

Summary
Este protocolo descreve um método não invasivo para identificar eficientemente células da fase S para estudos de microscopia a jusante, como medir o recrutamento de proteínas de reparação de DNA por micro-irradiação a laser.
Abstract
O reparo de danos no DNA mantém a integridade genética das células em um ambiente altamente reativo. As células podem acumular vários tipos de dano de DNA devido a fontes endógenas e exógenas, como atividades metabólicas ou radiação UV. Sem reparação de DNA, o código genético da célula fica comprometido, minando as estruturas e funções das proteínas e potencialmente causando doenças.
Compreender a dinâmica espostetemporal das diferentes vias de reparação de DNA em várias fases do ciclo celular é crucial no campo da reparação de danos no DNA. As técnicas atuais de microscopia fluorescente fornecem ótimas ferramentas para medir a cinética de recrutamento de diferentes proteínas de reparo após a indução de danos no DNA. A síntese de DNA durante a fase S do ciclo celular é um ponto peculiar no destino celular em relação à reparação do DNA. Ele fornece uma janela única para tela de todo o genoma em busca de erros. Ao mesmo tempo, os erros de síntese de DNA também representam uma ameaça à integridade do DNA que não é encontrada em células não-divisórias. Portanto, os processos de reparação de DNA diferem significativamente na fase S em comparação com outras fases do ciclo celular, e essas diferenças são mal compreendidas.
O protocolo a seguir descreve a preparação de linhas celulares e a medição da dinâmica das proteínas de reparação de DNA na fase S em locais de dano de DNA induzidos localmente, usando um microscópio confocal de varredura a laser equipado com uma linha laser de 405 nm. PcNA marcada (com mPlum) é usado como um marcador de ciclo celular combinado com uma proteína de reparação rotulada acgFP (ou seja, EXO1b) para medir o recrutamento de danos de DNA na fase S.
Introduction
Várias vias de reparação de DNA evoluíram para abordar os diferentes tipos de lesões de DNA que podem surgir nas células, todas altamente reguladas no espaço e no tempo. Um dos períodos mais vulneráveis do ciclo celular é a fase S, quando ocorre a síntese de DNA. Embora a proliferação seja fundamental para a vida, ela também oferece um grande desafio. As células precisam garantir a replicação fiel de seu genoma para evitar que mutações sejam transmitidas para as gerações futuras. Consequentemente, a proliferação proporciona um ponto terapêutico de intervenção que tem sido empregado para o desenvolvimento de abordagens terapêuticas no campo da oncologia.
Todas as principais técnicas utilizadas para o estudo do recrutamento de proteínas em lesões de DNA têm seus pontos fortes e limitações. A micro-irradiação tem melhor resolução espacial e temporal1 do que a maioria dos métodos alternativos como imagens imunofluorescentes de focos induzidos por radiação ionizante (IRIF), cromatina-imunoprecipitação (ChIP) ou fracionamento bioquímico. No entanto, a micro-irradiação mistura a robustez das técnicas acima mencionadas que podem amostrar um grande número de células ao mesmo tempo.
Para investigar o reparo de DNA na fase S, é preciso ser capaz de distinguir células da fase S em uma população de cultura celular assíncrona. Existem muitos métodos bem conhecidos para lidar com isso, envolvendo a sincronização das células ou a visualização das diferentes fases do ciclo celular. No entanto, ambas as abordagens introduzem desafios significativos e possíveis artefatos. Métodos de sincronização química amplamente utilizados para enriquecer células na fase S inicial (por exemplo, bloco de timmidina dupla, aphidicolina e tratamento de hidroxyurea) alcançam a sincronização através da indução do estresse de replicação e eventualmente danos do DNA em si. Isso limita o uso desses métodos para estudar processos de reparação de DNA na faseS 2. A sincronização através da fome e liberação de soro só é aplicável a um número limitado de linhas celulares, excluindo em grande parte linhas de células cancerígenas que dependem menos de fatores de crescimento para a progressão do ciclo celular em comparação com linhas celulares não transformadas. O sistema FUCCI (Fluorescence Ubiquitin Cell Cycle Indicator, indicador de ciclo celular de fluorescência) é uma ferramenta particularmente útil para estudar o ciclo celular, mas tem uma limitação fundamental ao diferenciar entre as fases do ciclo celular S e G23.
Aqui é demonstrado que o uso de PCNA fluorescentemente marcado como um marcador não invasivo para a fase S limita as desvantagens dos métodos de sincronização do ciclo celular químico, ao mesmo tempo em que permite mais especificidade e flexibilidade do que o sistema FUCCI. Como um único marcador, o PCNA não só pode destacar células de fase S em uma população assíncrona, mas também pode mostrar a progressão exata das células dentro da fase S (ou seja, precoce, média ou tardia fase S)4. Baixos níveis de expressão de PCNA exógeno e marcado garante interferência mínima tanto com a progressão do ciclo celular quanto com os processos de reparação de DNA. É importante ressaltar que o PCNA também serve como um controle interno para a indução adequada de danos ao DNA, pois está envolvido na reparação de várias lesões de DNA e é recrutado para locais de dano de DNA induzido localmente1,4.
Os experimentos aqui apresentados demonstram como medir a dinâmica de recrutamento do EXO1b na fase S e como isso é afetado pelo bem estabelecido inibidor parp, olaparibe. A atividade de nuclease EXO1b é relevante para uma ampla gama de vias de reparo de DNA, incluindo reparo incompatível (MMR), reparo de excisão de nucleotídeos (NER) e reparo de quebra de dois fios (DSB). Na fase S, o EXO1b desempenha um papel importante na recombinação homologous (RH) através da formação de ssDNA de 3' durante a ressecção de DNA5. EXO1b foi ainda mais implicado na replicação de DNA com papéis na ativação de checkpoint para reiniciar garfos de DNA parados parados, bem como remoção de primer e maturação de fragmentos okazaki no fio defasado durante o deslocamento do fio na replicação5. O recrutamento EXO1b para locais de DNA danificados é regulado pela interação direta com poli (ADP-ribose) (PAR)6,7. Devido às inúmeras implicações específicas do ciclo celular do EXO1b, é uma excelente escolha para estudos de recrutamento específicos da fase S usando PCNA.
Protocol
1. Cultivo de células derivadas de osteossarcoma humanas (U-2 OS)
NOTA: As células U-2 OS são ideais para esses estudos, pois possuem uma morfologia plana, núcleo grande e fortemente ligadas a várias superfícies, incluindo vidro. Outras linhas celulares com características semelhantes também poderiam ser usadas.
- Para o cultivo de linhas celulares U-2 OS, use o meio 5A da McCoy suplementado com 10% de soro bovino fetal (FBS) e antibióticos (penicilina de 100 U/mL e 100 μg/mL de estreptomicina). Incubar células a 37 °C em uma atmosfera umidificada contendo 5% de CO2. Para estudos de microscopia, mantenha a cultura celular em um prato de 10 cm para fornecer contagem celular suficiente.
- Quando as células se aproximam de 90% de confluência (~7 x 106 células/10 cm de prato), divida as células.
- Enxágüe células com PBS para lavar inibidores de trippsina contidos dentro do soro.
- Adicione 1 mL de Trypsin-EDTA e certifique-se de que a camada celular esteja igualmente coberta.
- Incubar a 37 °C até que a camada celular seja retirada da placa (aproximadamente 6 min).
- Resuspenque as células experimentpsinizadas no soro contendo mídia para inativar a trippsina e adicione 1/10 do volume (~0,7 x 106 células) em uma nova placa de 10 cm contendo 10 mL de meio de crescimento suplementar.
