

Summary
Этот протокол описывает неинвазивный метод эффективной идентификации клеток S-фазы для последующих микроскопических исследований, таких как измерение репарации белка репарации ДНК с помощью лазерного микролучения.
Abstract
Восстановление повреждений ДНК поддерживает генетическую целостность клеток в высокореактивной среде. Клетки могут накапливать различные типы повреждений ДНК из-за эндогенных и экзогенных источников, таких как метаболическая активность или ультрафиолетовое излучение. Без репарации ДНК генетический код клетки становится скомпрометированным, подрывая структуры и функции белков и потенциально вызывая заболевания.
Понимание пространственно-временной динамики различных путей репарации ДНК в различных фазах клеточного цикла имеет решающее значение в области восстановления повреждений ДНК. Современные методы флуоресцентной микроскопии предоставляют отличные инструменты для измерения кинетики рекрутирования различных белков репарации после индукции повреждения ДНК. Синтез ДНК во время S-фазы клеточного цикла является своеобразной точкой в судьбе клеток в отношении репарации ДНК. Он предоставляет уникальное окно для проверки всего генома на наличие ошибок. В то же время ошибки синтеза ДНК также представляют угрозу целостности ДНК, которая не встречается в неделяющихся клетках. Поэтому процессы репарации ДНК значительно отличаются в S-фазе по сравнению с другими фазами клеточного цикла, и эти различия плохо изучены.
Следующий протокол описывает подготовку клеточных линий и измерение динамики белков репарации ДНК в S-фазе в локально индуцированных местах повреждения ДНК с использованием лазерно-сканирующего конфокального микроскопа, оснащенного лазерной линией 405 нм. Помеченный PCNA (с mPlum) используется в качестве маркера клеточного цикла в сочетании с меченым AcGFP репараторным белком, представляющим интерес (т. Е. EXO1b), для измерения рекрутации повреждений ДНК в S-фазе.
Introduction
Несколько путей репарации ДНК эволюционировали для решения различных типов поражений ДНК, которые могут возникать в клетках, все из которых строго регулируются как в пространстве, так и во времени. Одним из наиболее уязвимых периодов клеточного цикла является S-фаза, когда происходит синтез ДНК. Хотя распространение имеет основополагающее значение для жизни, оно также представляет собой серьезную проблему. Клетки должны обеспечить верную репликацию своего генома, чтобы избежать мутаций, которые будут переданы будущим поколениям. Следовательно, пролиферация обеспечивает терапевтическую точку вмешательства, которая была использована для разработки терапевтических подходов в области онкологии.
Все основные методы, используемые для изучения рекрутирования белка при поражениях ДНК, имеют свои сильные и слабые стороны. Микроизлучение имеет лучшее пространственное и временное разрешение1, чем большинство альтернативных методов, таких как иммунофлуоресцентная визуализация ионизирующих радиационно-индуцированных очагов (IRIF), хроматин-иммунопреципитация (ChIP) или биохимическое фракционирование. Тем не менее, микроизлучение повышает надежность вышеупомянутых методов, которые могут одновременно отбирать большое количество клеток.
Чтобы исследовать репарации ДНК в S-фазе, необходимо уметь различать клетки S-фазы в асинхронной популяции клеточной культуры. Существует много известных методов решения этой проблемы, включая либо синхронизацию клеток, либо визуализацию различных фаз клеточного цикла. Однако оба подхода создают значительные проблемы и возможные артефакты. Методы химической синхронизации, широко используемые для обогащения клеток в ранней S-фазе (например, двойной блок тимидина, лечение афидиколином и гидроксимочевиной), достигают синхронизации путем индукции напряжения репликации и, в конечном итоге, повреждения самой ДНК. Это ограничивает использование этих методов для изучения процессов репарации ДНК в S фазе2. Синхронизация через сывороточное голодание и высвобождение применима только к ограниченному числу клеточных линий, в значительной степени исключая линии раковых клеток, которые меньше полагаются на факторы роста для прогрессирования клеточного цикла по сравнению с непереобощенными клеточными линиями. Система Fluorescence Ubiquitin Cell Cycle Indicator (FUCCI) является особенно полезным инструментом для изучения клеточного цикла, но она имеет фундаментальное ограничение при дифференциации между фазами клеточного цикла S и G23.
Здесь показано, что использование флуоресцентно помеченной PCNA в качестве неинвазивного маркера для S-фазы ограничивает недостатки методов синхронизации химического клеточного цикла, обеспечивая при этом большую специфичность и гибкость, чем система FUCCI. В качестве единого маркера PCNA не только может выделять клетки S-фазы в асинхронной популяции, но также может показывать точнуюпрогрессию клеток в S-фазе (т.е. ранней, средней или поздней S-фазе)4. Низкие уровни экспрессии экзогенной, помеченной PCNA обеспечивают минимальное вмешательство как в прогрессирование клеточного цикла, так и в процессы репарации ДНК. Важно отметить, что PCNA также служит внутренним контролем для правильной индукции повреждений ДНК, поскольку она участвует в восстановлении нескольких поражений ДНК и набирается в локально индуцированные участки повреждения ДНК1,4.
Эксперименты, представленные здесь, демонстрируют, как измерить динамику рекрутации EXO1b в S-фазе и как на это влияет хорошо заявляемый ингибитор PARP, олапариб. Активность нуклеазы EXO1b имеет отношение к широкому спектру путей репарации ДНК, включая восстановление несоответствия (MMR), репарации нуклеотидной эксцизии (NER) и двухцепочечное восстановление разрыва (DSB). В S-фазе EXO1b играет важную роль в гомологиозной рекомбинации (HR) путем образования 3'ssDNA свесов во время резекции ДНК5. EXO1b был дополнительно замешан в репликации ДНК с ролями в активации контрольных точек для перезапуска остановленных вил ДНК, а также удаления праймера и созревания фрагмента Окадзаки на запаздывающей цепи во время смещения нити в репликации5. Рекрутирования EXO1b к поврежденным участкам ДНК регулируется прямым взаимодействием с поли (АДФ-рибоза) (PAR)6,7. Из-за многочисленных специфических последствий EXO1b для клеточного цикла он является отличным выбором для исследований рекрутации S-фазы с использованием PCNA.
Protocol
1. Культивирование клеток, полученных из остеосаркомы человека (U-2 OS)
ПРИМЕЧАНИЕ: Клетки U-2 OS идеально подходят для этих исследований, поскольку они имеют плоскую морфологию, большое ядро и сильно прикрепляются к нескольким поверхностям, включая стекло. Другие клеточные линии с аналогичными характеристиками также могут быть использованы.
- Для культивирования клеточных линий U-2 OS используйте среду McCoy 5A, дополненную 10% фетальной бычим сывороткой (FBS) и антибиотиками (100 ЕД /мл пенициллина и 100 мкг/мл стрептомицина). Инкубировать клетки при 37 °C в увлажненной атмосфере, содержащей 5% CO2. Для микроскопических исследований поддерживайте культуру клеток в чашке 10 см, чтобы обеспечить достаточное количество клеток.
- Когда клетки приближаются к 90% слиянию (~7 х10 6 ячеек/10 см чашки), разделите ячейки.
- Промывайте клетки PBS, чтобы смыть ингибиторы трипсина, содержащиеся в сыворотке крови.
- Добавьте 1 мл трипсина-ЭДТА и убедитесь, что клеточный слой одинаково покрыт.
- Инкубировать при 37 °C до тех пор, пока слой клеток не будет снят с пластины (приблизительно 6 мин).
- Повторно суспендировать трипсинизированные клетки в сыворотке, содержащей среду, чтобы инактивировать трипсин и добавить1/10-ю часть объема (~ 0,7 х 106 клеток) в новую 10 см пластину, содержащую 10 мл дополненной питательной среды.
