

Summary
Detta protokoll beskriver en icke-invasiv metod för att effektivt identifiera S-fasceller för mikroskopistudier nedströms, såsom mätning av DNA-reparationsproteinrekrytering genom lasermikrålning.
Abstract
DNA-skadereparation upprätthåller cellernas genetiska integritet i en mycket reaktiv miljö. Celler kan ackumulera olika typer av DNA-skador på grund av både endogena och exogena källor som metabolisk aktivitet eller UV-strålning. Utan DNA-reparation äventyras cellens genetiska kod, vilket undergräver proteinens strukturer och funktioner och potentiellt orsakar sjukdom.
Att förstå den spatiotemporala dynamiken i de olika DNA-reparationsvägarna i olika cellcykelfaser är avgörande inom DNA-skadereparation. Nuvarande fluorescerande mikroskopitekniker ger bra verktyg för att mäta rekryteringskinetiken för olika reparationsproteiner efter DNA-skada induktion. DNA-syntes under cellcykelns S-fas är en märklig punkt i cellens öde när det gäller DNA-reparation. Det ger ett unikt fönster för att screena hela genomet för misstag. Samtidigt utgör DNA-syntesfel också ett hot mot DNA-integriteten som inte påträffas i icke-dela celler. Därför skiljer sig DNA-reparationsprocesserna avsevärt i S-fas jämfört med andra faser av cellcykeln, och dessa skillnader är dåligt förstådda.
Följande protokoll beskriver beredning av cellinjer och mätning av dynamiken hos DNA-reparationsproteiner i S-fas på lokalt inducerade DNA-skadeplatser, med hjälp av ett laserskanningskonfokalt mikroskop utrustat med en 405 nm laserlinje. Märkt PCNA (med mPlum) används som cellcykelmarkör kombinerat med ett AcGFP-märkt reparationsprotein av intresse (dvs EXO1b) för att mäta DNA-skaderekryteringen i S-fas.
Introduction
Flera DNA-reparationsvägar har utvecklats för att ta itu med de olika typer av DNA-lesioner som kan uppstå i celler, som alla är starkt reglerade i både tid och rum. En av de mest sårbara perioderna i cellcykeln är S-fasen, när DNA-syntes uppstår. Även om spridning är grundläggande för livet utgör det också en stor utmaning. Celler måste säkerställa trofast replikation av sitt arvsmassa för att undvika mutationer som ska överföras till framtida generationer. Följaktligen ger spridning en terapeutisk interventionspunkt som har använts för utveckling av terapeutiska metoder inom onkologiområdet.
Alla de viktigaste teknikerna som används för att studera proteinrekrytering vid DNA-lesioner har sina styrkor och begränsningar. Mikrostrålning har bättre rumslig och temporal upplösning1 än de flesta alternativa metoder som immunofluorescerande avbildning av joniserande strålningsinducerade högborgar (IRIF), kromatinimmunprecipitation (ChIP) eller biokemisk fraktionering. Mikro bestrålning är dock robustheten hos ovannämnda tekniker som kan ta prov på ett stort antal celler samtidigt.
För att undersöka DNA-reparation i S-fas måste man kunna urskilja S-fasceller i en asynkron cellkulturpopulation. Det finns många välkända metoder för att ta itu med detta, som antingen omfattar synkronisering av celler eller visualisering av de olika cellcykelfaserna. Båda tillvägagångssätten medför dock betydande utmaningar och möjliga artefakter. Kemiska synkroniseringsmetoder som ofta används för att berika celler i tidig S-fas (t.ex. dubbla tymidinblock, aphidicolin och hydroxyureabehandling) uppnår synkronisering genom induktion av replikeringsstress och så småningom DNA-skada själv. Detta begränsar användningen av dessa metoder för att studera DNA-reparationsprocesser i S fas2. Synkronisering genom serum svält och frisättning är endast tillämpligt på ett begränsat antal cellinjer, till stor del exklusive cancer cellinjer som förlitar sig mindre på tillväxtfaktorer för cellcykel progression jämfört med icke-omvandlade cellinjer. Fluorescens Ubiquitin Cell Cycle Indicator (FUCCI) system är ett särskilt användbart verktyg för att studera cellcykeln, men det har en grundläggande begränsning vid differentiering mellan S- och G2-cellcykelfaser3.
Här visas att användning av fluorescerande märkt PCNA som en icke-invasiv markör för S-fas begränsar nackdelarna med kemiska cellcykelsynkroniseringsmetoder, samtidigt som det möjliggör mer specificitet och flexibilitet än FUCCI-systemet. Som en enda markör kan PCNA inte bara markera S-fasceller i en asynkron population, utan det kan också visa den exakta utvecklingen av celler inom S-fasen (dvs. tidig, mitten eller sen S-fas)4. Låga uttrycksnivåer av exogena, taggade PCNA säkerställer minimal interferens med både cellcykelprogression och DNA-reparationsprocesser. Viktigt är att PCNA också fungerar som en intern kontroll för korrekt DNA-skada induktion eftersom det är involverat i reparation av flera DNA-skador och rekryteras till lokalt inducerade DNA-skadeplatser1,4.
De experiment som presenteras här visar hur man mäter rekryteringsdynamiken hos EXO1b i S-fas och hur detta påverkas av den väletablerade PARP-hämmaren olaparib. EXO1b nukleasaktivitet är relevant för ett brett spektrum av DNA-reparationsvägar, inklusive felmatchning reparation (MMR), nukleotid excision reparation (NER) och dubbelsträngad paus (DSB) reparation. I S-fas spelar EXO1b en viktig roll i homolog rekombination (HR) genom bildandet av 3' ssDNA-överhäng under DNA-samband5. EXO1b har varit ytterligare inblandad i DNA-replikering med roller i kontrollpunktsaktivering för att starta om avstannade DNA-gafflar samt primerborttagning och Okazaki fragmentmognad vid den eftersläpande strängen under strandförskjutning ireplikering 5. EXO1b-rekrytering till skadade DNA-platser regleras av den direkta interaktionen med poly (ADP-ribose) (PAR)6,7. På grund av de många cellcykelspecifika konsekvenserna av EXO1b är det ett utmärkt val för S-fasspecifika rekryteringsstudier med PCNA.
Protocol
1. Odling av humana osteosarkom-härledda celler (U-2 OS)
OBS: U-2 OS-celler är idealiska för dessa studier eftersom de har en platt morfologi, stor kärna och fästs starkt på flera ytor, inklusive glas. Andra cellinjer med liknande egenskaper kan också användas.
- För odling av U-2 OS cellinjer, använd McCoys 5A medium kompletterat med 10% fetala nötkreatur serum (FBS) och antibiotika (100 U/mL penicillin och 100 μg/mL streptomycin). Inkubera celler vid 37 °C i en fuktad atmosfär som innehåller 5 % CO2. För mikroskopistudier, behåll cellkulturen i en 10 cm maträtt för att ge tillräckligt med cellantal.
- När celler närmar sig 90% konfluens (~ 7 x 106 celler / 10 cm maträtt), dela cellerna.
- Skölj celler med PBS för att tvätta bort trypsinhämmare som finns i serumet.
- Tillsätt 1 ml Trypsin-EDTA och se till att cellskiktet är lika täckt.
- Inkubera vid 37 °C tills cellskiktet lyfts av plattan (ca 6 min).
- Återanvänd de trypsiniserade cellerna i serum som innehåller media för att inaktivera trypsin och tillsätt 1/10th av volymen (~ 0,7 x 106 celler) i en ny 10 cm platta som innehåller 10 mL kompletterat tillväxtmedium.