- Antes da experimentação, teste rotineiramente células para contaminação por mycoplasma usando o kit universal de detecção de mycoplasma seguindo a recomendação do fabricante.
2. Infecção retroviral
NOTA: Para medidas de segurança BSL-2 e enquanto trabalha com vírus recombinantes, consulte: Diretrizes do NIH, Seção III-D-3: Vírus recombinantes na cultura tecidual.
- Sementes 4 x 106 células HEK293T para alcançar ~60% de confluência dentro de 24 horas depois de emplacar em um prato de cultura de 10 cm.
- Para cultivar HEK293T siga os passos de cultivo do U-2 OS descritos em 1.1-1.3 deste protocolo. Para hek293T substituir o meio 5A da McCoy para DMEM. Certifique-se de sempre lavar suavemente as células HEK293T à medida que se prendem fracamente às placas de cultura tecidual.
- Células HEK293T transfectas usando um reagente de transfecção à base de lipídios para embalagem viral de plasmídeos.
- Para vetores retrovirais, combine 1,5 μg de vetores de embalagem VSV-G (Addgene #8454) e 1,5 μg de pUMVC (Addgene #8449) juntamente com vetores de embalagem de pUMVC (Addgene #8449) juntamente com 3 μg do vetor contendo o gene de interesse (em uma espinha dorsal vetorial retroviral com resistência à puramicina) em 250 μL de opti-MEM reduziu a mídia de soro em um tubo de microcentrifuuge. Adicione 1 μL de reagente P3000 para cada μg de DNA adicionado na mistura Opti-MEM/DNA (neste caso 6 μL) e misture suavemente tocando. Não vórtice ou pipeta para cima e para baixo.
- Em outro tubo de microcentrifuge, combine 2 μL por μg de DNA (neste caso 12 μL) de reagente de transfecção com 250 μL de mídia de soro reduzida Opti-MEM.
- Combine as duas misturas (500 μL combinadas, não vórtice, apenas misture por toque suave) e deixe incubar por 15 minutos à temperatura ambiente.
- Cuidadosamente, adicione a mistura dropwise às células HEK293T sem separar as células. Gire as placas suavemente.
- Infecção viral para gerar linhas celulares estáveis.
- Remova o vírus contendo supernasce das células HEK293T 72 h após a transfecção. Filtre cuidadosamente a solução com um filtro de 0,45 μm para remover detritos celulares e células separadas. Opcionalmente, adicione 8 μg/mL de polibrene ao supernatante viral para facilitar a infecção viral.
- Adicione vírus contendo supernascedores às células U-2 OS a ~50% de confluência em uma antena de 10 cm (~3 x 106 células). Semearam as células do Sistema Operacional U-2 no dia anterior.
- Infecte por 6-16 h antes de remover e descartar o supernanato contendo vírus.
NOTA: Para alcançar a quantidade desejada de superexpressão para o gene de interesse, incubar uma série de diluições virais por um período fixo de tempo. Verifique os níveis de expressão do transgene em cada linha celular recém-estabelecida com a mancha ocidental comparando-a com níveis endógenos. - Permitir que as células selecionem na presença de antibióticos apropriados (por 3-4 dias em caso de puramicina a 2 μg/mL de concentração final) e verifique a expressão do gene de interesse marcado pela proteína fluorescente sob um microscópio.
- Repita estas etapas para gerar linhas celulares duplamente rotuladas. Nos experimentos aqui apresentados, o mPlum-PCNA foi expresso a partir de um vetor retroviral (pBABE) combinado com EXO1B-AcGFP, também expresso a partir de um vetor retroviral (pRetroQ-AcGFP1-N1).
3. Preparação de células para micro-irradiação
- Células de revestimento: 24 h antes do experimento, placa um total de 8,0 x 104 células em um volume entre 500 μL-1 mL de mídia (para cerca de 70% de confluência) em uma tampa de quatro câmaras bem com um fundo de vidro nº 1,5 borossilicate que fornece resultados ideais para microscopia confocal de alta ampliação e micro-irradiação laser. Uma maior confluência celular permite mais células medidas em um único campo de visão (FOV); no entanto, slides totalmente confluentes introduzirão irregularidades no ciclo celular.
- Mídia de imagem: Uma hora antes da micro-irradiação, troque o meio de crescimento regular para FluoroBrite DMEM complementado com 10% de FBS, penicilina de 100 U/mL e 100 μg/mL streptomicina, 15 mM HEPES (pH=7,4) e 1 mM de piruvato de sódio. Esta mídia de imagem ajuda a maximizar a relação sinal-ruído permitindo a detecção de fluorescência muito fraca. Uma vez que contém HEPES, também estabiliza o pH na ausência de uma atmosfera de CO2 de 5%.
- Aplique qualquer tratamento adicional antes da imagem nesta etapa. Nos experimentos aqui apresentados, as células foram pré-tratadas uma hora antes da imagem com olaparibe (inibidor PARP, a 1 μM de concentração final) ou um controle de veículo (DMSO)1,8,9.
4. Preparar o microscópio e selecionar células de fase S para imagem.
- Use um sistema confocal que tenha as propriedades semelhantes às do sistema aqui delineadas para obter melhores resultados. Os experimentos aqui apresentados foram realizados utilizando um microscópio confocal montado em um suporte de microscópio invertido (ver Tabela de Materiais).
NOTA: O microscópio utilizado aqui foi equipado com um módulo laser FRAP de 50 mW 405 nm e um objetivo 60x 1.4 NA oil plan-apochromat. O scanhead confocal tinha duas opções de scanner: um scanner galvano (para alta resolução) e um scanner ressonante (para imagens de alta velocidade).- Introduza a recuperação da fluorescência após o laser de fotobleaching (FRAP) na amostra através de um dispositivo de galvano XY controlado por software. Use uma linha laser de 488 nm para excitar AcGFP e uma linha laser de 561 nm ou 594 nm para excitar o mPlum.
NOTA: A seguinte combinação de filtros dá ótimos resultados: usando um filtro de passagem longa de 560 nm, a luz de emissão com comprimento de onda inferior a 560 nm foi passada através de um filtro de emissão de 525/50 nm para AcGFP, enquanto a luz de emissão com comprimento de onda superior a 560 nm foi passada através de um filtro de emissão de 595/50 nm para mPlum. Qualquer conjunto de filtro apropriado (por exemplo, FITC/TRITC, GFP/mCherry, FITC/TxRed) que garanta o uso de sangramento mínimo de fluorescência.
- Introduza a recuperação da fluorescência após o laser de fotobleaching (FRAP) na amostra através de um dispositivo de galvano XY controlado por software. Use uma linha laser de 488 nm para excitar AcGFP e uma linha laser de 561 nm ou 594 nm para excitar o mPlum.
- Ligue a câmara ambiental e os componentes do microscópio.
- Ligue o aquecimento (etapa, objetivo e câmara ambiental quando possível), o fornecimento de CO2 e o regulador de umidade pelo menos 4h antes do início do experimento para garantir o equilíbrio térmico para aquisição estável de imagens.
- Inicialize fontes de luz junto com as linhas de laser pelo menos 1 h antes da transferência das células para o microscópio.
- Selecione células de fase S em uma população assíncrona usando PCNA marcada fluorescentemente como um marcador. Faça isso seguindo os passos abaixo.