- Перед экспериментом регулярно проверяйте клетки на загрязнение микоплазмой с помощью универсального набора для обнаружения микоплазмы в соответствии с рекомендацией производителя.
2. Ретровирусная инфекция
ПРИМЕЧАНИЕ: Для принятия мер безопасности BSL-2 и при работе с рекомбинантными вирусами, пожалуйста, обратитесь к: Руководство NIH, Раздел III-D-3: Рекомбинантные вирусы в культуре тканей.
- Посейте 4 x 106 клеток HEK293T для достижения ~ 60% слияния в течение 24 ч после нанесения на 10 см культурную посуду.
- Для выращивания HEK293T следуйте этапам культивирования ОС U-2, описанным в 1.1-1.3 настоящего протокола. Для HEK293T замените среду McCoy 5A на DMEM. Обязательно всегда осторожно промывайте клетки HEK293T, так как они слабо прикрепляются к пластинам тканевой культуры.
- Трансфектировать клетки HEK293T с использованием трансфектного реагента на основе липидов для вирусной упаковки плазмид.
- Для ретровирусных векторов объедините 1,5 мкг VSV-G (Addgene #8454) и 1,5 мкг упаковочных векторов pUMVC (Addgene #8449) вместе с 3 мкг вектора, содержащего интересующий ген (в ретровирусной векторной магистрали с устойчивостью к пуромицину), в 250 мкл восстановленной сывороточной среды Opti-MEM в микроцентрифужной пробирке. Добавьте 1 мкл реагента P3000 на каждый мкг ДНК, добавленной в смесь Opti-MEM/ДНК (в данном случае 6 мкл), и аккуратно перемешайте постукиванием. Не вихрь или пипетка вверх и вниз.
- В другой микроцентрифужной трубке соедините 2 мкл на мкг ДНК (в данном случае 12 мкл) трансфекционного реагента с 250 мкл восстановленной сывороточной среды Opti-MEM.
- Смешайте две смеси (500 мкл вместе, не вихрево, только перемешайте мягким постукиванием) и дайте ему инкубировать в течение 15 минут при комнатной температуре.
- Осторожно добавьте смесь по каплям в засеянные клетки HEK293T без отсоединения клеток. Аккуратно закрутите тарелки.
- Вирусная инфекция для генерации стабильных клеточных линий.
- Удаляют вирус, содержащий супернатант, из клеток HEK293T через 72 ч после трансфекции. Тщательно отфильтруйте раствор с помощью фильтра 0,45 мкм для удаления остатков клеток и отсоединенных клеток. Необязательно, добавьте 8 мкг/мл полибрена к вирусному супернатанту для облегчения вирусной инфекции.
- Добавьте вирус, содержащий супернатант, в клетки ОС U-2 при слиянии ~50% в чашке 10 см (~3 x 106 клеток). Засеять клетки U-2 OS накануне.
- Заражайте в течение 6-16 ч перед удалением и выбросом вируссодержащего супернатанта.
ПРИМЕЧАНИЕ: Чтобы достичь желаемого количества сверхэкспрессии для интересуемого гена, инкубируют серию вирусных разведений в течение фиксированного периода времени. Проверьте уровни экспрессии трансгена в каждой вновь созданной клеточной линии с помощью западного пятна, сравнивая его с эндогенными уровнями. - Позвольте клеткам выбрать в присутствии соответствующих антибиотиков (в течение 3-4 дней в случае пуромицина в конечной концентрации 2 мкг/мл) и проверить экспрессию флуоресцентного белка, помеченного интересующей геном под микроскопом.
- Повторите эти шаги, чтобы создать линии клеток с двойной меткой. В представленных здесь экспериментах mPlum-PCNA экспрессировали из ретровирусного вектора (pBABE) в сочетании с EXO1B-AcGFP, также экспрессируемого из ретровирусного вектора (pRetroQ-AcGFP1-N1).
3. Подготовка клеток к микрооблучению
- Гальванические ячейки: за 24 часа до эксперимента, пластинчатые в общей сложности8,0 х 10 4 ячейки в объем между 500 мкл-1 мл среды (для примерно 70% слияния) на четырехкамерном покрытом стекле с боросиликатным стеклянным дном No 1,5, которое обеспечивает идеальные результаты для конфокальной микроскопии с высоким увеличением и лазерного микролучения. Более высокая слияние клеток позволяет измерять больше клеток в одном поле зрения (FOV); однако полностью слиенные слайды будут вносить нарушения клеточного цикла.
- Носители визуализации: За час до микрооблучения обменная регулярная питательная среда для FluoroBrite DMEM с добавлением 10% FBS, 100 ЕД /мл пенициллина и 100 мкг/мл стрептомицина, 15 мМ HEPES (pH = 7,4) и 1 мМ натрия-пирувата. Этот носитель изображений помогает максимизировать отношение сигнал/шум, позволяя обнаруживать очень тусклую флуоресценцию. Поскольку он содержит HEPES, он также стабилизирует pH в отсутствие 5% атмосферы CO2.
- Примените любое дополнительное лечение перед визуализацией на этом этапе. В экспериментах, представленных здесь, клетки предварительно обрабатывали за час до визуализации либо олапарибом (ингибитор PARP, при конечной концентрации 1 мкМ), либо контролем транспортного средства (DMSO)1,8,9.
4. Подготовка микроскопа и выбор S-фазы клеток для визуализации.
- Используйте конфокальную систему, которая имеет те же свойства, что и система, описанная здесь, для достижения наилучших результатов. Представленные здесь эксперименты проводились с использованием конфокального микроскопа, установленного на подставке для перевернутого микроскопа (см. Таблицу материалов).
ПРИМЕЧАНИЕ: Используемый здесь микроскоп был оснащен лазерным модулем FRAP мощностью 50 мВт 405 нм и масляным планом-апохроматом 60x 1,4 NA. Конфокальная сканирующая головка имела два варианта сканера: гальваносканер (для высокого разрешения) и резонансный сканер (для высокоскоростной визуализации).- Введите флуоресцентное восстановление после фотоотбеливания (FRAP) лазера в образец с помощью программно управляемого устройства XY galvano. Используйте лазерную линию 488 нм для возбуждения AcGFP и лазерную линию 561 нм или 594 нм для возбуждения mPlum.
ПРИМЕЧАНИЕ: Следующая комбинация фильтров дает оптимальные результаты: используя фильтр длинных проходов 560 нм, эмиссионный свет с длиной волны ниже 560 нм пропускался через эмиссионный фильтр 525/50 нм для AcGFP, в то время как эмиссионный свет с длиной волны выше 560 нм пропускался через эмиссионный фильтр 595/50 нм для mPlum. Может быть использован любой соответствующий набор фильтров (например, FITC/TRITC, GFP/mCherry, FITC/TxRed), который обеспечивает минимальное прохождение флуоресцентной проточия.
- Введите флуоресцентное восстановление после фотоотбеливания (FRAP) лазера в образец с помощью программно управляемого устройства XY galvano. Используйте лазерную линию 488 нм для возбуждения AcGFP и лазерную линию 561 нм или 594 нм для возбуждения mPlum.
- Включите камеру окружающей среды и компоненты микроскопа.
- Включите нагрев (ступень, объектив и экологическую камеру, когда это возможно), подачу CO2 и регулятор влажности не менее чем за 4 часа до начала эксперимента, чтобы обеспечить тепловое равновесие для стабильного получения изображения.
- Инициализируют источники света вместе с лазерными линиями не менее чем за 1 ч до переноса клеток в микроскоп.