- Före experimentet testar du rutinmässigt celler för mykoplasmakontaminering med universalförpackningen Mycoplasma Detection enligt tillverkarens rekommendation.
2. Retroviral infektion
OBS: För BSL-2 säkerhetsåtgärder och medan du arbetar med rekombinanta virus, se: NIH-riktlinjer, avsnitt III-D-3: Rekombinanta virus i vävnadskultur.
- Frö 4 x 106 HEK293T celler för att uppnå ~60% sammanflöde inom 24 h efter plätering i en 10 cm kulturrätt.
- För odling av HEK293T, följ odlingsstegen i U-2 OS som beskrivs i 1.1-1.3 i detta protokoll. För HEK293T ersättning McCoys 5A medium för DMEM. Var noga med att alltid försiktigt tvätta HEK293T-celler när de fäster på vävnadskulturplattor svagt.
- Transfekta HEK293T-celler med hjälp av ett lipidbaserat transfection reagens för viral förpackning av plasmider.
- För retrovirala vektorer, kombinera 1,5 μg VSV-G (Addgene #8454) och 1,5 μg pUMVC (Addgene #8449) förpackningsvektorer tillsammans med 3 μg vektor som innehåller genen av intresse (i en retroviral vektor ryggrad med puromycinresistens) i 250 μL Opti-MEM reducerat serummedier i ett mikrocentrifugrör. Tillsätt 1 μL P3000-reagens för varje μg DNA som tillsätts i Opti-MEM/DNA-blandningen (i detta fall 6 μL) och blanda försiktigt genom att knacka. Virvel eller pipett upp och ner.
- Kombinera 2 μL per μg DNA (i detta fall 12 μL) transfektreagens i ett annat mikrocentrifugrör med 250 μL Opti-MEM-reducerat serummedium.
- Kombinera de två blandningarna (500 μL kombinerat, virvel inte, blanda bara genom skonsam gängning) och låt det inkubera i 15 minuter vid rumstemperatur.
- Tillsätt försiktigt blandningen droppvis till de sådda HEK293T-cellerna utan att lossa cellerna. Snurra plattorna försiktigt.
- Virusinfektion för att generera stabila cellinjer.
- Ta bort viruset som innehåller supernatant från HEK293T-cellerna 72 h efter transfektion. Filtrera försiktigt lösningen med ett filter på 0,45 μm för att avlägsna cellskräp och fristående celler. Som tillval, tillsätt 8 μg/ml polybren till virussplitanten för att underlätta virusinfektion.
- Tillsätt virus som innehåller supernatant till U-2 OS-celler vid ~ 50% sammanflöde i en 10 cm maträtt (~ 3 x 106 celler). Så U-2 OS-cellerna dagen innan.
- Infektera i 6-16 timmar innan du tar bort och kasserar det virusinnehållande supernatantet.
OBS: För att uppnå önskad mängd överuttryck för genen av intresse, inkubera en serie virala utspädningar under en fast tid. Kontrollera transgenens uttrycksnivåer i varje nyetablerad cellinje med western blot som jämför den med endogena nivåer. - Låt cellerna välja i närvaro av lämpliga antibiotika (i 3-4 dagar vid puromycin vid 2 μg/ml slutlig koncentration) och kontrollera att den fluorescerande proteinmärkta genen av intresse uttrycks under ett mikroskop.
- Upprepa dessa steg för att generera dubbelmärkta cellinjer. I de experiment som presenteras här uttrycktes mPlum-PCNA från en retroviral vektor (pBABE) i kombination med EXO1B-AcGFP, också uttryckt från en retroviral vektor (pRetroQ-AcGFP1-N1).
3. Beredning av celler för mikrostrålning
- Pläteringsceller: 24 timmar före försöket, plätera totalt 8,0 x 104 celler i en volym mellan 500 μL-1 ml media (för ungefär 70% sammanflöde) på ett fyra välkammade täckglas med en borosilikatglasbotten nr 1,5 som ger idealiska resultat för konfokal mikroskopi med hög förstoring och lasermikrålning. En högre cellkonfluens möjliggör fler celler mätta i ett enda synfält (FOV); Men helt sammanflytade bilder kommer att införa oegentligheter i cellcykeln.
- Bildmedier: En timme före mikrostrålning, utbyte av regelbundet tillväxtmedium mot FluoroBrite DMEM kompletterat med 10% FBS, 100 U/mL penicillin och 100 μg/mL streptomycin, 15 mM HEPES (pH=7,4) och 1 mM natrium-pyruvat. Detta bildmedier hjälper till att maximera signal-till-brus-förhållandet så att mycket dim fluorescens kan upptäckas. Eftersom den innehåller HEPES stabiliserar den också pH i avsaknad av en 5% CO2 atmosfär.
- Applicera eventuell ytterligare behandling före avbildningen i detta steg. I de experiment som presenteras här förbehandlades cellerna en timme före avbildning med antingen olaparib (PARP-hämmare, vid 1 μM slutlig koncentration) eller en fordonskontroll (DMSO)1,8,9.
4. Förbereda mikroskopet och välja S-fasceller för avbildning.
- Använd ett konfokalt system som har liknande egenskaper som systemet som beskrivs här för bästa resultat. Experimenten som presenteras här utfördes med hjälp av ett konfokalt mikroskop monterat på ett inverterat mikroskopstativ (se Materialförteckning ).
OBS: Mikroskopet som användes här var utrustat med en 50 mW 405 nm FRAP lasermodul och ett 60x 1,4 NA oljeplan-apochromat mål. Det konfokala scanhead hade två skanneralternativ: en galvanoskanner (för hög upplösning) och resonansskanner (för höghastighetsavbildning).- Introducera fluorescensåterställning efter att ha fotoblekat (FRAP) laser till provet via en programvarustyrd XY galvano-enhet. Använd en laserlinje på 488 nm för att excite AcGFP och en 561 nm eller 594 nm laserlinje för att excite mPlum.
OBS: Följande filterkombination ger optimala resultat: med hjälp av ett 560 nm långt passfilter passerades emissionsljus med en våglängd under 560 nm genom ett 525/50 nm utsläppsfilter för AcGFP, medan emissionsljus med en våglängd högre än 560 nm passerades genom ett 595/50 nm utsläppsfilter för mPlum. Alla lämpliga filteruppsättningar (t.ex. FITC/TRITC, GFP/mCherry, FITC/TxRed) som säkerställer minimal fluorescensblödning kan användas.
- Introducera fluorescensåterställning efter att ha fotoblekat (FRAP) laser till provet via en programvarustyrd XY galvano-enhet. Använd en laserlinje på 488 nm för att excite AcGFP och en 561 nm eller 594 nm laserlinje för att excite mPlum.
- Slå på miljökammaren och mikroskopkomponenterna.
- Slå på uppvärmningen (steg, mål och miljökammare när det är möjligt), CO2-tillförseln och fuktighetsregulatorn minst 4 timmar före experimentets början för att säkerställa termisk jämvikt för stabilt bildförvärv.
- Initiera ljuskällor tillsammans med laserlinjerna minst 1 timme före överföringen av cellerna till mikroskopet.
- Markera S-fasceller i en asynkron population med fluorescerande taggade PCNA som markör. Gör detta genom att följa stegen nedan.