- Procure o padrão de localização exclusivo do PCNA marcado por mPlum na fase S, tornando possível a identificação desta fase de ciclo celular. O PCNA possui uma distribuição completamente homogênea no núcleo nas fases G1 e G2 do ciclo celular, ao mesmo tempo em que é excluída do nucleoli. Na fase S, o PCNA forma focos na localização de replisomes no núcleo. A Figura 1 mostra os diferentes padrões de focos PCNA ao longo da fase S, o que torna possível até mesmo diferenciar a fase S precoce, média e tardia.
- Olhe através do ocular para selecionar um FOV que tenha células fase S suficientes para micro-irradiação. Assíncronsas células de SO U-2 geralmente têm 30-40% de sua população em fase S.
- Tente evitar extremos nos níveis de expressão (células brilhantes e fracas) tanto para PCNA quanto para a proteína de interesse (POI), neste caso EXO1b-AcGFP, o que poderia levar a artefatos experimentais.
- Ao encontrar um FOV adequado, tente evitar digitalizar o campo por um longo tempo para minimizar o esflicamento de fotos e danos indesejados do DNA.
- Defina a região de interesse desejada (ROI) para micro-irradiação. Utilizando o software associado (ver Tabela de Materiais),defina o ROI desejado inserindo primeiramente linhas binárias (defina o número desejado de linhas e espaçamento). Clique em Binário, em seguida, clique em Inserir linha | | círculo Elipse para desenhar o número desejado de linhas.
- Converta essas linhas binárias em ROIs e, finalmente, converta esses ROIs em ROIs de estimulação. Para fazer isso, primeiro clique no ROI, depois clique em Mover Binário para ROI, em seguida, clique com o botão direito do mouse em qualquer um dos ROIs e selecione Usar como ROI de estimulação: S1. Coloque essas linhas no FOV para passar pelo núcleo das células. ROIs com um comprimento de 1024 pixels que abrangeram todo o FOV foram usados em todo o protocolo.
5. Micro-irradiação para coloração de imunofluorescência ou imagem de lapso de tempo.
- Determinando as configurações ideais de micro-irradiação.
- Antes da micro-irradiação das células, tome uma imagem de maior resolução do FOV para identificar focos PCNA para análise posterior. Em vez de digitalização sequencial, registos ópticos utilizados simultaneamente (verde e vermelho), para evitar o movimento celular entre a varredura nos dois comprimentos de onda. Para a resolução adequada dos focos use pelo menos 1024 x 1024 pixels/resolução de campo com zoom de 1x (tamanho de pixel de 0,29 μm no sistema de imagem usado aqui), com velocidade de digitalização de 1/8 quadro/s (4,85 μs/pixel) com média de 2x. Uma vez definidos esses parâmetros na GUI compacta A1 LFOV e nas janelas da Área de Varredura A1 LFOV, aperte o botão Capture para gravar o FOV.
NOTA: É importante manter o mesmo tamanho de pixel ao longo dos experimentos para garantir resultados comparáveis. - Para configurar a micro-irradiação, abra a guia ND Stimulation no software de imagem para acessar a janela cronograma (A1 LFOV / Galvano Device). Isso usa os scanners galvano para adquirir uma série de imagens pré-estimulação, estimular (usando o laser LUN-F 50 mW 405 nm FRAP), e então adquirir uma série de imagens pós-estimulação novamente usando os scanners galvano. Primeiramente configurar três fases na janela de horários. Na coluna Acq/Stim selecione Aquisição | | branqueamento Aquisição para as três fases, respectivamente. Para a fase de branqueamento, defina S1 como o ROI.
NOTA: No experimento aqui apresentado, nenhuma imagem foi adquirida durante a fase de estimulação. - Na janela Galvano XY, configure os principais fatores para a micro-irradiação: saída de energia laser de 405 nm, tempo de moradia (iteração é 1 por padrão neste sistema). Nos experimentos aqui apresentados, as células foram irradiadas com o laser FRAP de 405 nm (50 mW na ponta da fibra) a 100% de potência com um tempo de 1000-3000 μs de moradia.
NOTA: Como o tempo de moradia do laser é em uma base por pixel, enquanto o tamanho do pixel permanecer o mesmo, a relação entre o tempo de moradia e a densidade de energia será comparável entre diferentes FOVs. A Figura 2A mostra o uso de proteínas específicas da via de resposta a danos de DNA (DDR) (FBXL10 para DSBs e NTHL1 para danos na base oxidativa) para otimizar as configurações de energia laser para indução específica de danos. Estas linhas celulares estáveis foram geradas com infecção viral seguindo a seção 2 do protocolo.
- Antes da micro-irradiação das células, tome uma imagem de maior resolução do FOV para identificar focos PCNA para análise posterior. Em vez de digitalização sequencial, registos ópticos utilizados simultaneamente (verde e vermelho), para evitar o movimento celular entre a varredura nos dois comprimentos de onda. Para a resolução adequada dos focos use pelo menos 1024 x 1024 pixels/resolução de campo com zoom de 1x (tamanho de pixel de 0,29 μm no sistema de imagem usado aqui), com velocidade de digitalização de 1/8 quadro/s (4,85 μs/pixel) com média de 2x. Uma vez definidos esses parâmetros na GUI compacta A1 LFOV e nas janelas da Área de Varredura A1 LFOV, aperte o botão Capture para gravar o FOV.
- Imagem de lapso de tempo.
- Configure imagens de lapso de tempo para a janela de tempo desejada e intervalos usando o cronograma de tempo, gui compacta A1 LFOV e as janelas A1 LFOV Scan Area. Nos experimentos aqui apresentados, o recrutamento de EXO1b e PCNA foi imageado por 12 min, escaneando o FOV a cada 5 segundos, em 1024 x 1024 pixels/campo, usando zoom de 1x (resultando em tamanho de pixel de 0,29 μm no sistema de imagem usado aqui) com velocidade de varredura de 0,35 quadro/s (1,45 μs/pixel) sem uma média para reduzir o branqueamento de fotos.
- Otimize a potência do laser %, obter e compensar as configurações para reduzir o branqueamento de fotos durante a imagem na janela GUI compacta A1 LFOV. Se alguém pretende medir tanto POI quanto PCNA, use digitalização simultânea em vez de varredura sequencial para evitar o movimento celular entre a varredura do campo para os dois fluoroforos separados.
- O sistema de imagem foi utilizado com as seguintes configurações. Para a linha laser de 488 nm (20 mW): 7% de potência laser, ganho: 45 (detector GaAsP) com e deslocamento de 2, para a linha laser de 561 nm (20 mW): 4% de potência laser, ganho de 40 (detector GaAsP) e deslocamento de 2.
- Dependendo da cinética da proteína, amplie ou encurte o intervalo entre as imagens ou a duração do lapso de tempo total. Na janela Cronograma de tempo, defina o intervalo e duração desejados para a linha de aquisição da terceira fase.
- Pressione Execute agora para executar a micro-irradiação e a imagem subsequente do lapso de tempo.
- No final da imagem de lapso de tempo, salve os ROIs de estimulação como imagens separadas, o que será um auxílio útil para identificar as coordenadas da micro-irradiação em qualquer software a jusante usado para análise.
- Mancha de imunofluorescência.
NOTA: O passo 5.1.3 e a Figura 2A demonstram o uso de proteínas conhecidas de reparação de DNA para avaliar os tipos de lesões de DNA introduzidas pela micro-irradiação. Certas lesões de DNA também podem ser detectadas usando anticorpos específicos após a fixação das células. Também é possível detectar o recrutamento do POI pela detecção de anticorpos da proteína endógena. A visualização de γH2A.X para verificar se há DSBs é demonstrada abaixo (Figura 2B). A Figura 3 mostra a consistência da localização e recrutamento do PCNA ao longo do ciclo celular tanto para PCNA endógena quanto exógena.- Após a etapa 5.1.3, pegue apenas uma imagem após a micro-irradiação para garantir o evento FRAP adequado com base no recrutamento de mPlum-PCNA. Tome nota das coordenadas exatas do FOV para encontrar o campo mais tarde após a rotulagem imunofluorescente.