- Выберите S-фазовые клетки в асинхронной популяции, используя флуоресцентно помеченную PCNA в качестве маркера. Для этого выполните следующие действия.
- Ищите уникальную схему локализации помеченной mPlum PCNA в S-фазе, что делает возможной идентификацию этой фазы клеточного цикла. PCNA имеет полностью однородное распределение в ядре в фазах G1 и G2 клеточного цикла, будучи исключенной из ядрышек. В S-фазе PCNA образует очаги в месте расположения реплисом в ядре. На рисунке 1 показаны различные паттерны очагов PCNA на протяжении всей S-фазы, что позволяет даже дифференцировать раннюю, среднюю и позднюю S-фазу.
- Посмотрите через глаз, чтобы выбрать FOV, который имеет достаточно S-фазных клеток для микролучения. Асинхронные клетки U-2 OS обычно имеют 30-40% своей популяции в S-фазе.
- Старайтесь избегать экстремальных уровней экспрессии (как ярких, так и тусклых клеток) как для PCNA, так и для интересуемого белка (POI), в данном случае EXO1b-AcGFP, что может привести к экспериментальным артефактам.
- При поиске подходящего FOV старайтесь избегать сканирования поля в течение длительного времени, чтобы свести к минимуму фотоотбеливание и нежелательное повреждение ДНК.
- Задайте нужную интересуемую область (ROI) для микроизлучения. Используя соответствующее программное обеспечение (см. Таблица материалов),установите желаемую рентабельность инвестиций, сначала вставив двоичные строки (установите желаемое количество строк и интервалов). Нажмите «Двоичный»,затем нажмите «Вставить строку | Круг | Эллипс, чтобы нарисовать нужное количество линий.
- Преобразуйте эти двоичные линии в ROI и, наконец, преобразуйте эти ROI в стимулирующие ROI. Для этого сначала нажмите ROI,затем нажмите Переместить двоичный файл в ROI,затем щелкните правой кнопкой мыши на любом из ROI и выберите Использовать в качестве стимуляции ROI: S1. Поместите эти линии в FOV, чтобы пройти через ядро клеток. ROI длиной 1024 пикселя, которые охватывали весь FOV, использовались во всем протоколе.
5. Микролучение для иммунофлуоресцентного окрашивания или покадровой визуализации.
- Определение оптимальных параметров микроизлучения.
- Перед микрооблучением клеток сделайте изображение FOV с более высоким разрешением, чтобы идентифицировать очаги PCNA для последующего анализа. Вместо последовательного сканирования одновременно записывайте оба используемых оптических канала (зеленый и красный), чтобы избежать движения клеток между сканированием на двух длинах волн. Для правильного разрешения фокусов используйте разрешение не менее 1024 x 1024 пикселей/поле с 1-кратным зумом (размер пикселя 0,29 мкм на используемой здесь системе визуализации), со скоростью сканирования 1/8 кадра/с (4,85 мкс/пиксель) с 2-кратным усреднением. После того, как эти параметры установлены в окнах A1 LFOV Compact GUI и A1 LFOV Scan Area, нажмите кнопку Capture, чтобы записать FOV.
ПРИМЕЧАНИЕ: Важно поддерживать один и тот же размер пикселя во всех экспериментах, чтобы обеспечить сопоставимые результаты. - Чтобы настроить микроизлучение, откройте вкладку ND Stimulation в программном обеспечении для визуализации, чтобы получить доступ к окну Расписание (A1 LFOV / Galvano Device). При этом гальваносканеры используются для получения серии изображений предварительной стимуляции, стимуляции (с использованием ЛАЗЕРА FRAP LUN-F 50 мВт 405 нм), а затем снова получают серию изображений после стимуляции с помощью гальваносканеров. Сначала настройте три этапа в окне Расписание времени. В столбце Acq/Stim выберите Приобретение | Отбеливающие | Приобретение для трех этапов соответственно. Для фазы отбеливания установите S1 в качестве ROI.
ПРИМЕЧАНИЕ: В эксперименте, представленном здесь, не было получено никаких изображений во время фазы стимуляции. - В окне Galvano XYнастройте ключевые факторы для микроизлучения: выходная мощность лазера 405 нм, время выдержки (итерация по умолчанию в этой системе равна 1). В экспериментах, представленных здесь, клетки облучали 405-нм FRAP-лазером (50 мВт на кончике волокна) при 100% выходной мощности со временем выдержки 1000-3000 мкс.
ПРИМЕЧАНИЕ: Поскольку время ожидания лазера определяется на пиксель, пока размер пикселя остается неизменным, соотношение между временем выдержки и плотностью мощности будет сопоставимо между различными FOV. На рисунке 2A показано использование специфических белков пути реакции на повреждение ДНК (DDR) (FBXL10 для DSB и NTHL1 для повреждения окислительного основания) для оптимизации настроек мощности лазера для индукции конкретных повреждений. Эти стабильные клеточные линии были сгенерированы вирусной инфекцией в соответствии с разделом 2 протокола.
- Перед микрооблучением клеток сделайте изображение FOV с более высоким разрешением, чтобы идентифицировать очаги PCNA для последующего анализа. Вместо последовательного сканирования одновременно записывайте оба используемых оптических канала (зеленый и красный), чтобы избежать движения клеток между сканированием на двух длинах волн. Для правильного разрешения фокусов используйте разрешение не менее 1024 x 1024 пикселей/поле с 1-кратным зумом (размер пикселя 0,29 мкм на используемой здесь системе визуализации), со скоростью сканирования 1/8 кадра/с (4,85 мкс/пиксель) с 2-кратным усреднением. После того, как эти параметры установлены в окнах A1 LFOV Compact GUI и A1 LFOV Scan Area, нажмите кнопку Capture, чтобы записать FOV.
- Покадровая визуализация.
- Настройте покадровое изображение для требуемого временного окна и интервалов с помощью расписания времени, компактного графического интерфейса A1 LFOV и окон области сканирования A1 LFOV. В экспериментах, представленных здесь, набор EXO1b и PCNA был визуалирован в течение 12 минут, сканируя FOV каждые 5 секунд, со скоростью 1024 x 1024 пикселей / поле, с использованием 1-кратного зума (что приводит к размеру пикселя 0,29 мкм в используемой здесь системе визуализации) со скоростью сканирования 0,35 кадра / с (1,45 мкс / пиксель) без усреднения для уменьшения отбеливания фотографий.
- Оптимизируйте параметры мощности лазера, усиления и смещения для уменьшения фотоотбеливания во время съемки в окне A1 LFOV Compact GUI. Если кто-то стремится измерить как POI, так и PCNA, используйте одновременное сканирование вместо последовательного сканирования, чтобы избежать движения клеток между сканированием поля для двух отдельных флуорофоров.
- Система обработки изображений использовалась со следующими настройками. Для лазерной линии 488 нм (20 мВт): мощность лазера 7%, коэффициент усиления: 45 (детектор GaAsP) со смещением 2 и смещением 2, для лазерной линии 561 нм (20 мВт): мощность лазера 4%, коэффициент усиления 40 (детектор GaAsP) с и смещением 2.
- В зависимости от кинетики белка, удлиняйте или сокращайте интервал между изображениями или продолжительность общего временного интервала. В окне Расписание времени задайте требуемые Интервал и Длительность для третьей строки получения фазы.
- Нажмите кнопку Выполнить, чтобы выполнить микроизлучение и последующую покадровую визуализацию.
- В конце покадровой визуализации сохраните ROI стимуляции в виде отдельных изображений, что будет полезным подспорьем для определения координат микролучения в любом последующем программном обеспечении, используемом для анализа.
- Иммунофлуоресцентное окрашивание.