- Leta efter det unika lokaliseringsmönstret för den mPlum-taggade PCNA i S-fasen, vilket gör identifiering av denna cellcykelfas möjlig. PCNA har en helt homogen fördelning i kärnan i G1- och G2-faserna i cellcykeln, samtidigt som den utesluts från kärnan. I S-fas bildar PCNA högborgar på platsen för replisomes i kärnan. Figur 1 visar de olika mönstren för PCNA-högborgar under hela S-fasen, vilket gör det möjligt att till och med differentiera tidig, medel- och sen S-fas.
- Titta igenom okulär för att välja en FOV som har tillräckligt med S-fasceller för mikro bestrålning. Asynkrona U-2 OS-celler har vanligtvis 30-40% av sin befolkning i S-fas.
- Försök att undvika ytterligheter i uttrycksnivåer (både ljusa och dunkla celler) för både PCNA och proteinet av intresse (POI), i detta fall EXO1b-AcGFP, vilket kan leda till experimentella artefakter.
- När du hittar en lämplig FOV, försök att undvika att skanna fältet under lång tid för att minimera fotoblekning och oönskade DNA-skador.
- Ställ in önskad intresseregion (ROI) för mikro bestrålning. Använd tillhörande programvara (se Materialförteckning ), ställin önskad ROI genom att först infoga binära linjer (ange önskat antal linjer och avstånd). Klicka på Binäroch sedan på Infoga rad | Cirkel | Ellips för att rita önskat antal linjer.
- Konvertera dessa binära linjer till ROM och konvertera slutligen dessa ROM till stimulerings-ROM. För att göra det, klicka först på ROI, klicka sedan på Flytta binärtill ROI , högerklicka sedan på någon av ROIs och välj Använd som Stimulering ROI: S1. Placera dessa linjer i FOV för att passera genom cellernas kärna. ROM med en längd på 1024 pixlar som sträckte sig över hela FOV användes i hela protokollet.
5. Mikrostrålning för immunofluorescensfärgning eller tidsfördröjningsavbildning.
- Bestämma optimala inställningar för mikro bestrålning.
- Innan mikrostrålning av cellerna, ta en högre upplösning bild av FOV för att identifiera PCNA högborgar för senare analys. Istället för sekventiell skanning spelar du samtidigt in båda optiska kanalerna som används (grönt och rött), för att undvika cellrörelser mellan skanning vid de två våglängderna. För korrekt upplösning av foci använd minst 1024 x 1024 pixlar / fältupplösning med 1x zoom (0,29 μm pixelstorlek på bildsystemet som används här), med 1/8 bildruta / skanningshastighet (4,85 μs / pixel) med 2x i genomsnitt. När dessa parametrar har angetts i A1 LFOV Compact GUI och A1 LFOV Scan Area-fönstren trycker du på capture-knappen för att spela in FOV.
Obs: Det är viktigt att behålla samma pixelstorlek under experiment för att säkerställa jämförbara resultat. - Om du vill ställa in mikrostrålningen öppnar du fliken ND-stimulering i bildframställningsprogrammet för att komma åt fönstret Tidsschema (A1 LFOV / Galvano Device). Detta använder galvanoskannrarna för att förvärva en serie förstimuleringsbilder, stimulera (med lun-F 50 mW 405 nm FRAP-laser) och sedan förvärva en serie efterstimuleringsbilder igen med hjälp av galvanoskannrarna. Ställ först in tre faser i tidsschemafönstret. Välj Förvärvs- och anskaffnings- och | i kolumnen Acq/Stim Blekning | Förvärv för de tre faserna. För blekningsfasen ställer du in S1 som ROI.
OBS: I experimentet som presenteras här förvärvades inga bilder under stimuleringsfasen. - I Galvano XY-fönstretställer du in de viktigaste faktorerna för mikrostrålning: 405 nm lasereffekt, uppehållstid (iteration är 1 som standard på detta system). I experimenten som presenteras här bestrålades celler med 405 nm FRAP-laser (50 mW vid fiberspetsen) vid 100% uteffekt med en 1000-3000 μs uppehållstid.
OBS: Eftersom laser uppehållstiden är per pixel, så länge pixelstorleken förblir densamma, kommer förhållandet mellan uppehållstiden och effekttätheten att vara jämförbart mellan olika FOV: er. Figur 2A visar användningen av DDR-spårspecifika proteiner (FBXL10 för DSB och NTHL1 för oxidativ basskada) för att optimera lasereffektinställningarna för specifik induktion av skador. Dessa stabila cellinjer genererades med viral infektion efter avsnitt 2 i protokollet.
- Innan mikrostrålning av cellerna, ta en högre upplösning bild av FOV för att identifiera PCNA högborgar för senare analys. Istället för sekventiell skanning spelar du samtidigt in båda optiska kanalerna som används (grönt och rött), för att undvika cellrörelser mellan skanning vid de två våglängderna. För korrekt upplösning av foci använd minst 1024 x 1024 pixlar / fältupplösning med 1x zoom (0,29 μm pixelstorlek på bildsystemet som används här), med 1/8 bildruta / skanningshastighet (4,85 μs / pixel) med 2x i genomsnitt. När dessa parametrar har angetts i A1 LFOV Compact GUI och A1 LFOV Scan Area-fönstren trycker du på capture-knappen för att spela in FOV.
- Avbildning av tidsfördröjning.
- Ställ in time lapse-avbildning för önskat tidsfönster och intervall med hjälp av tidsschemat, A1 LFOV Compact GUI och A1 LFOV Scan Area-fönstren. I experimenten som presenteras här, rekryteringen av EXO1b och PCNA var bild i 12 min, skannade FOV var 5: e sekund, vid 1024 x 1024 pixlar / fält, med 1x zoom (vilket resulterade i 0,29 μm pixelstorlek på bildsystemet som används här) med 0,35 bildruta / s skanningshastighet (1,45 μs / pixel) utan att i genomsnitt minska fotoblekning.
- Optimera lasereffekten %, få och förskjuta inställningar för att minska fotoblekningen under avbildningen i A1 LFOV Compact GUI-fönstret. Om man strävar efter att mäta både POI och PCNA, använd samtidig skanning istället för sekventiell skanning för att undvika cellrörelser mellan skanning av fältet för de två separata fluorforerna.
- Bildsystemet användes med följande inställningar. För laserlinjen på 488 nm (20 mW): 7% lasereffekt, förstärkning: 45 (GaAsP-detektor) med och förskjutning av 2, för 561 nm laserlinje (20 mW): 4% lasereffekt, få 40 (GaAsP-detektor) med och förskjutning på 2.
- Beroende på proteinets kinetik, förläng eller förkorta intervallet mellan bilderna eller varaktigheten av den totala tidsförfall. I fönstret Tidsschema anger du önskat intervall och varaktighet för anskaffningsraden för tredje fasen.
- Tryck på Kör nu för att köra mikrostrålningen och den efterföljande time lapse-avbildningen.
- I slutet av tidsfördröjningen, spara stimulering ROIs som separata bilder, vilket kommer att vara ett användbart hjälpmedel för att identifiera koordinaterna för mikro bestrålning i någon nedströms programvara som används för analys.
- Immunofluorescens färgning.
OBS: Steg 5.1.3 och figur 2A visar användningen av kända DNA-reparationsproteiner för att bedöma de typer av DNA-lesioner som införs genom mikrostrålning. Vissa DNA-lesioner kan också detekteras genom att använda specifika antikroppar efter fixering av cellerna. Det är också möjligt att upptäcka rekryteringen av POI genom antikroppsdetektering av det endogena proteinet. Visualiseringen av γH2A.X för att kontrollera om DSB visas nedan (figur 2B). Figur 3 visar konsekvensen i PCNA lokalisering och rekrytering under hela cellcykeln för både endogena och exogena taggade PCNA.- Efter steg 5.1.3, ta bara en bild efter mikrostrålning för att säkerställa korrekt FRAP-händelse baserat på rekrytering av mPlum-PCNA. Notera de exakta koordinaterna för FOV för att hitta fältet senare efter den immunofluorescerande märkningen.