- Tire a câmara de cultura celular do microscópio e incuba as células a 37 °C em uma atmosfera umidificada contendo 5% de CO2 por 5-10 min.
NOTA: Paraformaldeído (PFA) é tóxico, e o trabalho deve ser feito em uma área bem ventilada ou em um capô de fumaça. Toda a lavagem e incubação subsequentes será feita com volumes de 0,5 mL no slide de câmara de 4 poços. Após o tempo de incubação, lave as células com 0,5 mL de PBS (137 mM NaCl, 2,7 mM KCl, 8 mM Na2HPO4e 2 mM KH2PO4) e fixe com 0,5 mL de 4% PFA em PBS para 10 min em temperatura ambiente (RT). - Lave as células uma vez com PBS, depois lave-as com 50 mM NH4Cl para saciar o PFA residual.
- Permeabilize as células por 15 min em RT com Triton X-100 de 0,1% na PBS.
- Bloqueie as amostras por 1h com tampão de bloqueio (5% FBS, 3% BSA, 0,05% Triton X-100 em PBS).
- Remova a solução de bloqueio e adicione o anticorpo primário diluído (anti-γH2A.X, 1:2000) no buffer de bloqueio por 1 h no RT.
- Lave os poços com tampão de bloqueio 3 x 10 min.
- Adicione anticorpo secundário diluído (anticorpo Alexa 488 Plus conjugado, 1:2000) no buffer de bloqueio por 1 h no RT.
- Lave os poços com tampão de bloqueio 3 x 10 min.
- Contratente o núcleo com solução DAPI de 1 μg/mL em PBS por 15 min.
- Lave as células uma vez com PBS. A imagem pode ser realizada diretamente em PBS ou uma solução PBS com reagentes antifade (por exemplo, AFR3) para reduzir o fotobleaching.
6. Análise de recrutamento
NOTA: A Figura 4A mostra imagens representativas do recrutamento do Exo1b e pcna na presença de DMSO ou olaparibe. A Figura 4B mostra uma imagem representativa para análise de dados. Os valores médios de fluorescência foram calculados medindo intensidades médias de AcGFP usando um retângulo ao longo da faixa laser destacada pelo mPlum-PCNA (A, retângulos amarelos) em diferentes pontos de tempo usando Fiji. O PCNA pode servir como um controle interno para destacar a irradiação bem sucedida ao longo das coordenadas do ROI. Da mesma forma, os valores médios de fluorescência acgfp também foram calculados para regiões não danificadas do núcleo (B, retângulos azuis). A intensidade do sinal de fundo foi medida em áreas não povoadas (C, retângulos vermelhos) e foi subtraída dos valores fluorescentes médios(Figura A e B). Assim, a unidade fluorescente média relativa (RFU) para cada ponto de coleta de dados foi calculada pela equação RFU = (A − C)/ (B − C)8,9. Os valores resultantes da RFU da região micro-irradiada são normalizados para os valores da RFU antes da micro-irradiação.
- Para definir a região A do sítio micro-irradiado, exclua regiões nucleolares, focos de replicação e regiões nucleares irregulares da célula da medida. Segure a chave de mudança entre o desenho de dois ROIs em Fiji para agrupar duas regiões separadas como uma.
NOTA: O recrutamento de proteínas variará entre diferentes genes e condições de irradiação; assim, o tamanho da região A deve ser determinado individualmente. Uma vez determinada a largura do pixel da região A, ela deve permanecer constante para quaisquer recrutamentos comparativos. Nos experimentos aqui apresentados, foram utilizados retângulos de largura de 7 pixels. - Exclua células que se moveram durante a duração dos vídeos gravados da análise. Para incluir células altamente móveis, a análise descrita deve ser realizada quadro a quadro.
- Para visualizar o perfil de recrutamento, plote os valores de RFU normalizados contra o tempo usando um software estatístico.
- Calcule a diferença em um ponto de tempo indicado entre o tratamento DMSO e olaparibe (n=31) usando um teste mann-whitney.
Representative Results
As células abordam cada tipo de lesão de DNA de forma específica que também depende de qual fase de ciclo celular elas estão. Por exemplo, após a microdiarração, as quebras duplas encalhadas (DSB) serão processadas por junção final não homólogo (NHEJ) ou RH, dependendo da fase do ciclo celular. Os núcleos que atuam mais extensivamente durante as fases S e G2 do ciclo celular criam saliências de DNA que são cruciais para o RH adequado. Para promover a avaliação das células na fase S, o PCNA foi utilizado como marcador de ciclo celular de cor única. A Figura 1A mostra o perfil de localização do mPlum-PCNA durante a progressão do ciclo celular. O PCNA possui uma distribuição completamente homogênea no núcleo na fase G1 e G2 (ao mesmo tempo em que é excluída principalmente do nucleoli). Na fase S, o PCNA localiza-se em locais de replicação de DNA, que podem ser visualizados como pontos brilhantes no núcleo. Nas primeiras células da fase S, as manchas são relativamente pequenas e igualmente distribuídas por todo o núcleo da célula. Progredindo para a fase média S, as manchas ficam borradas e se localizam mais em direção ao perímetro do núcleo e do nucleoli. Na fase final de S, as manchas reduzem em números, mas se tornam cada vez maiores à medida que o PCNA se concentra em locais de replicação tardios(Figura 1B). É importante ressaltar que a expressão pcna exógena da espinha dorsal do vetor pBABE foi menor que os níveis endógenos, mas foi suficiente para detecção por microscopia que minimiza artefatos potenciais na progressão do ciclo celular e DDR. A Figura 1C mostra a extensão da superexpressão do PCNA em comparação com os níveis endógenos. Observe que a banda correspondente ao mPlum-PCNA migra mais lentamente devido ao seu tamanho maior.
Nosso objetivo era introduzir DSBs durante a microdiesão para investigar o recrutamento parp1/2 dependente do EXO1b a essas lesões na fase S. A Figura 2A mostra que baixas doses de energia (1000 μs de moradia) não induzem o recrutamento de EGFP-FBXL10, um respondente DSB (componente do complexo FRUCC 8),enquanto foi suficiente para induzir o recrutamento de NTHL1-mCherry, uma proteína de caminho de reparo de excisão base (BER), recrutando para locais de dano oxidativo de DNA10,11,11. Com 3000 μs de moradia, tanto o EGFP-FBXL10 quanto o NTHL1-mCherry recrutam, demonstrando uma saída de laser que gera lesões oxidativas e DSBs. Fortalecendo esses resultados, a Figura 2B mostra a coloração da imunofluorescência contra γH2A.X (marcador DSB), que é claramente mais aparente ao usar doses de energia mais altas. O PCNA serve tanto como um marcador de ciclo celular quanto um marcador para micro-irradiação bem-sucedida, pois recruta adequadamente com as configurações de tempo de moradia a laser. É importante ressaltar que tanto o PCNA com proteína exógena e/ou endógena fluorescente pode ser usado para esta função repórter, pois eles se comportam da mesma forma(Figura 3). PCNA etiquetado endogenously foi projetado inserindo mRuby em quadro com o primeiro exon em um alelo do lócus PCNA13 (a linha celular era uma espécie de presente de Jörg Mansfeld).