ПРИМЕЧАНИЕ: Этап 5.1.3 и рисунок 2А демонстрируют использование известных белков репарации ДНК для оценки типов поражений ДНК, введенных микролучением. Некоторые поражения ДНК также могут быть обнаружены с помощью специфических антител после фиксации клеток. Также можно обнаружить рекрутирование POI путем обнаружения антител эндогенного белка. Визуализация γH2A.X для проверки DSB демонстрируется ниже(рисунок 2B). На рисунке 3 показана согласованность локализации и рекрутации PCNA на протяжении всего клеточного цикла как для эндогенной, так и для экзогенной помеченной PCNA.- После шага 5.1.3 сделайте только одно изображение после микрооблучения, чтобы обеспечить надлежащее событие FRAP на основе набора mPlum-PCNA. Обратите внимание на точные координаты FOV, чтобы найти поле позже после иммунофлуоресцентной маркировки.
- Выньте камеру клеточной культуры из микроскопа и инкубируют клетки при 37 °C в увлажненной атмосфере, содержащей 5% CO2 в течение 5-10 мин.
ПРИМЕЧАНИЕ: Параформальдегид (ПФА) токсичен, и работа должна проводиться в хорошо проветриваемом помещении или вытяжке. Вся последующая промывка и инкубация будут выполнены с объемами 0,5 мл в 4-х скважинной камере. После инкубационного времени промыть клетки 0,5 мл PBS (137 мМ NaCl, 2,7 мМ KCl, 8 мМ Na2HPO4и 2 мМ KH2PO4)и зафиксировать 0,5 мл 4% PFA в PBS в течение 10 мин при комнатной температуре (RT). - Промыть ячейки один раз с PBS, затем промыть их 50 мМ NH4Cl для закалки остаточного PFA.
- Пермеабилизируют клетки в течение 15 мин при RT с 0,1% Triton X-100 в PBS.
- Блокируйте образцы в течение 1 ч с блокирующим буфером (5% FBS, 3% BSA, 0,05% Triton X-100 в PBS).
- Удалить блокирующий раствор и добавить разбавленные первичные антитела (анти-γH2A.X, 1:2000) в блокирующий буфер в течение 1 ч при РТ.
- Промывайте колодцы с блокирующим буфером 3 х 10 мин.
- Добавьте разбавленное вторичное антитело (конъюгат против мыши Alexa 488 Plus, 1:2000) в блокирующий буфер на 1 ч при RT.
- Промывайте колодцы с блокирующим буфером 3 х 10 мин.
- Противопоставляем ядро 1 мкг/мл раствора DAPI в PBS в течение 15 мин.
- Вымойте ячейки один раз с помощью PBS. Визуализация может быть выполнена непосредственно в PBS или растворе PBS с антитемеющими реагентами (например, AFR3) для уменьшения фотоотбеливания.
6. Рекрутинговый анализ
ПРИМЕЧАНИЕ: На рисунке 4А показаны репрезентативные изображения набора Exo1b и PCNA в присутствии DMSO или олапариба. На рисунке 4B показано репрезентативное изображение для анализа данных. Средние значения флуоресценции были рассчитаны путем измерения средней интенсивности AcGFP с использованием прямоугольника вдоль лазерной дорожки, выделенной mPlum-PCNA (A, желтые прямоугольники) в разных временных точках с использованием Фиджи. PCNA может служить внутренним контролем для выделения успешного облучения вдоль координат ROI. Аналогичным образом, средние значения флуоресценции AcGFP были также рассчитаны для неповрежденных областей ядра (B, синие прямоугольники). Интенсивность фонового сигнала измеряли в незаполненных районах (С, красные прямоугольники) и вычитали из средних флуоресцентных значений(рис. А и В). Таким образом, относительная средняя флуоресцентная единица (РФС) для каждой точки сбора данных рассчитывалась по уравнению РФС = (A − C)/ (B − C)8,9. Полученные значения РФС микрооблучаемой области нормализуются до значений РФС до микрооблучения.
- Для определения области А микрооблучаемого участка исключить из измерения нуклеолярные области, очаги репликации и нерегулярные ядерные области клетки. Удерживайте клавишу shift между рисованием двух ROI на Фиджи, чтобы сгруппировать два отдельных региона как один.
ПРИМЕЧАНИЕ: Рекрутирование белка будет варьироваться в зависимости от различных генов и условий облучения; таким образом, размер области А должен определяться индивидуально. Как только ширина пикселя области A определена, она должна оставаться постоянной для любых сравнительных наборов. В представленных здесь экспериментах использовались прямоугольники шириной 7 пикселей. - Исключить из анализа ячейки, которые перемещались в течение всего времени записи видео. Чтобы включить высокомобильные ячейки, описанный анализ должен проводиться покадровно.
- Чтобы визуализировать профиль найма, отобразим нормализованные значения РФС по времени с помощью статистического программного обеспечения.
- Рассчитайте разницу в указанной временной точке между ДМСО и лечением олапарибом (n=31) с помощью теста Манна-Уитни.
Representative Results
Клетки обращаются к каждому типу поражения ДНК определенным образом, который также зависит от того, в какой фазе клеточного цикла они находятся. Например, после микроизлучения двухцепочечные разрывы (DSB) будут обрабатываться либо негомологичным концевой соединением (NHEJ), либо HR в зависимости от фазы клеточного цикла. Нуклеазы, действующие наиболее широко во время фаз S и G2 клеточного цикла, создают навесы ДНК, которые имеют решающее значение для правильного ЧСС. Чтобы способствовать оценке клеток в S-фазе, PCNA использовался в качестве одноцветного маркера клеточного цикла. На рисунке 1А показан профиль локализации mPlum-PCNA во время прогрессирования клеточного цикла. PCNA имеет полностью однородное распределение в ядре в фазе G1 и G2 (при этом также в основном исключается из ядрышков). В S-фазе PCNA локализуется в местах репликации ДНК, которые можно визуализировать как яркие пятна в ядре. В клетках ранней S-фазы пятна относительно невелики и равномерно распределены по всему ядру клетки. Прогрессируя в середину S-фазы, пятна становятся размытыми и локализуются больше по периметру ядра и ядрышка. В поздней S-фазе количество пятен уменьшается, но становится все более большим, поскольку PCNA концентрируется в поздних местах репликации(рисунок 1B). Важно отметить, что экзогенная экспрессия PCNA из векторной основы pBABE была меньше эндогенных уровней, но была достаточной для обнаружения с помощью микроскопии, которая минимизирует потенциальные артефакты в прогрессировании клеточного цикла и DDR. На рисунке 1С показана степень сверхэкспрессии ПКНА по сравнению с эндогенными уровнями. Обратите внимание, что полоса, соответствующая mPlum-PCNA, мигрирует медленнее из-за своего большего размера.
Мы стремились ввести DSB во время микролучения, чтобы исследовать PARP1/2-зависимый набор EXO1b к этим поражениям в S-фазе. На фиг.2А показано, что низкие дозы энергии (время выдержки 1000 мкс) не индуцируют рекрутирование EGFP-FBXL10, ответчика DSB (компонент комплекса FRUCC 8),в то время как этого было достаточно, чтобы индуцировать рекрутирование NTHL1-mCherry, белка пути репарации основания (BER), рекрутируя в места окислительного повреждения ДНК10,11,12. При времени выдержки 3000 мкс как EGFP-FBXL10, так и NTHL1-mCherry набирают, демонстрируя лазерный выход, который генерирует как окислительный очаг, так и DSB. Усиливая эти результаты, рисунок 2B показывает иммунофлуоресцентное окрашивание против γH2A.X (маркер DSB), что явно более очевидно при использовании более высоких доз энергии. PCNA служит как маркером клеточного цикла, так и маркером для успешного микроизлучения, поскольку он адекватно набирает с обоими настройками времени пребывания лазера. Важно отметить, что для этой репортерной функции могут использоваться как экзогенные, так и/или эндогенные флуоресцентные белки, помеченные PCNA, поскольку они ведут себя одинаково(рисунок 3). Эндогенно помеченная PCNA была спроектирована путем вставки mRuby в рамку с первым экзоном в один аллель локуса PCNA13 (клеточная линия была своеобразным подарком Йорга Мансфельда).