- Ta ut cellkulturkammaren ur mikroskopet och inkubera celler vid 37 °C i en fuktad atmosfär som innehåller 5 % CO2 i 5-10 min.
OBS: Paraformaldehyd (PFA) är giftigt och arbete bör utföras i ett välventilerat område eller en rökhuv. All efterföljande tvätt och inkubation kommer att göras med 0,5 ml volymer i 4 brunnskammare. Efter inkubationstiden, tvätta cellerna med 0,5 ml PBS (137 mM NaCl, 2,7 mM KCl, 8 mM Na2HPO4och 2 mM KH2PO4) och fixera med 0,5 mL 4% PFA i PBS i 10 min vid rumstemperatur (RT). - Tvätta cellerna en gång med PBS och tvätta dem sedan med 50 mM NH4Cl för att släcka kvarvarande PFA.
- Permeabilisera cellerna i 15 minuter på RT med 0,1% Triton X-100 i PBS.
- Blockera proverna i 1 h med blockeringsbuffert (5% FBS, 3% BSA, 0,05% Triton X-100 i PBS).
- Ta bort blockeringslösningen och tillsätt den utspädda primära antikroppen (anti-γH2A.X, 1:2000) i blockeringsbufferten i 1 timme vid RT.
- Tvätta brunnarna med blockeringsbuffert 3 x 10 min.
- Tillsätt utspädd sekundär antikropp (antimus Alexa 488 Plus konjugat, 1:2000) i blockeringsbuffert i 1 timme på RT.
- Tvätta brunnarna med blockeringsbuffert 3 x 10 min.
- Kontrahåll kärnan med 1 μg/ml DAPI-lösning i PBS i 15 minuter.
- Tvätta cellerna en gång med PBS. Avbildningen kan utföras direkt i PBS eller en PBS-lösning med antifadereagenser (t.ex. AFR3) för att minska fotoblekningen.
6. Rekryteringsanalys
OBS: Figur 4A visar representativa bilder av Exo1b- och PCNA-rekrytering i närvaro av DMSO eller olaparib. Figur 4B visar en representativ bild för dataanalys. Medelvärdet för fluorescensvärden beräknades med hjälp av genomsnittliga AcGFP-intensiteter med hjälp av en rektangel längs laserspåret som markerats av mPlum-PCNA (A, gula rektanglar) över olika tidspunkter med Fiji. PCNA kan fungera som en intern kontroll för att markera framgångsrik bestrålning längs ROI-koordinaterna. På samma sätt beräknades genomsnittliga AcGFP fluorescensvärden också för oskadade regioner i kärnan (B, blå rektanglar). Bakgrundssignalens intensitet mättes i obefolkade områden (C, röda rektanglar) och subtraherades från de genomsnittliga fluorescerande värdena (figur A och B). Således beräknades den relativa genomsnittliga fluorescerande enheten (RFU) för varje datainsamlingspunkt med ekvationen RFU = (A − C)/ (B − C)8,9. De resulterande RFU-värdena i det mikrostrålade området normaliseras till RFU-värdena före mikrostrålning.
- För att definiera region A i det mikrostrålade området, exkludera nukleolära regioner, replikeringsfoci och oregelbundna kärnområden i cellen från mätning. Håll skiftnyckeln mellan att rita två ROI i Fiji för att gruppera två separata regioner som en.
OBS: Proteinrekryteringen varierar mellan olika gener och bestrålningsförhållanden; Storleken på region A måste därför fastställas individuellt. När pixelbredden i region A har fastställts bör den förbli konstant för eventuella jämförande rekryteringar. I experimenten som presenterades här användes rektanglar med 7 pixlars bredd. - Exkludera celler som har flyttats under de inspelade videorna från analysen. För att inkludera mycket mobila celler måste den beskrivna analysen utföras bildruta för bildruta.
- Om du vill visualisera rekryteringsprofilen ritar du de normaliserade RFU-värdena mot tiden med hjälp av en statistisk programvara.
- Beräkna skillnaden vid en angiven tidpunkt mellan DMSO- och olaparibbehandling (n=31) med hjälp av ett Mann-Whitney-test.
Representative Results
Celler adresserar varje typ av DNA-lesion på ett specifikt sätt som också beror på vilken cellcykelfas de befinner sig i. Efter mikro bestrålning bearbetas till exempel dubbelsträngade raster (DSB) antingen genom icke-homolog end joining (NHEJ) eller HR beroende på cellcykelfasen. Nukleaser som verkar mest omfattande under S- och G2-faserna i cellcykeln skapar DNA-överhäng som är avgörande för korrekt HR. För att främja utvärderingen av celler i S-fasen användes PCNA som en enfärgs cellcykelmarkör. Figur 1A visar lokaliseringsprofilen för mPlum-PCNA under cellcykelprogression. PCNA har en helt homogen fördelning i kärnan i G1- och G2-fasen (samtidigt som den till största delen är utesluten från kärnan). I S-fas lokaliserar PCNA till platser för DNA-replikering, som kan visualiseras som ljusa fläckar i kärnan. I tidiga S-fasceller är fläckarna relativt små och jämnt fördelade över cellens kärna. Utvecklas in i mitten av S-fasen blir fläckarna suddiga och lokaliserar mer mot kärnans och kärnans omkrets. I sen S-fas minskar fläckarna i antal men blir allt större i och med att PCNA koncentrerar sig på sena replikeringsplatser (Figur 1B). Viktigt, exogena PCNA uttryck från pBABE vektor ryggraden var mindre än de endogena nivåerna men var tillräckligt för upptäckt av mikroskopi som minimerar potentiella artefakter i cell cykel progression och DDR. Figur 1C visar omfattningen av PCNA överuttryck jämfört med endogena nivåer. Observera att bandet som motsvarar mPlum-PCNA migrerar långsammare på grund av dess större storlek.
Vi syftade till att införa DSB under mikro-bestrålning för att undersöka PARP1/2-beroende rekrytering av EXO1b till dessa skador i S fas. Figur 2A visar att låga doser av energi (1000 μs uppehållstid) inte inducerar rekryteringen av EGFP-FBXL10, en DSB-svarande (komponent i FRUCC-komplexet 8), medan det var tillräckligt för att inducera rekrytering av NTHL1-mCherry, ett basexcisionsreparationsprotein (BER) och rekryterade till platser med oxidativ DNA-skada10,11,12. Vid 3000 μs uppehållstid, både EGFP-FBXL10 och NTHL1-mCherry rekryt, visar en laserproduktion som genererar både oxidativa skador och DSB. Förstärkning av dessa resultat, figur 2B visar immunofluorescensfärgning mot γH2A.X (DSB-markör), vilket är klart tydligare vid användning av högre energidoser. PCNA fungerar både som cellcykelmarkör och markör för framgångsrik mikrostrålning, eftersom den rekryterar tillräckligt med både laserboende tidsinställningar. Viktigt är att både exogena och/eller endogena fluorescerande proteinmärkta PCNA kan användas för denna reporterfunktion eftersom de beter sig på samma sätt (figur 3). Endogent taggade PCNA konstruerades genom att sätta in mRuby i ram med den första exon i en allel av PCNA locus13 (cellinjen var en snäll gåva av Jörg Mansfeld).