A Figura 4A e a Figura 4C mostram o recrutamento de células de fase EXO1b marcadas pela AcGFP em S. EXO1b atinge o nível máximo de acumulação em locais de micro-irradiação em torno de 1 minuto e, em seguida, lentamente começa a se desengajar das lesões de DNA depois. Os enriquecimentos em locais de micro-irradiação são denotados por uma unidade de fluorescência relativa de 1 > no gráfico. Na presença de olaparibe, o acúmulo de EXO1b na faixa laser em 1 minuto é significativamente menor em comparação com o controle do veículo. Esses resultados estão de acordo com a literatura6,7. A Figura 4B demonstra regiões representativas para quantificação (áreas A, B e C) conforme descrito no ponto 6 do protocolo. A Figura 4D mostra os níveis de expressão comparáveis do EXO1b endógeno e exógeno EXO1b-AcGFP em células utilizadas para micro-irradiação.
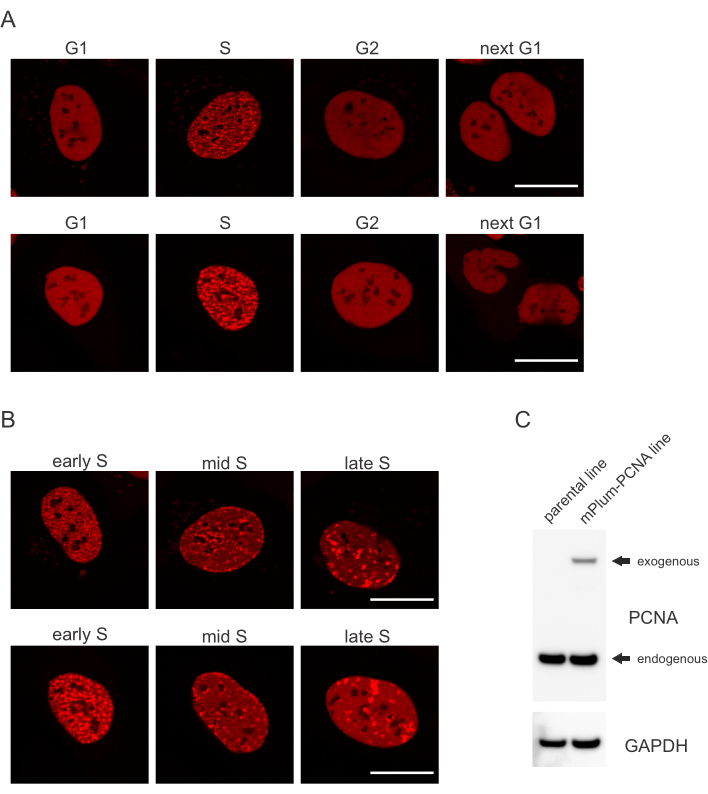
Figura 1: Padrão de localização do PCNA. (A) As imagens mostram o padrão de localização de PCNA estávelmente integrada e exógena durante todo o ciclo celular em células U-2 OS. (B) As imagens mostram padrões de focos PCNA em diferentes estágios da fase S (precoce, médio e tardio) em células de SO U-2. (C) Mancha ocidental mostrando níveis endógenos e exógenos de PCNA nas células U-2 OS utilizadas para imagem. A barra de escala representa 20 μm. Por favor, clique aqui para ver uma versão maior desta figura.

Figura 2: Indução de DSBs através de saída de energia laser otimizada. (A) As configurações a laser podem ser otimizadas para induzir diferentes formas de dano de DNA. As células de SO U-2 expressando tanto egfp-FBXL10 quanto NTHL1-mCherry foram utilizadas para identificar DSBs e locais de lesões oxidativas, respectivamente. A micro-irradiação com uma linha laser de 405 nm foi realizada em células assíncronsas U-2 OS com 1000 μs ou 3000 μs de tempo de moradia. Barra de escala representa 20 μm. (B) A coloração imunofluorescente contra γH2A.X foi feita em células epiteliais de pigmento de retina humana (hTERT RPE-1) tendo PCNA endógena marcada por mRuby. As células foram fixadas e processadas 5 minutos após a micro-irradiação com 1000 μs ou 3000 μs de tempo de moradia. A barra de escala representa 20 μm. Por favor, clique aqui para ver uma versão maior desta figura.

Figura 3: Recrutamento comparável de mRuby-PCNA endógeno e exógeno mPlum-PCNA para locais de micro-irradiação a 1000 μs ou 3000 μs laser dwell time. Tanto focos de replicação de forma PCNA endógeno quanto exógeno marcados durante a fase S. Clique aqui para ver uma versão maior desta figura.

Figura 4: PARP1/2-dependent recrutamento do EXO1b na fase S. As células de so U-2 expressando stably EXO1b-AcGFP e mPlum-PCNA foram micro-irradiadas com linha laser FRAP de 405 nm usando 3000 μs de tempo de moradia. (A) Imagens representativas de células microdirratadas nos pontos de tempo indicados após o pré-tratamento com controle de veículo (DMSO) ou olaparibe (1 μM). Barra de escala representa 20 μm. (B) Imagens representativas de regiões definidas das áreas A, B e C para a análise de recrutamento. A barra de escala representa 20 μm. (C) A dinâmica de recrutamento de danos de DNA foi capturada por imagens de células vivas. Valores e imagens de fluorescência média relativa foram adquiridos a cada 5 s por 12 min. Para cada condição, foram avaliadas ≥30 células. Os valores médios de fluorescência relativa (linhas pretas sólidas) e erro padrão (alcance visualizado por uma área sombreada) foram plotados contra o tempo. A linha tracejada mostra valores de recrutamento em 1 min após a micro-irradiação. A diferença entre o tratamento de DMSO (n=32) e olaparibe (n=31) foi calculada por meio de um teste de Mann-Whitney. Astérix denota p<0.0001. (D) A mancha ocidental compara os níveis de expressão do exogenous EXO1b e exógeno EXO1b-AcGFP em células utilizadas para micro-irradiação. Clique aqui para ver uma versão maior desta figura.
Discussion
Etapas críticas e possíveis problemas de solução de problemas/modificações do protocolo
O vaso de cultura tecidual adequado para micro-irradiação é fundamental para o sucesso. A maioria dos sistemas de imagem de alta resolução são otimizados para espessura de vidro de cobertura de 0,17 mm. O uso de câmaras de imagem de espessura maior ou inferior ou feitas de polímeros plásticos (não otimizados para imagens de 405 nm), pode reduzir significativamente a qualidade da imagem. Ao usar superfícies de vidro, certifique-se de que elas são de cultura tecidual tratada para aumentar a adesão celular. Se não forem tratadas com cultura tecidual, essas câmaras precisarão ser revestidas, por exemplo, com poli-d-lysina antes de semear as células. Ao emplacar células no vidro de cobertura câmara, a densidade celular ideal é primordial para evitar irregularidades no ciclo celular e estresse adicional para as células. O equilíbrio térmico adequado dos componentes do microscópio antes da experimentação para manter uma temperatura estável é crucial tanto para manter o foco durante todo o lapso de tempo quanto também é necessário para garantir um DDR homogêneo através do tempo e amostras.