На рисунках 4A и 4C показан набор ПОМЕЧЕННЫХ AcGFP EXO1b в S-фазовых ячейках. EXO1b достигает максимального уровня накопления в местах микрооблучения около 1 минуты, а затем медленно начинает отстраиваться от поражений ДНК. Обогащение в местах микроизлучения обозначается > 1 единицей относительной флуоресценции на графике. В присутствии олапариба накопление EXO1b на лазерной полосе через 1 минуту значительно меньше по сравнению с управлением транспортным средством. Эти результаты согласуются с литературой6,7. На рисунке 4В показаны репрезентативные регионы для количественной оценки (области А, В и С), как описано в пункте 6 протокола. На рисунке 4D показаны сопоставимые уровни экспрессии эндогенного EXO1b и экзогенного EXO1b-AcGFP в клетках, используемых для микроизлучения.
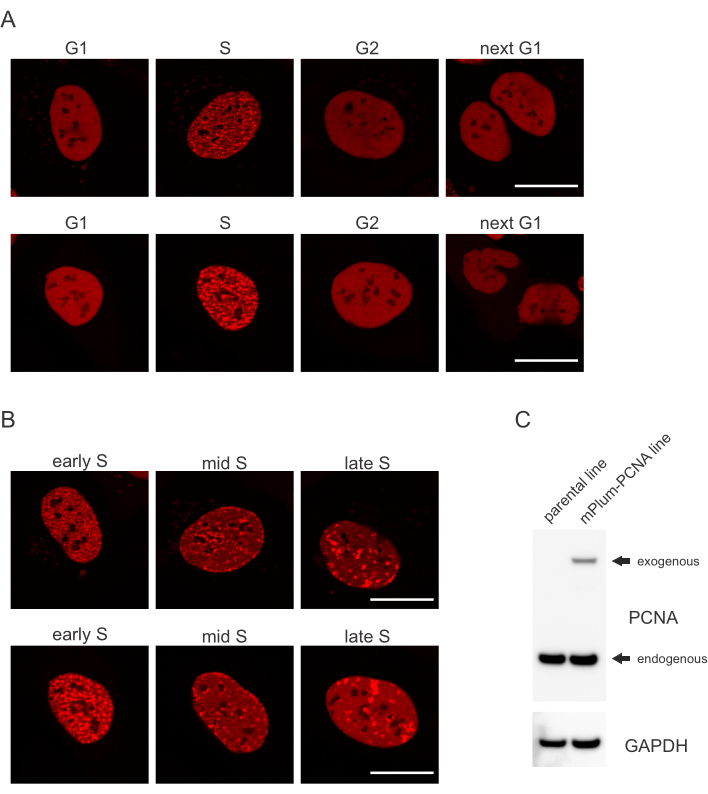
Рисунок 1:Схема локализации PCNA. (A)Изображения показывают паттерн локализации стабильно интегрированной, экзогенной PCNA на протяжении всего клеточного цикла в клетках U-2 OS. (B) Изображения показывают паттерны фокусов PCNA на разных стадиях S-фазы (ранняя, средняя и поздняя) в клетках U-2 OS. (C)Вестерн-блот, показывающий эндогенные и экзогенные уровни PCNA в клетках U-2 OS, используемых для визуализации. Шкала представляет 20 мкм. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.

Рисунок 2:Индукция DSB через оптимизированную выходную мощность лазера. (A) Настройки лазера могут быть оптимизированы для индуцирования различных форм повреждения ДНК. Клетки U-2 OS, стабильно экспрессирующие как EGFP-FBXL10, так и NTHL1-mCherry, использовались для идентификации DSB и участков окислительных поражений соответственно. Микроизлучение лазерной линией 405 нм проводили на асинхронных ячейках ОС U-2 со временем выдержки 1000 мкс или 3000 мкс. Шкала представляет собой 20 мкм.(B)Иммунофлуоресцентное окрашивание против γH2A.X было выполнено на пигментных эпителиальных клетках сетчатки человека (hTERT RPE-1), имеющих эндогенную PCNA, помеченную mRuby. Клетки фиксировали и обрабатывали через 5 минут после микрооблучения с временем выдержки 1000 мкс или 3000 мкс. Шкала представляет 20 мкм. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.

Рисунок 3:Сопоставимая рекрутирование эндогенной mRuby-PCNA и экзогенной mPlum-PCNA в места микролучения при времени выдержки лазера 1000 мкс или 3000 мкс. Как эндогенные, так и экзогенные метки PCNA образуют очаги репликации во время S-фазы. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.

Рисунок 4:ПАРП1/2-зависимый набор EXO1b на S-фазе. Клетки U-2 ОС, стабильно экспрессирующие EXO1b-AcGFP и mPlum-PCNA, были микрооблучены лазерной линией FRAP 405 нм с использованием времени выдержки 3000 мкс. (A)Репрезентативные изображения микрооблученных клеток в указанные моменты времени после предварительной обработки либо контролем транспортного средства (DMSO), либо олапарибом (1 мкМ). Шкала представляет собой 20 мкм. (B) Репрезентативные изображения определенных областей областей A, B и C для анализа набора персонала. Шкала представляет собой 20 мкм.(C)динамика рекрутации повреждений ДНК была зафиксирована с помощью визуализации живых клеток. Относительные средние значения флуоресценции и изображения получались каждые 5 с в течение 12 мин. Для каждого состояния оценивалось ≥30 клеток. Средние относительные значения флуоресценции (сплошные черные линии) и стандартная погрешность (диапазон, визуализируемый затененной областью) были построены со временем. Пунктирная линия показывает значения набора через 1 мин после микролучения. Разница между лечением ДМСО (n=32) и олапарибом (n=31) была рассчитана с использованием теста Манна-Уитни. Астерикс обозначает p<0.0001. (D)Вестерн-блот сравнивает уровни экспрессии эндогенного EXO1b и экзогенного EXO1b-AcGFP в клетках, используемых для микролучения. Пожалуйста, нажмите здесь, чтобы просмотреть увеличенную версию этого рисунка.
Discussion
Критические шаги и потенциальные способы устранения неполадок/модификаций протокола
Правильный сосуд для посева тканей для микролучедиации имеет решающее значение для успеха. Большинство систем визуализации с высоким разрешением оптимизированы для толщины покровного стекла 0,17 мм. Использование камер визуализации большей или меньшей толщины или камер, изготовленных из пластиковых полимеров (не оптимизированных для визуализации 405 нм), может значительно снизить качество изображения. При использовании стеклянных поверхностей убедитесь, что они обработаны культурой тканей для усиления клеточной адгезии. Если они не обработаны тканевой культурой, эти камеры должны быть покрыты, например, поли-D-лизином перед посевом клеток. При покрытии клеток в камерное покровное стекло идеальная плотность клеток имеет первостепенное значение, чтобы избежать нарушений клеточного цикла и дополнительного стресса для клеток. Правильное тепловое равновесие компонентов микроскопа перед экспериментированием для поддержания стабильной температуры имеет решающее значение как для поддержания фокуса на протяжении всего замедленного изображения, так и для обеспечения однородной DDR во времени и образцах.