Figur 4A och figur 4C visar rekryteringen av acgfp-märkt EXO1b i S-fasceller. EXO1b når maximal ackumuleringsnivå på mikrostrålningsplatser cirka 1 minut och börjar sedan långsamt kopplas ur från DNA-lesionerna efteråt. Berikningar på mikrostrålningsplatser betecknas med en > 1 relativ fluorescensenhet i diagrammet. I närvaro av olaparib är ackumuleringen av EXO1b vid laserremsan vid 1 minut betydligt mindre jämfört med fordonskontrollen. Dessa resultat är i överenskommelsen medlitteraturen 6,7. Figur 4B visar representativa regioner för kvantifiering (områdena A, B och C) enligt beskrivningen i punkt 6 i protokollet. Figur 4D visar de jämförbara uttrycksnivåerna för endogen EXO1b och exogen EXO1b-AcGFP i celler som används för mikrostrålning.
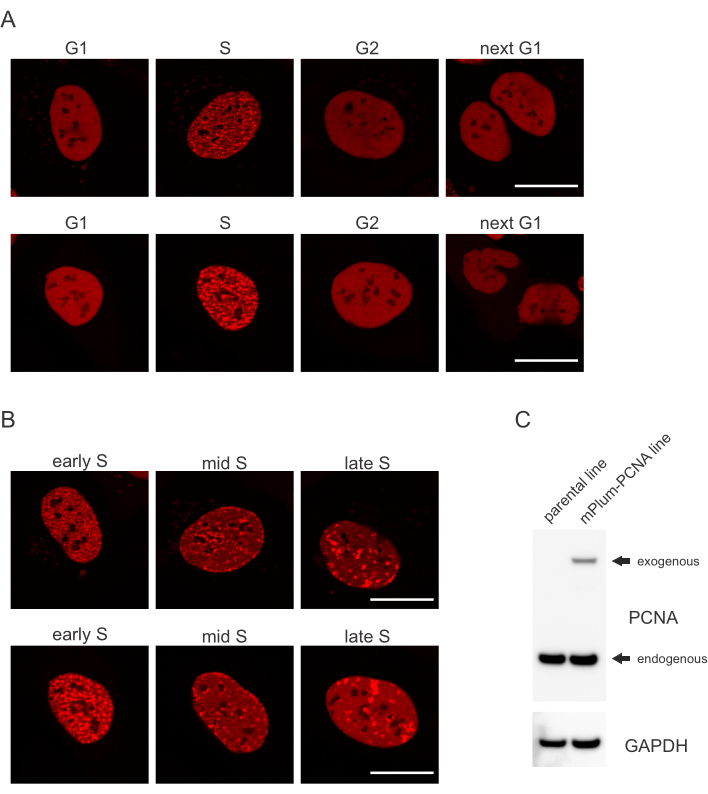
Bild 1: Lokaliseringsmönster för PCNA. (A) Bilder visar lokaliseringsmönster av stabilt integrerade, exogena PCNA under hela cellcykeln i U-2 OS-celler. (B) Bilder visar PCNA foci mönster i olika stadier av S fas (tidigt, mitten och sent) i U-2 OS celler. C)Western blot som visar endogena och exogena nivåer av PCNA i U-2 OS-cellerna som används för avbildning. Skalbar representerar 20 μm. Klicka här för att se en större version av denna siffra.

Bild 2:Induktion av DSB genom optimerad lasereffekt. (A) Laserinställningar kan optimeras för att inducera olika former av DNA-skador. U-2 OS celler stabilt uttrycker både EGFP-FBXL10 och NTHL1-mCherry användes för att identifiera DSB och platser för oxidativa skador, respektive. Mikrostrålning med en laserlinje på 405 nm utfördes på asynkrona U-2 OS-celler med antingen 1000 μs eller 3000 μs uppehållstid. Skala stången representerar 20 μm. (B) Immunofluorescent färgning mot γH2A.X gjordes på mänskliga retinal pigment epitelial celler (hTERT RPE-1) med mRuby-märkta endogena PCNA. Cellerna fixerades och bearbetades 5 minuter efter mikrostrålning med antingen 1000 μs eller 3000 μs uppehållstid. Skalbar representerar 20 μm. Klicka här för att se en större version av denna siffra.

Figur 3:Jämförbar rekrytering av endogen mRuby-PCNA och exogen mPlum-PCNA till mikrostrålningsplatser vid 1000 μs eller 3000 μs laserboendetid. Både endogena och exogena taggade PCNA form replikering högborgar under S fas. Klicka här om du vill visa en större version av den här figuren.

Figur 4:PARP1/2-beroende rekrytering av EXO1b i S-fas. U-2 OS celler stabilt uttrycker EXO1b-AcGFP och mPlum-PCNA var mikro-bestrålade med 405 nm FRAP laser linje med 3000 μs uppehållstid. a)Representativa bilder av mikrostrålade celler vid de angivna tidpunkterna efter förbehandling med antingen fordonskontroll (DMSO) eller olaparib (1 μM). Skalstrecket representerar 20 μm. (B) Representativa bilder av definierade regioner i A-, B- och C-områden för rekryteringsanalysen. Skala bommar för föreställer 20 μm. (C) DNA-skadarekryteringsdynamik fångades av levande cellavbildning. Relativa genomsnittliga fluorescensvärden och bilder förvärvades var 5: e s i 12 min. För varje villkor utvärderades ≥30-celler. Genomsnittliga relativa fluorescensvärden (helsvarta linjer) och standardfel (intervall visualiserat av ett skuggat område) ritades mot tiden. Streckad linje visar rekryteringsvärden på 1 min efter mikrostrålning. Skillnaden mellan DMSO (n=32) och olaparib (n=31) behandling beräknades med hjälp av ett Mann-Whitney test. Asterix betecknar p<0.0001. D)Western blot jämför uttrycksnivåerna för endogen EXO1b och exogen EXO1b-AcGFP i celler som används för mikrostrålning. Klicka här om du vill visa en större version av den här figuren.
Discussion
Kritiska steg och felsökning/modifieringar av potentiella protokoll
Korrekt vävnadskulturkärl för mikrostrålning är avgörande för framgång. De flesta högupplösta bildsystem är optimerade för 0,17 mm täckglastjocklek. Att använda bildkammare med högre eller lägre tjocklek eller sådana tillverkade av plastpolymerer (inte optimerade för 405 nm avbildning) kan avsevärt minska bildkvaliteten. När du använder glasytor, se till att de är vävnadskultur behandlade för att förbättra cellens vidhäftning. Om de inte behandlas med vävnadskultur måste dessa kamrar beläggas, till exempel med poly-D-lysin innan cellerna sås. Vid plätering av celler i det kammade täckglaset är idealisk celltäthet av största vikt för att undvika oegentligheter i cellcykeln och ytterligare stress i cellerna. Korrekt termisk jämvikt av mikroskopkomponenterna före experiment för att upprätthålla en stabil temperatur är avgörande för att både upprätthålla fokus under hela tidsfördröjningen och är också nödvändig för att säkerställa ett homogent DDR över tid och prover.
Det är viktigt att cellerna är i ett hälsosamt tillstånd före mikrostrålning för att minska artefaktuella data. Om celler har oregelbunden morfologi efter infektion/markering, låt cellerna gå igenom flera passager tills morfologin återgår till det normala. Se alltid till att de celler som används är fria från mykoplasmaförorening. Bland de många negativa effekterna av mykoplasmainfektion orsakar det också DNA-skador på värdcellerna och kan påverka derasDDR-vägar 14,15. Det känsligaste sättet att upptäcka mykoplasma i cellkulturen är genom PCR (jämfört med detektering med DAPI eller Hoechst).
