglass (4 well) | Thermo Fisher Scientific | 155382PK | Live cell microscopy cell culture chamber |
| Olaparib | Selleck Chemicals | S1060 | PARP inhibitor |
| Opti-MEM reduced serum media | Thermo Fisher Scientific | 31985062 | Dilution medium for transient transfection |
| Paraformaldehyde aqueous solution (32%) | Thermo Fisher Scientific | 50-980-494 | Fixative |
| pBABE (hygro) | Addgene | #1765 | retroviral expression vector (for low expression levels) |
| pBABE (neo) | Addgene | #1767 | retroviral expression vector (for low expression levels) |
| pBABE (puro) | Addgene | #1764 | retroviral expression vector (for low expression levels) |
| pBABE (zeo) | Addgene | #1766 | retroviral expression vector (for low expression levels) |
| PCNA Antibody (PC10) | Santa Cruz | sc-56 | primary antibody |
| Penicillin-Streptomycin-Glutamine (100x) | Gibco | 10378016 | Media supplement |
| polybrene | Sigma-Aldrich | TR-1003 | Increase viral infection efficiency |
| pRetroQ-AcGFP-C1 | Takara | 632506 | retroviral expression vector |
| pRetroQ-AcGFP-N1 | Takara | 632505 | retroviral expression vector |
| pRetroQ-mCherry-C1 | Takara | 632567 | retroviral expression vector |
| pRetroQ-mCherry-N1 | Takara | 632568 | retroviral expression vector |
| pUMVC | Addgene | #8449 | Viral packaging vector |
| Sodium-pyruvate | Thermo Fisher Scientific | 11360070 | Supplement for live cell imaging medium |
| Triton X-100 aqueous solution (10%) | Sigma-Aldrich | 11332481001 | Dilute in PBS for cell permeabilization buffer |
| Trypsin-EDTA Solution 10X | Sigma-Aldrich | 59418C-100ML | Dilute in PBS to split cells |
| U-2 OS Cells | ATCC | HTB-96 | Optimal cell line for microscopy experiments |
| Universal Mycoplasma Detection Kit | ATCC | 30-1012K | PCR based Mycoplasma detection kit |
| VSV-G | Addgene | #8454 | Viral protein envelope vector |
References
- Aleksandrov, R., et al.
- Darzynkiewicz, Z., Halicka, H. D., Zhao, H., Podhorecka, M. Cell synchronization by inhibitors of DNA replication induces replication stress and DNA damage response: Analysis by flow cytometry. Methods in Molecular Biology. 761, 85-96 (2011).
- Sakaue-Sawano, A., et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 132 (3), 487-498 (2008).
- Herce, H. D., Rajan, M., Lattig-Tunnemann, G., Fillies, M., Cardoso, M. C. A novel cell permeable DNA replication and repair marker. Nucleus. 5 (6), 590-600 (2014).
- Keijzers, G., et al. Human exonuclease 1 (EXO1) regulatory functions in dna replication with putative roles in cancer. International Journal of Molecular Sciences. 20 (1), (2018).
- Cheruiyot, A., et al. Poly(ADP-ribose)-binding promotes Exo1 damage recruitment and suppresses its nuclease activities. DNA Repair (Amsterdam). 35, 106-115 (2015).
- Zhang, F., Shi, J., Chen, S. H., Bian, C., Yu, X. The PIN domain of EXO1 recognizes poly(ADP-ribose) in DNA damage response. Nucleic Acids Research. 43 (22), 10782-10794 (2015).
- Rona, G., et al. PARP1-dependent recruitment of the FBXL10-RNF68-RNF2 ubiquitin ligase to sites of DNA damage controls H2A.Z loading. elife. 7, (2018).
- Young, L. M., et al. TIMELESS forms a complex with PARP1 distinct from its complex with TIPIN and plays a role in the dna damage response. Cell Reports. 13 (3), 451-459 (2015).
- Kong, X., et al. Laser microirradiation to study in vivo cellular responses to simple and complex dna damage. Journal of Visualized Experiments. (131), e56213 (2018).
- Kong, X., et al. Condensin I recruitment to base damage-enriched DNA lesions is modulated by PARP1. PLoS One. 6 (8), 23548 (2011).
- Lan, L., et al. Novel method for site-specific induction of oxidative DNA damage reveals differences in recruitment of repair proteins to heterochromatin and euchromatin. Nucleic Acids Research. 42 (4), 2330-2345 (2014).
- Zerjatke, T., et al. Quantitative cell cycle analysis based on an endogenous all-in-one reporter for cell tracking and classification. Cell Reports. 19 (9), 1953-1966 (2017).
- Ji, Y., Karbaschi, M., Cooke, M. S. Mycoplasma infection of cultured cells induces oxidative stress and attenuates cellular base excision repair activity. Mutation Research. 845, 403054 (2019).
- Sun, G., et al. Mycoplasma pneumoniae infection induces reactive oxygen species and DNA damage in A549 human lung carcinoma cells. Infection and Immunity. 76 (10), 4405-4413 (2008).
- Gassman, N. R., Wilson, S. H. Micro-irradiation tools to visualize base excision repair and single-strand break repair. DNA Repair (Amsterdam). 31, 52-63 (2015).
- Muster, B., Rapp, A., Cardoso, M. C. Systematic analysis of DNA damage induction and DNA repair pathway activation by continuous wave visible light laser micro-irradiation. AIMS Genetics. 4 (1), 47-68 (2017).
- Ikeda, S., et al. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. Journal of Biological Chemistry. 273 (34), 21585-21593 (1998).
- Rosenquist, T. A., Zharkov, D. O., Grollman, A. P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proceedings of the National Academy of Science U. S. A. 94 (14), 7429-7434 (1997).
- Reid, D. A., et al. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proceedings of the National Academy of Science U. S. A. 112 (20), 2575-2584 (2015).
- Taccioli, G. E., et al. Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science. 265 (5177), 1442-1445 (1994).
- Marsin, S., et al. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. Journal of Biological Chemistry. 278 (45), 44068-44074 (2003).
- Thompson, L. H., Brookman, K. W., Jones, N. J., Allen, S. A., Carrano, A. V. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Molecular and Cell Biology. 10 (12), 6160-6171 (1990).
- Scharer, O. D.
- Oeck, S., et al. High-throughput evaluation of protein migration and localization after laser micro-irradiation. Science Reports. 9 (1), 3148 (2019).
- Mistrik, M., et al. Cells and stripes: A novel quantitative photo-manipulation technique. Science Reports. 6, 19567 (2016).
- Durand, R. E., Olive, P. L. Cytotoxicity, mutagenicity and dna damage by hoechst 33342. Journal of Histochemistry and Cytochemistry. 30 (2), 111-116 (1982).
- Tobey, R. A., Oishi, N., Crissman, H. A. Cell cycle synchronization: reversible induction of G2 synchrony in cultured rodent and human diploid fibroblasts. Proceedings of the National Academy of Science U. S. A. 87 (13), 5104-5108 (1990).
- Podhorecka, M., Skladanowski, A., Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. Journal of Nucleic Acids. 2010, (2010).