Крайне важно, чтобы клетки были в здоровом состоянии до микролучения, чтобы уменьшить артефактные данные. Если клетки имеют неправильную морфологию после заражения / отбора, позвольте клеткам прогрессировать через несколько проходов, пока морфология не вернется к норме. Всегда следите за тем, чтобы используемые клеточные линии не были загрязнены микоплазмой. Среди многих побочных эффектов микоплазменной инфекции она также вызывает повреждение ДНК клеток-хозяев и может повлиять на их пути DDR14,15. Наиболее чувствительным способом обнаружения микоплазмы в клеточной культуре является ПЦР (по сравнению с обнаружением с помощью DAPI или Hoechst).
Оптимальная сверхэкспрессия интересующей вас восстановительного белка должна быть сопоставима с эндогенными уровнями, однако достаточно высокой для обнаружения. Промотор, используемый на вирусных векторах, вирусный титр во время инфекции и продолжительность времени заражения могут быть скорректированы для идеальных уровней экспрессии. Для получения последовательных результатов изолируйте отдельные клоны клеток, чтобы обеспечить однородные уровни экспрессии и нормальную морфологию клеток. Рекомендуется использовать векторные конструкции, которые не переэкспрессируют помеченную PCNA на более высоких, чем эндогенные уровни, для правильной функции клеточного цикла и маркера репарации ДНК. Даже низкие уровни сверхэкспрессии PCNA достаточны для различения клеток S-фазы. Для этой цели успешно используются ретровирусные векторы pBABE (Addgene #1764, #1765, #1766, #1767). PCNA может быть помечена любыми мономерными красными(например, mPlum, mCherry, mRuby и т. Д.) Или мономерными зелеными флуоресцентными белками (например, mEGFP, AcGFP, mWasabi, mNeonGreen, mEmerald и т. Д.), Которые затем могут быть объединены с попеременно помеченными POI. Сверхэкспрессия флуоресцентно помеченного POI имеет некоторые ограничения и соображения. Флуоресцентные метки могут нарушить нормальную функцию и локализацию белка. Таким образом, необходимо учитывать местоположение метки (N или C-терминал). Всегда используйте мономерные флуоресцентные белки, так как олигомеризация немономерных вариантов может повлиять на функцию POI.
Настройки лазера должны быть определены для каждой системы визуализации, так как многие компоненты оптического пути будут влиять на фактическую мощность, доставляемую в ячейки. Лазерное микроизлучение может вызвать несколько типов поражений ДНК в зависимости от длины волны возбуждения, выходной мощности лазера FRAP и если использовались какие-либо предсенсибилизирующие агенты (такие как бромодезоксиуридин или Hoechst). 405 нм лазеры могут вызывать окислительное повреждение ДНК, одно- и двухцепочечные разрывы16,17. При использовании более высоких настроек выхода лазера количество DSB увеличивается. В этом протоколе методы предварительной сенсибилизации не использовались, но эти методы широко освещаются в литературе и вновь ограничиваются в обсуждении ниже. По нашему мнению, лучший способ проверить, генерируется ли желаемое поражение, - это тестирование на набор известных генов, специфичных для пути повреждения ДНК. Рекрутирование NTHL1 или OGG1, компонентов пути BER, предполагает индукцию окислено-окисленых оснований ДНК10,11,17,18,19,в то время как FBXL10 или XRCC5 указывают на наличие DSB8,20,21. Рекрутация XRCC1 может указывать как на наличие окисленные основания ДНК, так и на одноцепочечные разрывы (SSB)22,23. XPC (т.е. RAD4) является хорошим индикатором NER, который удаляет громоздкие аддукты ДНК, генерируемые ультрафиолетовым светом (UV)17,24. Поскольку рекрутирование экзогенных белков может привести к определенным нарушениям, иммунофлуоресцентное окрашивание эндогенных белков или маркеров репарации ДНК (таких как γH2A.X для двухцепочечных разрывов) может подтвердить наличие специфических поражений ДНК. В качестве альтернативы также могут быть использованы антитела, поднятые против определенных типов поражений ДНК. Чтобы отрегулировать мощность поставляемого лазера, можно изменять как время выдержки, так и мощность лазера.
С помощью математического моделирования может быть выполнен подробный кинетический анализ, который может дать ценную информацию о рекрутируемых свойствах POI (например, вклад нескольких доменов связывания ДНК, чувствительность к различным сигнальным событиям и т. Д.). Автоматизированная оценка набора персонала и отслеживание ячеек могут быть объединены для создания надежных рабочих процессов 1,25.
Преимущества и ограничения пресенсибилизации ДНК
Предварительная сенсибилизация ДНК перед микрооблучением является широко используемым инструментом для репарации ДНК белкового набора16,17. Сенсибилизация ДНК перед микролучением делает ее более восприимчивой к DSB. Двумя наиболее распространенными методами пресенсибилизации ДНК являются предварительная обработка клеток либо бромодезоксиуридином (BrdU), либо красителем Hoechst. Для систем, не способных к микролучению при высокой мощности лазера, эти методы могут быть необходимы для индуцирования поражений ДНК, таких как DSB. Кроме того, в отсутствие детектора пропускаемого света или флуоресцентного сигнала, выделяющего ядро клетки (например, при изучении рекрутирования непомеченных эндогенных белков репарации ДНК), Hoechst действует как предсенсибилизирующий инструмент и флуоресцентное ядерное пятно. Тем не менее, предварительная сенсибилизация ДНК может привести к значительным осложнениям. BrdU (используется в конечной концентрации 10 мкМ) должен быть добавлен к клеткам 24 часа (или время, эквивалентное полному клеточный цикл в используемой клеточной линии) для правильного включения в ДНК и может вызвать интерференцию клеточного цикла26. Hoechst 33342 (используется в конечной концентрации 1 мкг/мл) является цитотоксичным после длительных инкубационных периодов, но требует достаточного времени для насыщения ядра красителем. Поэтому его следует наносить только за 15-20 минут до микрооблучения; в противном случае данные о наборе персонала не будут согласованными. Окрашенные таким образом клетки нельзя держать в культуре более нескольких часов27,28. Убедитесь, что вы не используете Hoechst 33358, который не так проницаем в клетках, как краситель Hoechst 33342. Предварительная сенсибилизация также может внести ненужную дисперсию между экспериментами и делает эксперимент еще более чувствительным к различиям в плотности клеток (поскольку это повлияет на количество включенного красителя / клетки).
Преимущества и ограничения конфокальной микроскопии
Скорость визуализации конфокальной микроскопии может быть ограниченной по сравнению с широкоугольной микроскопией. Тем не менее, конфокальный микроскоп, оснащенный резонансным сканером, может значительно улучшить скорость изображения (за счет разрешения), приближаяся к скоростям вращательно-дисковой микроскопии. Три особенности делают конфокальную систему A1R HD25 отличным выбором для представленного здесь протокола. Во-первых, 25-миллиметровый FOV системы позволяет визуалировать от 15 до 20 клеток в одном сканируемом поле (против 5-10 ячеек в обычных установках), ограничивая количество приобретений, необходимых для получения достаточного количества клеток для статистического анализа. Во-вторых, модуль FRAP и две сканируемые головки позволяют визуалировать и микрооблучать клетки одновременно, а не только последовательно. Наконец, гибкость наличия как резонансных, так и гальваносканеров обеспечивает возможность легко переключаться между изображениями с высоким временным разрешением с исключительной скоростью, которая сводит к минимуму закалку флуорофоров, и изображениями с высоким пространственным разрешением, которые используют более медленные скорости сканирования для получения изображений с более высоким соотношением сигнал/шум. В то время как используемая система позволяла вышеупомянутой гибкости, напоминать более широко доступные конфигурации конфокального микроскопа, в представленных экспериментах использовался только гальваносканер (как для микроизлучения, так и для последующей визуализации).