É fundamental que as células estejam em uma condição saudável antes da micro-irradiação para reduzir os dados artefatos. Se as células tiverem morfologia irregular pós-infecção/seleção, permitam que as células progridam através de múltiplas passagens até que a morfologia volte ao normal. Certifique-se sempre de que as linhas de células utilizadas estão livres de contaminação por mycoplasma. Entre os muitos efeitos adversos da infecção por micoplasma, também causa danos ao DNA das células hospedeiras e pode afetar suas vias DDR14,15. A maneira mais sensível de detectar mycoplasma na cultura celular é através do PCR (versus. detecção com DAPI ou Hoechst).
A superexpressão ideal da proteína de reparação de interesse deve ser comparável aos níveis endógenos, no entanto, alto o suficiente para detecção. O promotor usado nos vetores virais, no título viral durante a infecção e no tempo de infecção podem ser ajustados para níveis ideais de expressão. Para resultados consistentes, isole clones de células individuais para garantir níveis de expressão homogêneos e morfologia celular normal. Recomenda-se o uso de construções vetoriais que não expressam pcna etiquetada em níveis superiores aos níveis endógenos para a função adequada do ciclo celular e do marcador de reparo de DNA. Mesmo os baixos níveis de superexpressão do PCNA são suficientes para discriminar células da fase S. Os vetores retrovirais pBABE foram usados com sucesso para este fim (Addgene #1764, #1765, #1766, #1767). PCNA pode ser marcado com qualquer vermelho monomérico(por exemplo, mPlum, mCherry, mRuby, etc.) ou proteínas fluorescentes verdes monoméricas (por exemplo, mEGFP, AcGFP, mWasabi, mNeonGreen, mEmerald, etc.) que poderiam então ser combinadas com um POI marcado alternadamente. Superexpressar um POI marcado fluorescentemente tem algumas limitações e considerações. As etiquetas fluorescentes podem interromper a função normal da proteína e a localização. Assim, deve-se considerar a localização da tag (N ou terminal C). Use sempre proteínas fluorescentes monoméricas, pois a oligomerização de variantes não monoméricas pode afetar a função do POI.
As configurações de laser devem ser determinadas para cada sistema de imagem, pois muitos componentes do caminho óptico afetarão a potência real entregue nas células. A micro-irradiação a laser pode causar vários tipos de lesões de DNA dependendo do comprimento de onda de excitação, da saída de energia do laser FRAP e se algum agente pré-sensibilizador (como Bromodeoxyuridine ou Hoechst) foi usado. Lasers de 405 nm podem causar danos oxidativos ao DNA, quebras de um único e duplo encalhados16,17. Usando configurações de saída de laser mais altas, a quantidade de DSBs aumenta. Neste protocolo, não foram utilizados métodos de pré-sensibilização, mas essas técnicas são muito abordadas na literatura e recapitudas na discussão abaixo. Em nossa opinião, a melhor maneira de testar se a lesão desejada é gerada é através de testes para o recrutamento de genes específicos da via de dano de DNA conhecidos. O recrutamento de NTHL1 ou OGG1, componentes da via BER, sugere a indução de bases de DNA oxidadas10,11,17,18,19, enquanto FBXL10 ou XRCC5 indicam a presença de DSBs8,20,21. O recrutamento do XRCC1 pode indicar tanto a presença de bases de DNA oxidadas quanto quebras únicas encalhadas (SSB)22,23. XPC (i.e., RAD4) é um bom indicador de NER que remove os adutos de DNA volumosos gerados pela luz ultravioleta (UV)17,24. Como o recrutamento de proteínas exógenas pode introduzir certas irregularidades, a coloração imunofluorescente de proteínas ou marcadores de reparação de DNA endógeno (como γH2A.X para quebras duplas) pode confirmar a presença de lesões específicas de DNA. Alternativamente, anticorpos levantados contra tipos específicos de lesões de DNA também poderiam ser usados. Para ajustar a potência do laser entregue, tanto o tempo de moradia quanto a potência do laser podem ser alterados.
Com a ajuda da modelagem matemática, poderia ser realizada uma análise cinética detalhada que pode fornecer insights valiosos sobre as propriedades de recrutamento do POI (por exemplo, contribuição de múltiplos domínios de ligação de DNA, sensibilidade para diferentes eventos de sinalização, etc.). A avaliação automatizada de recrutamento e o rastreamento de células podem ser combinados para criar fluxos de trabalho robustos 1,25.
Vantagens e limitações da pré-sensibilização do DNA
A pré-sensibilização do DNA antes da micro-irradiação é uma ferramenta comumente usada para recrutamento de proteínas de reparação de DNA16,17. Sensibilizar o DNA antes da micro-irradiação o deixa mais suscetível aos DSBs. Os dois métodos mais comuns para pré-sensibilização do DNA são o pré-tratamento de células com bromodeoxyuridina (BrdU) ou corante hoechst. Para sistemas não capazes de micro-irradiação em altas potências laser, esses métodos podem ser necessários para induzir lesões de DNA como DSBs. Além disso, na ausência de um detector de luz transmitido ou de um sinal fluorescente destacando o núcleo celular (por exemplo, ao estudar o recrutamento de proteínas de reparação de DNA endógenas não sinalizadas), Hoechst atua como uma ferramenta pré-sensibilizadora e uma mancha fluorescente nuclear. No entanto, a pré-sensibilização do DNA pode introduzir complicações significativas. A BRDU (usada em uma concentração final de 10 μM) deve ser adicionada às células 24 horas (ou tempo equivalente a um ciclo celular completo na linha celular utilizada) para incorporar adequadamente no DNA e pode causar interferência no ciclo celular26. Hoechst 33342 (usado em uma concentração final de 1 μg/mL) é citotóxico após longos períodos de incubação, mas requer tempo suficiente para saturar o núcleo com o corante. Portanto, só deve ser aplicado 15-20 minutos antes da microdiarração; caso contrário, os dados de recrutamento não serão consistentes. As células manchadas dessa forma não podem ser mantidas na cultura por mais de algumas horas27,28. Certifique-se de não usar hoechst 33358, que não é tão permeável celular quanto o corante Hoechst 33342. A pré-sensibilização também pode introduzir variância desnecessária entre os experimentos e torna o experimento ainda mais sensível às diferenças na densidade celular (pois isso afetará a quantidade de corante/célula incorporada).
Vantagens e limitações da microscopia confocal
A velocidade de imagem da microscopia confocal pode ser limitante quando comparada à microcopia de campo largo. No entanto, um microscópio confocal equipado com um scanner ressonante pode melhorar tremendamente a velocidade de imagem (ao custo da resolução) chegando perto das velocidades de microscopia de disco giratório. Três recursos fazem do sistema confocal A1R HD25 uma excelente escolha para o protocolo apresentado aqui. Primeiro, o FOV de 25 mm do sistema permite a imagem entre 15-20 células em um único campo digitalizado (vs. 5-10 células em configurações regulares), limitando o número de aquisições necessárias para obter células suficientes para análise estatística. Em segundo lugar, o módulo FRAP e dois scanheads tornam possível a imagem e micro-irradiação das células simultaneamente, não apenas sequencialmente. Por fim, a flexibilidade de ter tanto os scanners ressonantes quanto os galvanos fornece a capacidade de alternar facilmente entre imagens de alta resolução temporal com velocidade excepcional, o que minimiza a sacieçação de fluoroforos, e imagens de alta resolução espacial que utilizam velocidades de varredura mais lentas para produzir imagens com uma maior relação de sinal para ruído. Embora o sistema utilizado permitisse que a flexibilidade acima mencionada, se assemelhasse a configurações de microscópio confocal mais amplamente disponíveis, apenas o scanner galvano foi usado nos experimentos apresentados (tanto para micro-irradiação quanto para imagens subsequentes).