Optimal överuttryck av reparationsproteinet av intresse bör vara jämförbart med endogena nivåer, men tillräckligt hög för detektion. Promotorn som används på virusvektorerna, virustitern under infektionen och infektionstidens längd kan alla justeras för idealiska uttrycksnivåer. För konsekventa resultat isolerar du enskilda cellkloner för att säkerställa homogena uttrycksnivåer och normal cellmorfologi. Det rekommenderas att använda vektorkonstruktioner som inte överuttrycker taggade PCNA på högre än endogena nivåer för korrekt cellcykel- och DNA-reparationsmarkörfunktion. Även låga nivåer av PCNA överuttryck är tillräckliga för att diskriminera S-fas celler. Retrovirala pBABE-vektorer har framgångsrikt använts för detta ändamål (Addgene #1764, #1765, #1766, #1767). PCNA kan märkas med alla monomeriska röda(t.ex. mPlum, mCherry, mRuby, etc.) eller monomeriska gröna fluorescerande proteiner (t.ex. mEGFP, AcGFP, mWasabi, mNeonGreen, mEmerald, etc.) som sedan kan kombineras med en växelvis märkt POI. Att överuttrycka en fluorescerande taggad POI har vissa begränsningar och överväganden. Fluorescerande taggar kan störa normal proteinfunktion och lokalisering. Således måste taggens placering (N eller C-terminal) beaktas. Använd alltid monomeriska fluorescerande proteiner, eftersom oligomerisering av icke-monomeriska varianter kan påverka POI: s funktion.
Laserinställningarna måste bestämmas för varje bildsystem eftersom många komponenter i den optiska banan kommer att påverka den faktiska effekt som levereras till cellerna. Laser mikrostrålning kan orsaka flera typer av DNA-skador beroende på excitation våglängd, uteffekten av FRAP laser och om några pre-sensibiliserande medel (som Bromodeoxyuridine eller Hoechst) användes. 405 nm lasrar kan orsaka oxidativ DNA-skada, enkla och dubbla strandadebrott 16,17. Genom att använda högre laserutgångsinställningar ökar mängden DSB. I detta protokoll användes inte pre-sensibiliseringsmetoder, men dessa tekniker är kraftigt täckta i litteraturen och omappade i diskussionen nedan. Enligt vår mening är det bästa sättet att testa om den önskade lesionen genereras genom att testa för rekrytering av kända DNA-skador utbildningsavsnitt specifika gener. Rekrytering av NTHL1 eller OGG1, komponenter i BER-vägen, tyder på induktion av oxiderade DNA-baser10,11,17,18,19, medan FBXL10 eller XRCC5 indikerar närvaron av DSB8,20,21. Rekrytering av XRCC1 kan indikera både förekomsten av oxiderade DNA-baser och enstaka strandade pauser (SSB)22,23. XPC (dvs. RAD4) är en bra indikator på NER som tar bort de skrymmande DNA-kanalerna som genereras av ultraviolett ljus (UV)17,24. Eftersom rekrytering exogena proteiner kan införa vissa oegentligheter, immunfluorescerande färgning av endogena DNA reparation proteiner eller markörer (som γH2A.X för dubbla strandade pauser) kan bekräfta förekomsten av specifika DNA-skador. Alternativt kan antikroppar som höjs mot specifika typer av DNA-lesioner också användas. För att justera den levererade lasereffekten kan både uppehållstiden och lasereffekten ändras.
Med hjälp av matematisk modellering kan en detaljerad kinetisk analys utföras som kan ge värdefulla insikter om POI:s rekryteringsegenskaper (t.ex. bidrag från flera DNA-bindande domäner, känslighet för olika signalhändelser etc.). Automatiserad rekryteringsutvärdering och cellspårning kan kombineras för att skapa robusta arbetsflöden 1,25.
Fördelar och begränsningar av DNA-presensibilisering
Presensibilisering av DNA före mikrostrålning är ett vanligt verktyg för DNA-reparation av proteinrekrytering16,17. Sensibiliserande DNA före mikrostrålning gör det mer mottagligt för DSB. De två vanligaste metoderna för DNA-presensibilisering är förbehandling av celler med antingen Bromodeoxyuridin (BrdU) eller Hoechst färgämne. För system som inte kan mikrostråla vid höga laserkrafter kan dessa metoder vara nödvändiga för att inducera DNA-lesioner som DSB. Dessutom, i avsaknad av en överförd ljusdetektor eller en fluorescerande signal som belyser cellkärnan (till exempel när man studerar rekrytering av ouppklarade endogena DNA-reparationsproteiner), fungerar Hoechst både som ett förkänsligt verktyg och en fluorescerande nukleär fläck. DNA pre-sensibilisering kan dock införa betydande komplikationer. BrdU (som används vid en slutlig koncentration på 10 μM) måste tillsättas celler 24 timmar (eller tid som motsvarar en fullständig cellcykel i den använda cellinjen) för att korrekt införlivas i DNA och kan orsaka cellcykelstörningar26. Hoechst 33342 (används vid en slutlig koncentration av 1 μg/ml) är cytotoxisk efter långa inkubationstider men kräver tillräckligt med tid för att mätta kärnan med färgämnet. Därför bör den endast appliceras 15-20 minuter före mikrostrålning. Annars kommer rekryteringsuppgifterna inte att vara konsekventa. Cellerna som färgas på detta sätt kan inte hållas i kultur i mer än några timmar27,28. Se till att inte använda Hoechst 33358, som inte är lika cellpermeabel som Hoechst 33342 färgämne. Pre-sensibilisering kan också införa onödig varians bland experiment och gör experimentet ännu känsligare för skillnader i celltäthet (eftersom detta kommer att påverka mängden inkorporerat färgämne / cell).
Fördelar och begränsningar av konfokal mikroskopi
Bildhastigheten för konfokal mikroskopi kan vara begränsande jämfört med widefieldmikroskopi. Ett konfokalt mikroskop utrustat med en resonansskanner kan dock avsevärt förbättra bildhastigheten (på bekostnad av upplösningen) som närmar sig hastigheter för spinndiskmikroskopi. Tre funktioner gör A1R HD25 confocal-systemet till ett utmärkt val för protokollet som presenteras här. För det första gör systemets 25 mm FOV det möjligt att avbilda mellan 15-20 celler i ett enda skannat fält (jämfört med 5-10 celler i regelbundna inställningar), vilket begränsar antalet förvärv som krävs för att få tillräckligt med celler för statistisk analys. För det andra gör FRAP-modulen och två scanheads det möjligt att avbilda och mikrostråla cellerna samtidigt, inte bara sekventiellt. Slutligen ger flexibiliteten i att ha både resonans- och galvanoskannrarna möjligheten att enkelt växla mellan högupplöst bildbehandling med exceptionell hastighet som minimerar släckning av fluorforer och högupplöst bildbehandling med hög rumslig upplösning som använder långsammare skanningshastigheter för att producera bilder med ett högre signal-till-brusförhållande. Medan det använda systemet tillät ovannämnda flexibilitet, för att likna mer allmänt tillgängliga konfokala mikroskopkonfigurationer, användes endast galvanoskannern i de presenterade experimenten (för både mikrostrålning och efterföljande avbildning).