Преимущества и ограничения микроизлучения
Хотя микроизлучение обеспечивает непревзойденное пространственное и временное разрешение, оно не лишено ограничений. Повреждение ДНК лазерным микролучением сильно кластеризовано на определенные части ядра по сравнению с естественными повреждающими агентами. Таким образом, реакция хроматина на микролучение может отличаться по сравнению с гомогенно распределенным повреждением. Кроме того, микроизлучение занимает много времени и может проводиться только на нескольких десятках клеток, в то время как крупные популяционные биохимические методы (фракционирование хроматина, иммунопреципитация, ChIP) могут обеспечить повышенную надежность путем изучения тысяч клеток одновременно. Проверка наблюдений, сделанных путем микролучения, с помощью традиционных биохимических методов является эффективной стратегией для надежных выводов. Хотя одновременное микролучение многих клеток в определенном FOV возможно, системе визуализации потребуется больше времени для выполнения задачи. Поэтому измерение динамики белков, которые очень быстро рекрутируется к поражениям ДНК, ограничивает количество возможных ROI для микрооблучения, используемых одновременно. В системе визуализации, используемой для этого протокола, микроизлучение одной рентабельности инвестиций длиной 1024 пикселя занимает 1032 мс при длительности 1000 мкс и 3088 мс при использовании времени ожидания 3000 мкс. Использование нескольких линий ROI значительно увеличит время, необходимое для завершения микроизлучения (например, roi длиной 7 x 1024 пикселей занимает 14402 мс при длительности 1000 мкс и 21598 мс при времени ожидания 3000 мкс). Это время теряется при получении изображения и должно быть принято во внимание. При визуализации событий быстрого набора используйте кратчайший возможный ROI и облучайте только одну клетку за раз.
Преимущества и ограничения по сравнению с методами синхронизации
Для исследований, специфичных для клеточного цикла, существующие методы включают либо синхронизацию клеток в определенные фазы клеточного цикла, либо использование флуоресцентных репортеров для идентификации конкретной фазы клеточного цикла клетки. Однако каждый из этих методов предоставляет свои собственные проблемы и ограничения.
СистемаFUCCI 3 (основанная на флуоресцентных белках, помеченных усеченными формами CDT1 и Geminin) является особенно полезным инструментом для исследований клеточного цикла, но имеет ограничения, когда дело доходит до дифференциации между фазами S и G2 клеточного цикла. Уровни геминина уже высоки с середины S-фазы и остаются высокими до фазы M, что затрудняет разделение этих фаз. Использование системы FUCCI также означает, что два оптических канала микроскопа не могут быть использованы для визуализации POI.
Нераковые клеточные линии могут быть синхронизированы в G0 путем удаления факторов роста, обнаруженных в сыворотке (сывороточное голодание), вызывая небольшое повреждение или отсутствие повреждения ДНК клеток. Тем не менее, большинство линий раковых клеток будут частично продолжать прогрессировать через клеточный цикл даже без достаточного количества сыворотки в их средах. Кроме того, клетки частично начинают терять синхронизацию к концу G1, ранней S-фазе. В дополнение к сывороточному голоданию, существует множество химических методов для достижения синхронизации клеточного цикла. Блоки гидроксимочевины, афидиколина и тимидина являются методами остановки репликации ДНК для синхронизации клеток в раннюю S-фазу. Хотя эти методы дешевы и просты, они вводят стресс репликации, который приводит к повреждению ДНК. Было показано, что эти ингибиторы репликации ДНК индуцируют фосфорилирование H2A. X, известный маркер DSB2,29. Метод использования метки PCNA в качестве маркера для S-фазовых клеток снижает потенциал артефактов, вызванных химической синхронизацией, и может быть применен к широкому спектру клеточных линий по сравнению с сывороточным голоданием.
Заключение
Повреждение ДНК является движущей силой генетических заболеваний, где мутагенные поражения могут привести к злокачественной трансформации клеток. Нацеливание на механизм синтеза ДНК является фундаментальной терапевтической стратегией в лечении гиперпролиферативных заболеваний, таких как рак. Чтобы лечить эти заболевания более целенаправленно, нам нужно лучше понять белки, которые восстанавливают повреждения ДНК. Протокол, описанный здесь, помогает исследованиям на основе микролучения в S-фазе, сводя к минимуму проблемы, связанные с традиционными методами синхронизации, для уменьшения возможных артефактов и повышения воспроизводимости экспериментов.
Disclosures
Авторы утверждают, что публикация представленной работы была спонсирована корпорацией Nikon. Авторы заявляют, что конкурирующих интересов не существует.
Acknowledgments
Авторы благодарят М. Пагано за постоянную поддержку, а также Д. Симонески, А. Марцио и Г. Танга за критический обзор рукописи. Б. Миватани-Минтер благодарит Р. Миватани и Б. Минтера за их постоянную поддержку. Г. Рона благодарит К. Ронане Юраша и Г. Рону за их постоянную поддержку.
Materials
| Name | Company | Catalog Number | Comments |
| Ammonium chloride | Sigma-Aldrich | A9434-500G | For quenching formaldehyde |
| Anti-EXO1 Rabbit Polyclonal Antibody | Proteintech | 16253-1-AP | primary antibody |
| Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301 | Millipore | 05-636 | primary antibody |
| Bovine Serum Albumin | Sigma-Aldrich | 3117332001 | BSA for blocking |
| BrdU (5-Bromo-2'-deoxyuridine) | Merck | 19-160 | pre-sensitizing agent |
| Citifluor™ Mountant Solution AFR3 | Electron Microscopy Sciences | 17973-10 | antifade containing PBS solution for imaging |
| DAPI | Sigma-Aldrich | D9542-1MG | nucleic acid stain |
| DMEM Medium | Thermo Fisher Scientific | 10569010 | Cell culture medium for HEK293T cells |
| DMSO | Sigma-Aldrich | D2650-100ML | Vehichle control and dissolution solvent |
| EGFP-FBXL10 | Addgene | #126542 | viral expression vector for EGFP-FBXL10 |
| EXO1b-AcGFP (in pRetroQ) | custom cloning | na | EXO1b cDNA was cloned in the NheI, BamHI sites of pRetroQ-AcGFP1-N1 vector. |
| Fetal Bovine Serum | Gibco | 16140071 | Media supplement |
| FluoroBrite DMEM | Thermo Fisher Scientific | A1896701 | Phenol red free medium for microscopy |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | A32723 | secondary antibody |
| HEK293T cells | ATCC | ATCC CRL-3216 | Cell line for viral packaging |
| HEPES | Sigma-Aldrich | H0887-100ML | Buffering agent to supplement live cell imaging medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | pre-sensitizing agent |
| Lipofectamine 3000 | Thermo Fisher Scientific | L3000015 | Transfection reagent |
| McCoy’s 5A (Modified) Medium | Life Technologies | 16600-108 | Cell culture medium for U-2 OS cells |
| mCherry-PCNA | Addgene | #55117 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA | Addgene | #55994 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA (in pBABE) | custom cloning | na | mPlum-PCNA cDNA was cloned from Addgene #55994 in the BamHI, SalI sites of pBABE (puro) |