Vantagens e limitações da micro-irradiação
Embora a micro-irradiação forneça resolução espacial e temporal incomparável, não é sem limitações. O dano de DNA por micro-irradiação a laser é altamente agrupado a partes específicas do núcleo em comparação com agentes prejudiciais que ocorrem naturalmente. Assim, a resposta da cromatina devido à microdiaração pode diferir em comparação com danos distribuídos homogêneamente. Além disso, a micro-irradiação é demorada e só pode ser conduzida em algumas dezenas de células, enquanto grandes métodos bioquímicos de base populacional (fracionamento de cromatina, imunoprecipitação, ChIP) podem fornecer maior robustez estudando milhares de células ao mesmo tempo. Verificar observações feitas por micro-irradiação com técnicas bioquímicas tradicionais é uma estratégia eficaz para conclusões confiáveis. Embora a micro-irradiação simultânea de muitas células em um determinado FOV seja possível, o sistema de imagem precisará de mais tempo para executar a tarefa. Portanto, medir a dinâmica das proteínas que recrutam muito rapidamente para lesões de DNA limita o número de possíveis ROIs para micro-irradiação utilizada simultaneamente. No sistema de imagem usado para este protocolo, a microdiadistração de um único ROI de 1024 pixels de comprimento leva 1032 ms usando 1000 μs de tempo de moradia e 3088 ms usando 3000 μs tempo de moradia para completar. O uso de várias linhas de ROIs aumentará significativamente o tempo necessário para terminar a micro-irradiação (por exemplo, ROI de 7 x 1024 pixels leva 14402 ms usando 1000 μs de tempo de moradia e 21598 ms usando 3000 μs de tempo de moradia). Este tempo é perdido pela aquisição de imagens e deve ser levado em consideração. Ao fotografar eventos rápidos de recrutamento, use o ROI mais curto possível e apenas micro-irradie uma célula de cada vez.
Vantagens e limitações sobre métodos de sincronização
Para estudos específicos do ciclo celular, os métodos existentes envolvem a sincronização das células em fases específicas de ciclo celular ou o uso de repórteres fluorescentes para identificar a fase específica do ciclo celular da célula. No entanto, cada um desses métodos fornece seus próprios desafios e limitações.
O sistema FUCCI3 (contando com as formas truncadas de proteína fluorescente de CDT1 e Geminin) é uma ferramenta particularmente útil para estudos de ciclo celular, mas tem limitações quando se trata de diferenciar entre as fases S e G2 do ciclo celular. Os níveis geminianos já estão altos desde a fase S média e permanecem altos até a fase M, dificultando a separação dessas fases. O uso do sistema FUCCI também significa que dois canais ópticos do microscópio não podem ser usados para a imagem do POI.
As linhas celulares não cancerígenas podem ser sincronizadas em G0 pela remoção de fatores de crescimento encontrados no soro (fome de soro) causando pouco ou nenhum dano de DNA às células. No entanto, a maioria das linhas de células cancerígenas continuará parcialmente progredindo através do ciclo celular, mesmo sem quantidades adequadas de soro em seus meios de comunicação. Além disso, as células começam parcialmente a perder a sincronização pelo G1 final, fase S inicial. Além da fome de soro, existem inúmeros métodos químicos para alcançar a sincronização do ciclo celular. Blocos de hidroxiureia, afiicolina e timidina são métodos de parar a replicação do DNA para sincronizar as células na fase S primitiva. Embora esses métodos sejam baratos e simples, eles introduzem estresse de replicação que resulta em danos ao DNA. Estes inibidores de replicação de DNA têm sido mostrados para induzir a fosforilação de H2A. X, um marcador bem conhecido de DSBs2,29. O método de usar o PCNA marcado como um marcador para células da fase S reduz o potencial para artefatos causados pela sincronização química e pode ser aplicado a uma ampla gama de linhas celulares em comparação com a fome de soro.
Conclusão
O dano ao DNA é uma força motriz para doenças genéticas onde lesões mutagênicas podem levar à transformação maligna das células. Direcionar o maquinário de síntese de DNA é uma estratégia terapêutica fundamental no tratamento de doenças hiperproliferativas como o câncer. Para tratar essas doenças de forma mais direcionada, precisamos de uma melhor compreensão das proteínas que reparam lesões de DNA. O protocolo descrito aqui ajuda estudos baseados em micro-irradiação na fase S, minimizando os desafios apresentados pelos métodos tradicionais de sincronização para reduzir possíveis artefatos e aumentar a reprodutibilidade dos experimentos.
Disclosures
Os autores afirmam que a publicação do trabalho apresentado foi patrocinada pela Nikon Corporation. Os autores declaram que não existem interesses concorrentes.
Acknowledgments
Os autores agradecem a M. Pagano por seu apoio contínuo, bem como D. Simoneschi, A. Marzio e G. Tang por sua revisão crítica do manuscrito. B. Miwatani-Minter agradece R. Miwatani e B. Minter por seu apoio contínuo. G. Rona agradece K. Ronane Jurasz e G. Rona pelo apoio contínuo.
Materials
| Name | Company | Catalog Number | Comments |
| Ammonium chloride | Sigma-Aldrich | A9434-500G | For quenching formaldehyde |
| Anti-EXO1 Rabbit Polyclonal Antibody | Proteintech | 16253-1-AP | primary antibody |
| Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301 | Millipore | 05-636 | primary antibody |
| Bovine Serum Albumin | Sigma-Aldrich | 3117332001 | BSA for blocking |
| BrdU (5-Bromo-2'-deoxyuridine) | Merck | 19-160 | pre-sensitizing agent |
| Citifluor™ Mountant Solution AFR3 | Electron Microscopy Sciences | 17973-10 | antifade containing PBS solution for imaging |
| DAPI | Sigma-Aldrich | D9542-1MG | nucleic acid stain |
| DMEM Medium | Thermo Fisher Scientific | 10569010 | Cell culture medium for HEK293T cells |
| DMSO | Sigma-Aldrich | D2650-100ML | Vehichle control and dissolution solvent |
| EGFP-FBXL10 | Addgene | #126542 | viral expression vector for EGFP-FBXL10 |
| EXO1b-AcGFP (in pRetroQ) | custom cloning | na | EXO1b cDNA was cloned in the NheI, BamHI sites of pRetroQ-AcGFP1-N1 vector. |
| Fetal Bovine Serum | Gibco | 16140071 | Media supplement |
| FluoroBrite DMEM | Thermo Fisher Scientific | A1896701 | Phenol red free medium for microscopy |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | A32723 | secondary antibody |
| HEK293T cells | ATCC | ATCC CRL-3216 | Cell line for viral packaging |
| HEPES | Sigma-Aldrich | H0887-100ML | Buffering agent to supplement live cell imaging medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | pre-sensitizing agent |
| Lipofectamine 3000 | Thermo Fisher Scientific | L3000015 | Transfection reagent |
| McCoy’s 5A (Modified) Medium | Life Technologies | 16600-108 | Cell culture medium for U-2 OS cells |
| mCherry-PCNA | Addgene | #55117 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA | Addgene | #55994 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA (in pBABE) | custom cloning | na | mPlum-PCNA cDNA was cloned from Addgene #55994 in the BamHI, SalI sites of pBABE (puro) |