Fördelar och begränsningar av mikro bestrålning
Även om mikrostrålning ger oöverträffad rumslig och temporal upplösning, är det inte utan begränsningar. DNA-skador genom lasermikrostrålning är mycket grupperade med specifika delar av kärnan jämfört med naturligt förekommande skadliga medel. Kromatinsvar på grund av mikrostrålning kan således skilja sig åt jämfört med homogent fördelade skador. Dessutom är mikrostrålning tidskrävande och kan endast utföras på några dussin celler, medan stora populationsbaserade biokemiska metoder (kromatinfraktion, immunprecipitation, ChIP) kan ge ökad robusthet genom att studera tusentals celler åt gången. Att verifiera observationer gjorda genom mikrostrålning med traditionella biokemiska tekniker är en effektiv strategi för tillförlitliga slutsatser. Även om samtidig mikrostrålning av många celler i en viss FOV är möjlig, behöver bildsystemet mer tid för att utföra uppgiften. Att mäta dynamiken hos proteiner som rekryterar mycket snabbt till DNA-lesioner begränsar därför antalet möjliga ROI för mikrostrålning som används samtidigt. På det bildsystem som används för detta protokoll tar mikrostrålningen av en enda 1024 pixel lång ROI 1032 ms med 1000 μs uppehållstid och 3088 ms med 3000 μs uppehållstid att slutföra. Att använda flera rader av ROI ökar avsevärt den tid som behövs för att avsluta mikrostrålning (t.ex. 7 x 1024 pixel lång ROI tar 14402 ms med 1000 μs uppehållstid och 21598 ms med 3000 μs uppehållstid). Denna tid går förlorad från bildförvärv och måste beaktas. Vid snabb rekrytering av bilder, använd kortast möjliga ROI och endast mikrostråla en cell i taget.
Fördelar och begränsningar jämfört med synkroniseringsmetoder
För cellcykelspecifika studier innebär de befintliga metoderna antingen synkronisering av celler i specifika cellcykelfaser eller användning av fluorescerande reportrar för att identifiera cellens specifika cellcykelfas. Var och en av dessa metoder ger dock sina egna utmaningar och begränsningar.
FUCCI-systemet3 (som förlitar sig på fluorescerande proteinmärkta trunkerade former av CDT1 och Geminin) är ett särskilt användbart verktyg för cellcykelstudier men har begränsningar när det gäller att skilja mellan S- och G2-faser i cellcykeln. Gemininnivåerna är redan höga från mitten av S-fasen och förblir höga fram till M-fasen, vilket gör dessa faser svåra att separera. Att använda FUCCI-systemet innebär också att två optiska kanaler i mikroskopet inte kan användas för avbildning av POI.
Icke-cancer cellinjer kan synkroniseras till G0 genom avlägsnande av tillväxtfaktorer som finns i serumet (serum svält) orsakar liten eller ingen DNA-skada på cellerna. De flesta cancercelllinjer kommer dock delvis att fortsätta att utvecklas genom cellcykeln även utan tillräckliga mängder serum i sina medier. Dessutom börjar celler delvis förlora synkroniseringen i slutet av G1, tidig S-fas. Förutom serum svält finns det många kemiska metoder för att uppnå cellcykelsynkronisering. Hydroxyurea, aphidicolin och tymidinblock är metoder för att stoppa DNA-replikation för att synkronisera celler i tidig S-fas. Även om dessa metoder är billiga och enkla, introducerar de replikeringsstress som resulterar i DNA-skador. Dessa DNA-replikationshämmare har visat sig inducera fosforylering av H2A. X, en välkänd markör för DSB2,29. Metoden att använda taggad-PCNA som markör för S-fasceller minskar potentialen för artefakter orsakade av kemisk synkronisering och kan appliceras på ett brett spektrum av cellinjer jämfört med serum svält.
Slutsats
DNA-skador är en drivkraft för genetiska sjukdomar där mutagena lesioner kan leda till malign omvandling av celler. Att rikta in sig på DNA-syntesmaskineriet är en grundläggande terapeutisk strategi vid behandling av hyperproliferativa sjukdomar som cancer. För att behandla dessa sjukdomar på ett mer målinriktat sätt behöver vi en bättre förståelse för de proteiner som reparerar DNA-lesioner. Protokollet som beskrivs här hjälper mikrostrålningsbaserade studier i S-fas genom att minimera de utmaningar som traditionella synkroniseringsmetoder innebär för att minska möjliga artefakter och öka experimentens reproducerbarhet.
Disclosures
Författarna uppger att publiceringen av det presenterade verket sponsrades av Nikon Corporation. Författarna förklarar att det inte finns några konkurrerande intressen.
Acknowledgments
Författarna tackar M. Pagano för hans kontinuerliga stöd samt D. Simoneschi, A. Marzio och G. Tang för deras kritiska granskning av manuskriptet. B. Miwatani-Minter tackar R. Miwatani och B. Minter för deras fortsatta stöd. G. Rona tackar K. Ronane Jurasz och G. Rona för deras fortsatta stöd.
Materials
| Name | Company | Catalog Number | Comments |
| Ammonium chloride | Sigma-Aldrich | A9434-500G | For quenching formaldehyde |
| Anti-EXO1 Rabbit Polyclonal Antibody | Proteintech | 16253-1-AP | primary antibody |
| Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301 | Millipore | 05-636 | primary antibody |
| Bovine Serum Albumin | Sigma-Aldrich | 3117332001 | BSA for blocking |
| BrdU (5-Bromo-2'-deoxyuridine) | Merck | 19-160 | pre-sensitizing agent |
| Citifluor™ Mountant Solution AFR3 | Electron Microscopy Sciences | 17973-10 | antifade containing PBS solution for imaging |
| DAPI | Sigma-Aldrich | D9542-1MG | nucleic acid stain |
| DMEM Medium | Thermo Fisher Scientific | 10569010 | Cell culture medium for HEK293T cells |
| DMSO | Sigma-Aldrich | D2650-100ML | Vehichle control and dissolution solvent |
| EGFP-FBXL10 | Addgene | #126542 | viral expression vector for EGFP-FBXL10 |
| EXO1b-AcGFP (in pRetroQ) | custom cloning | na | EXO1b cDNA was cloned in the NheI, BamHI sites of pRetroQ-AcGFP1-N1 vector. |
| Fetal Bovine Serum | Gibco | 16140071 | Media supplement |
| FluoroBrite DMEM | Thermo Fisher Scientific | A1896701 | Phenol red free medium for microscopy |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | A32723 | secondary antibody |
| HEK293T cells | ATCC | ATCC CRL-3216 | Cell line for viral packaging |
| HEPES | Sigma-Aldrich | H0887-100ML | Buffering agent to supplement live cell imaging medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | pre-sensitizing agent |
| Lipofectamine 3000 | Thermo Fisher Scientific | L3000015 | Transfection reagent |
| McCoy’s 5A (Modified) Medium | Life Technologies | 16600-108 | Cell culture medium for U-2 OS cells |
| mCherry-PCNA | Addgene | #55117 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA | Addgene | #55994 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA (in pBABE) | custom cloning | na | mPlum-PCNA cDNA was cloned from Addgene #55994 in the BamHI, SalI sites of pBABE (puro) |
| Nikon A1R-HD25 Confocal Scanhead and Controller | Nikon | na | confocal imaging system |
| Nikon LUN4 laser unit | Nikon | na | excitation system |
| Nikon LUN-F 50 mW 405 nm FRAP laser unit | Nikon | na | FRAP laser unit |