| Nikon A1R-HD25 Confocal Scanhead and Controller | Nikon | na | confocal imaging system |
| Nikon LUN4 laser unit | Nikon | na | excitation system |
| Nikon LUN-F 50 mW 405 nm FRAP laser unit | Nikon | na | FRAP laser unit |
| Nikon NIS Elements Confocal Controller Software | Nikon | na | Confocal controlling software |
| Nikon Ti2-E Inverted Microscope | Nikon | na | inverted epifluorescent microscope base |
| Nikon Ti2-LAPP Modular Illumination System | Nikon | na | illumination system |
| NTHL1-mCherry (in pRetroQ) | custom cloning | na | NTHL1 cDNA was cloned in the NheI, SalI sites of pRetroQ-mCherry-N1 vector. |
| Nunc Lab-Tek II Chambered Coverglass (4 well) | Thermo Fisher Scientific | 155382PK | Live cell microscopy cell culture chamber |
| Olaparib | Selleck Chemicals | S1060 | PARP inhibitor |
| Opti-MEM reduced serum media | Thermo Fisher Scientific | 31985062 | Dilution medium for transient transfection |
| Paraformaldehyde aqueous solution (32%) | Thermo Fisher Scientific | 50-980-494 | Fixative |
| pBABE (hygro) | Addgene | #1765 | retroviral expression vector (for low expression levels) |
| pBABE (neo) | Addgene | #1767 | retroviral expression vector (for low expression levels) |
| pBABE (puro) | Addgene | #1764 | retroviral expression vector (for low expression levels) |
| pBABE (zeo) | Addgene | #1766 | retroviral expression vector (for low expression levels) |
| PCNA Antibody (PC10) | Santa Cruz | sc-56 | primary antibody |
| Penicillin-Streptomycin-Glutamine (100x) | Gibco | 10378016 | Media supplement |
| polybrene | Sigma-Aldrich | TR-1003 | Increase viral infection efficiency |
| pRetroQ-AcGFP-C1 | Takara | 632506 | retroviral expression vector |
| pRetroQ-AcGFP-N1 | Takara | 632505 | retroviral expression vector |
| pRetroQ-mCherry-C1 | Takara | 632567 | retroviral expression vector |
| pRetroQ-mCherry-N1 | Takara | 632568 | retroviral expression vector |
| pUMVC | Addgene | #8449 | Viral packaging vector |
| Sodium-pyruvate | Thermo Fisher Scientific | 11360070 | Supplement for live cell imaging medium |
| Triton X-100 aqueous solution (10%) | Sigma-Aldrich | 11332481001 | Dilute in PBS for cell permeabilization buffer |
| Trypsin-EDTA Solution 10X | Sigma-Aldrich | 59418C-100ML | Dilute in PBS to split cells |
| U-2 OS Cells | ATCC | HTB-96 | Optimal cell line for microscopy experiments |
| Universal Mycoplasma Detection Kit | ATCC | 30-1012K | PCR based Mycoplasma detection kit |
| VSV-G | Addgene | #8454 | Viral protein envelope vector |
References
- Aleksandrov, R., et al.
- Darzynkiewicz, Z., Halicka, H. D., Zhao, H., Podhorecka, M. Cell synchronization by inhibitors of DNA replication induces replication stress and DNA damage response: Analysis by flow cytometry. Methods in Molecular Biology. 761, 85-96 (2011).
- Sakaue-Sawano, A., et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 132 (3), 487-498 (2008).
- Herce, H. D., Rajan, M., Lattig-Tunnemann, G., Fillies, M., Cardoso, M. C. A novel cell permeable DNA replication and repair marker. Nucleus. 5 (6), 590-600 (2014).
- Keijzers, G., et al. Human exonuclease 1 (EXO1) regulatory functions in dna replication with putative roles in cancer. International Journal of Molecular Sciences. 20 (1), (2018).
- Cheruiyot, A., et al. Poly(ADP-ribose)-binding promotes Exo1 damage recruitment and suppresses its nuclease activities. DNA Repair (Amsterdam). 35, 106-115 (2015).
- Zhang, F., Shi, J., Chen, S. H., Bian, C., Yu, X. The PIN domain of EXO1 recognizes poly(ADP-ribose) in DNA damage response. Nucleic Acids Research. 43 (22), 10782-10794 (2015).
- Rona, G., et al. PARP1-dependent recruitment of the FBXL10-RNF68-RNF2 ubiquitin ligase to sites of DNA damage controls H2A.Z loading. elife. 7, (2018).
- Young, L. M., et al. TIMELESS forms a complex with PARP1 distinct from its complex with TIPIN and plays a role in the dna damage response. Cell Reports. 13 (3), 451-459 (2015).
- Kong, X., et al. Laser microirradiation to study in vivo cellular responses to simple and complex dna damage. Journal of Visualized Experiments. (131), e56213 (2018).
- Kong, X., et al. Condensin I recruitment to base damage-enriched DNA lesions is modulated by PARP1. PLoS One. 6 (8), 23548 (2011).
- Lan, L., et al. Novel method for site-specific induction of oxidative DNA damage reveals differences in recruitment of repair proteins to heterochromatin and euchromatin. Nucleic Acids Research. 42 (4), 2330-2345 (2014).
- Zerjatke, T., et al. Quantitative cell cycle analysis based on an endogenous all-in-one reporter for cell tracking and classification. Cell Reports. 19 (9), 1953-1966 (2017).
- Ji, Y., Karbaschi, M., Cooke, M. S. Mycoplasma infection of cultured cells induces oxidative stress and attenuates cellular base excision repair activity. Mutation Research. 845, 403054 (2019).
- Sun, G., et al. Mycoplasma pneumoniae infection induces reactive oxygen species and DNA damage in A549 human lung carcinoma cells. Infection and Immunity. 76 (10), 4405-4413 (2008).
- Gassman, N. R., Wilson, S. H. Micro-irradiation tools to visualize base excision repair and single-strand break repair. DNA Repair (Amsterdam). 31, 52-63 (2015).
- Muster, B., Rapp, A., Cardoso, M. C. Systematic analysis of DNA damage induction and DNA repair pathway activation by continuous wave visible light laser micro-irradiation. AIMS Genetics. 4 (1), 47-68 (2017).
- Ikeda, S., et al. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. Journal of Biological Chemistry. 273 (34), 21585-21593 (1998).
- Rosenquist, T. A., Zharkov, D. O., Grollman, A. P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proceedings of the National Academy of Science U. S. A. 94 (14), 7429-7434 (1997).
- Reid, D. A., et al. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proceedings of the National Academy of Science U. S. A. 112 (20), 2575-2584 (2015).
- Taccioli, G. E., et al. Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science. 265 (5177), 1442-1445 (1994).
- Marsin, S., et al. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. Journal of Biological Chemistry. 278 (45), 44068-44074 (2003).
- Thompson, L. H., Brookman, K. W., Jones, N. J., Allen, S. A., Carrano, A. V. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Molecular and Cell Biology. 10 (12), 6160-6171 (1990).
- Scharer, O. D.
- Oeck, S., et al. High-throughput evaluation of protein migration and localization after laser micro-irradiation. Science Reports. 9 (1), 3148 (2019).
- Mistrik, M., et al. Cells and stripes: A novel quantitative photo-manipulation technique. Science Reports. 6, 19567 (2016).
- Durand, R. E., Olive, P. L. Cytotoxicity, mutagenicity and dna damage by hoechst 33342. Journal of Histochemistry and Cytochemistry. 30 (2), 111-116 (1982).
- Tobey, R. A., Oishi, N., Crissman, H. A. Cell cycle synchronization: reversible induction of G2 synchrony in cultured rodent and human diploid fibroblasts. Proceedings of the National Academy of Science U. S. A. 87 (13), 5104-5108 (1990).
- Podhorecka, M., Skladanowski, A., Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. Journal of Nucleic Acids. 2010, (2010).