| Nikon A1R-HD25 Confocal Scanhead and Controller | Nikon | na | confocal imaging system |
| Nikon LUN4 laser unit | Nikon | na | excitation system |
| Nikon LUN-F 50 mW 405 nm FRAP laser unit | Nikon | na | FRAP laser unit |
| Nikon NIS Elements Confocal Controller Software | Nikon | na | Confocal controlling software |
| Nikon Ti2-E Inverted Microscope | Nikon | na | inverted epifluorescent microscope base |
| Nikon Ti2-LAPP Modular Illumination System | Nikon | na | illumination system |
| NTHL1-mCherry (in pRetroQ) | custom cloning | na | NTHL1 cDNA was cloned in the NheI, SalI sites of pRetroQ-mCherry-N1 vector. |
| Nunc Lab-Tek II Chambered Coverglass (4 well) | Thermo Fisher Scientific | 155382PK | Live cell microscopy cell culture chamber |
| Olaparib | Selleck Chemicals | S1060 | PARP inhibitor |
| Opti-MEM reduced serum media | Thermo Fisher Scientific | 31985062 | Dilution medium for transient transfection |
| Paraformaldehyde aqueous solution (32%) | Thermo Fisher Scientific | 50-980-494 | Fixative |
| pBABE (hygro) | Addgene | #1765 | retroviral expression vector (for low expression levels) |
| pBABE (neo) | Addgene | #1767 | retroviral expression vector (for low expression levels) |
| pBABE (puro) | Addgene | #1764 | retroviral expression vector (for low expression levels) |
| pBABE (zeo) | Addgene | #1766 | retroviral expression vector (for low expression levels) |
| PCNA Antibody (PC10) | Santa Cruz | sc-56 | primary antibody |
| Penicillin-Streptomycin-Glutamine (100x) | Gibco | 10378016 | Media supplement |
| polybrene | Sigma-Aldrich | TR-1003 | Increase viral infection efficiency |
| pRetroQ-AcGFP-C1 | Takara | 632506 | retroviral expression vector |
| pRetroQ-AcGFP-N1 | Takara | 632505 | retroviral expression vector |
| pRetroQ-mCherry-C1 | Takara | 632567 | retroviral expression vector |
| pRetroQ-mCherry-N1 | Takara | 632568 | retroviral expression vector |
| pUMVC | Addgene | #8449 | Viral packaging vector |
| Sodium-pyruvate | Thermo Fisher Scientific | 11360070 | Supplement for live cell imaging medium |
| Triton X-100 aqueous solution (10%) | Sigma-Aldrich | 11332481001 | Dilute in PBS for cell permeabilization buffer |
| Trypsin-EDTA Solution 10X | Sigma-Aldrich | 59418C-100ML | Dilute in PBS to split cells |
| U-2 OS Cells | ATCC | HTB-96 | Optimal cell line for microscopy experiments |
| Universal Mycoplasma Detection Kit | ATCC | 30-1012K | PCR based Mycoplasma detection kit |
| VSV-G | Addgene | #8454 | Viral protein envelope vector |
References
- Aleksandrov, R., et al.
- Darzynkiewicz, Z., Halicka, H. D., Zhao, H., Podhorecka, M. Cell synchronization by inhibitors of DNA replication induces replication stress and DNA damage response: Analysis by flow cytometry. Methods in Molecular Biology. 761, 85-96 (2011).
- Sakaue-Sawano, A., et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 132 (3), 487-498 (2008).
- Herce, H. D., Rajan, M., Lattig-Tunnemann, G., Fillies, M., Cardoso, M. C. A novel cell permeable DNA replication and repair marker. Nucleus. 5 (6), 590-600 (2014).
- Keijzers, G., et al. Human exonuclease 1 (EXO1) regulatory functions in dna replication with putative roles in cancer. International Journal of Molecular Sciences. 20 (1), (2018).
- Cheruiyot, A., et al. Poly(ADP-ribose)-binding promotes Exo1 damage recruitment and suppresses its nuclease activities. DNA Repair (Amsterdam). 35, 106-115 (2015).
- Zhang, F., Shi, J., Chen, S. H., Bian, C., Yu, X. The PIN domain of EXO1 recognizes poly(ADP-ribose) in DNA damage response. Nucleic Acids Research. 43 (22), 10782-10794 (2015).
- Rona, G., et al. PARP1-dependent recruitment of the FBXL10-RNF68-RNF2 ubiquitin ligase to sites of DNA damage controls H2A.Z loading. elife. 7, (2018).
- Young, L. M., et al. TIMELESS forms a complex with PARP1 distinct from its complex with TIPIN and plays a role in the dna damage response. Cell Reports. 13 (3), 451-459 (2015).
- Kong, X., et al. Laser microirradiation to study in vivo cellular responses to simple and complex dna damage. Journal of Visualized Experiments. (131), e56213 (2018).
- Kong, X., et al. Condensin I recruitment to base damage-enriched DNA lesions is modulated by PARP1. PLoS One. 6 (8), 23548 (2011).
- Lan, L., et al. Novel method for site-specific induction of oxidative DNA damage reveals differences in recruitment of repair proteins to heterochromatin and euchromatin. Nucleic Acids Research. 42 (4), 2330-2345 (2014).
- Zerjatke, T., et al. Quantitative cell cycle analysis based on an endogenous all-in-one reporter for cell tracking and classification. Cell Reports. 19 (9), 1953-1966 (2017).
- Ji, Y., Karbaschi, M., Cooke, M. S. Mycoplasma infection of cultured cells induces oxidative stress and attenuates cellular base excision repair activity. Mutation Research. 845, 403054 (2019).
- Sun, G., et al. Mycoplasma pneumoniae infection induces reactive oxygen species and DNA damage in A549 human lung carcinoma cells. Infection and Immunity. 76 (10), 4405-4413 (2008).
- Gassman, N. R., Wilson, S. H. Micro-irradiation tools to visualize base excision repair and single-strand break repair. DNA Repair (Amsterdam). 31, 52-63 (2015).
- Muster, B., Rapp, A., Cardoso, M. C. Systematic analysis of DNA damage induction and DNA repair pathway activation by continuous wave visible light laser micro-irradiation. AIMS Genetics. 4 (1), 47-68 (2017).
- Ikeda, S., et al. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. Journal of Biological Chemistry. 273 (34), 21585-21593 (1998).
- Rosenquist, T. A., Zharkov, D. O., Grollman, A. P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proceedings of the National Academy of Science U. S. A. 94 (14), 7429-7434 (1997).
- Reid, D. A., et al. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proceedings of the National Academy of Science U. S. A. 112 (20), 2575-2584 (2015).
- Taccioli, G. E., et al. Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science. 265 (5177), 1442-1445 (1994).
- Marsin, S., et al. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. Journal of Biological Chemistry. 278 (45), 44068-44074 (2003).
- Thompson, L. H., Brookman, K. W., Jones, N. J., Allen, S. A., Carrano, A. V. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Molecular and Cell Biology. 10 (12), 6160-6171 (1990).
- Scharer, O. D.
- Oeck, S., et al. High-throughput evaluation of protein migration and localization after laser micro-irradiation. Science Reports. 9 (1), 3148 (2019).
- Mistrik, M., et al. Cells and stripes: A novel quantitative photo-manipulation technique. Science Reports. 6, 19567 (2016).
- Durand, R. E., Olive, P. L. Cytotoxicity, mutagenicity and dna damage by hoechst 33342. Journal of Histochemistry and Cytochemistry. 30 (2), 111-116 (1982).
- Tobey, R. A., Oishi, N., Crissman, H. A. Cell cycle synchronization: reversible induction of G2 synchrony in cultured rodent and human diploid fibroblasts. Proceedings of the National Academy of Science U. S. A. 87 (13), 5104-5108 (1990).
- Podhorecka, M., Skladanowski, A., Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. Journal of Nucleic Acids. 2010, (2010).