| Nikon NIS Elements Confocal Controller Software | Nikon | na | Confocal controlling software |
| Nikon Ti2-E Inverted Microscope | Nikon | na | inverted epifluorescent microscope base |
| Nikon Ti2-LAPP Modular Illumination System | Nikon | na | illumination system |
| NTHL1-mCherry (in pRetroQ) | custom cloning | na | NTHL1 cDNA was cloned in the NheI, SalI sites of pRetroQ-mCherry-N1 vector. |
| Nunc Lab-Tek II Chambered Coverglass (4 well) | Thermo Fisher Scientific | 155382PK | Live cell microscopy cell culture chamber |
| Olaparib | Selleck Chemicals | S1060 | PARP inhibitor |
| Opti-MEM reduced serum media | Thermo Fisher Scientific | 31985062 | Dilution medium for transient transfection |
| Paraformaldehyde aqueous solution (32%) | Thermo Fisher Scientific | 50-980-494 | Fixative |
| pBABE (hygro) | Addgene | #1765 | retroviral expression vector (for low expression levels) |
| pBABE (neo) | Addgene | #1767 | retroviral expression vector (for low expression levels) |
| pBABE (puro) | Addgene | #1764 | retroviral expression vector (for low expression levels) |
| pBABE (zeo) | Addgene | #1766 | retroviral expression vector (for low expression levels) |
| PCNA Antibody (PC10) | Santa Cruz | sc-56 | primary antibody |
| Penicillin-Streptomycin-Glutamine (100x) | Gibco | 10378016 | Media supplement |
| polybrene | Sigma-Aldrich | TR-1003 | Increase viral infection efficiency |
| pRetroQ-AcGFP-C1 | Takara | 632506 | retroviral expression vector |
| pRetroQ-AcGFP-N1 | Takara | 632505 | retroviral expression vector |
| pRetroQ-mCherry-C1 | Takara | 632567 | retroviral expression vector |
| pRetroQ-mCherry-N1 | Takara | 632568 | retroviral expression vector |
| pUMVC | Addgene | #8449 | Viral packaging vector |
| Sodium-pyruvate | Thermo Fisher Scientific | 11360070 | Supplement for live cell imaging medium |
| Triton X-100 aqueous solution (10%) | Sigma-Aldrich | 11332481001 | Dilute in PBS for cell permeabilization buffer |
| Trypsin-EDTA Solution 10X | Sigma-Aldrich | 59418C-100ML | Dilute in PBS to split cells |
| U-2 OS Cells | ATCC | HTB-96 | Optimal cell line for microscopy experiments |
| Universal Mycoplasma Detection Kit | ATCC | 30-1012K | PCR based Mycoplasma detection kit |
| VSV-G | Addgene | #8454 | Viral protein envelope vector |
References
- Aleksandrov, R., et al.
- Darzynkiewicz, Z., Halicka, H. D., Zhao, H., Podhorecka, M. Cell synchronization by inhibitors of DNA replication induces replication stress and DNA damage response: Analysis by flow cytometry. Methods in Molecular Biology. 761, 85-96 (2011).
- Sakaue-Sawano, A., et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 132 (3), 487-498 (2008).
- Herce, H. D., Rajan, M., Lattig-Tunnemann, G., Fillies, M., Cardoso, M. C. A novel cell permeable DNA replication and repair marker. Nucleus. 5 (6), 590-600 (2014).
- Keijzers, G., et al. Human exonuclease 1 (EXO1) regulatory functions in dna replication with putative roles in cancer. International Journal of Molecular Sciences. 20 (1), (2018).
- Cheruiyot, A., et al. Poly(ADP-ribose)-binding promotes Exo1 damage recruitment and suppresses its nuclease activities. DNA Repair (Amsterdam). 35, 106-115 (2015).
- Zhang, F., Shi, J., Chen, S. H., Bian, C., Yu, X. The PIN domain of EXO1 recognizes poly(ADP-ribose) in DNA damage response. Nucleic Acids Research. 43 (22), 10782-10794 (2015).
- Rona, G., et al. PARP1-dependent recruitment of the FBXL10-RNF68-RNF2 ubiquitin ligase to sites of DNA damage controls H2A.Z loading. elife. 7, (2018).
- Young, L. M., et al. TIMELESS forms a complex with PARP1 distinct from its complex with TIPIN and plays a role in the dna damage response. Cell Reports. 13 (3), 451-459 (2015).
- Kong, X., et al. Laser microirradiation to study in vivo cellular responses to simple and complex dna damage. Journal of Visualized Experiments. (131), e56213 (2018).
- Kong, X., et al. Condensin I recruitment to base damage-enriched DNA lesions is modulated by PARP1. PLoS One. 6 (8), 23548 (2011).
- Lan, L., et al. Novel method for site-specific induction of oxidative DNA damage reveals differences in recruitment of repair proteins to heterochromatin and euchromatin. Nucleic Acids Research. 42 (4), 2330-2345 (2014).
- Zerjatke, T., et al. Quantitative cell cycle analysis based on an endogenous all-in-one reporter for cell tracking and classification. Cell Reports. 19 (9), 1953-1966 (2017).
- Ji, Y., Karbaschi, M., Cooke, M. S. Mycoplasma infection of cultured cells induces oxidative stress and attenuates cellular base excision repair activity. Mutation Research. 845, 403054 (2019).
- Sun, G., et al. Mycoplasma pneumoniae infection induces reactive oxygen species and DNA damage in A549 human lung carcinoma cells. Infection and Immunity. 76 (10), 4405-4413 (2008).
- Gassman, N. R., Wilson, S. H. Micro-irradiation tools to visualize base excision repair and single-strand break repair. DNA Repair (Amsterdam). 31, 52-63 (2015).
- Muster, B., Rapp, A., Cardoso, M. C. Systematic analysis of DNA damage induction and DNA repair pathway activation by continuous wave visible light laser micro-irradiation. AIMS Genetics. 4 (1), 47-68 (2017).
- Ikeda, S., et al. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. Journal of Biological Chemistry. 273 (34), 21585-21593 (1998).
- Rosenquist, T. A., Zharkov, D. O., Grollman, A. P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proceedings of the National Academy of Science U. S. A. 94 (14), 7429-7434 (1997).
- Reid, D. A., et al. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proceedings of the National Academy of Science U. S. A. 112 (20), 2575-2584 (2015).
- Taccioli, G. E., et al. Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science. 265 (5177), 1442-1445 (1994).
- Marsin, S., et al. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. Journal of Biological Chemistry. 278 (45), 44068-44074 (2003).
- Thompson, L. H., Brookman, K. W., Jones, N. J., Allen, S. A., Carrano, A. V. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Molecular and Cell Biology. 10 (12), 6160-6171 (1990).
- Scharer, O. D.
- Oeck, S., et al. High-throughput evaluation of protein migration and localization after laser micro-irradiation. Science Reports. 9 (1), 3148 (2019).
- Mistrik, M., et al. Cells and stripes: A novel quantitative photo-manipulation technique. Science Reports. 6, 19567 (2016).
- Durand, R. E., Olive, P. L. Cytotoxicity, mutagenicity and dna damage by hoechst 33342. Journal of Histochemistry and Cytochemistry. 30 (2), 111-116 (1982).
- Tobey, R. A., Oishi, N., Crissman, H. A. Cell cycle synchronization: reversible induction of G2 synchrony in cultured rodent and human diploid fibroblasts. Proceedings of the National Academy of Science U. S. A. 87 (13), 5104-5108 (1990).
- Podhorecka, M., Skladanowski, A., Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. Journal of Nucleic Acids. 2010, (2010).
