

Summary
Dieses Protokoll beschreibt eine nicht-invasive Methode zur effizienten Identifizierung von S-Phasen-Zellen für nachgelagerte Mikroskopiestudien, wie z. B. die Messung der Rekrutierung von DNA-Reparaturproteinen durch Lasermikrobestrahlung.
Abstract
Die Reparatur von DNA-Schäden erhält die genetische Integrität der Zellen in einer hochreaktiven Umgebung. Zellen können verschiedene Arten von DNA-Schäden aufgrund endogener und exogener Quellen wie Stoffwechselaktivitäten oder UV-Strahlung ansammeln. Ohne DNA-Reparatur wird der genetische Code der Zelle beeinträchtigt, was die Strukturen und Funktionen von Proteinen untergräbt und möglicherweise Krankheiten verursacht.
Das Verständnis der raumzeitlichen Dynamik der verschiedenen DNA-Reparaturwege in verschiedenen Zellzyklusphasen ist im Bereich der DNA-Schadensreparatur von entscheidender Bedeutung. Aktuelle Fluoreszenzmikroskopie-Techniken bieten großartige Werkzeuge, um die Rekrutierungskinetik verschiedener Reparaturproteine nach DNA-Schadensinduktion zu messen. Die DNA-Synthese während der S-Phase des Zellzyklus ist ein besonderer Punkt im Zellschicksal in Bezug auf die DNA-Reparatur. Es bietet ein einzigartiges Fenster, um das gesamte Genom auf Fehler zu überprüfen. Gleichzeitig stellen DNA-Synthesefehler auch eine Bedrohung für die DNA-Integrität dar, die in nicht teilenden Zellen nicht anzutreffen ist. Daher unterscheiden sich DNA-Reparaturprozesse in der S-Phase signifikant von anderen Phasen des Zellzyklus, und diese Unterschiede sind schlecht verstanden.
Das folgende Protokoll beschreibt die Herstellung von Zelllinien und die Messung der Dynamik von DNA-Reparaturproteinen in der S-Phase an lokal induzierten DNA-Schadensstellen mit einem laserscannenden konfokalen Mikroskop, das mit einer 405-nm-Laserlinie ausgestattet ist. Tagged PCNA (mit mPlum) wird als Zellzyklusmarker in Kombination mit einem AcGFP-markierten Reparaturprotein von Interesse (dh EXO1b) verwendet, um die DNA-Schadensrekrutierung in der S-Phase zu messen.
Introduction
Mehrere DNA-Reparaturwege haben sich entwickelt, um die verschiedenen Arten von DNA-Läsionen anzugehen, die in Zellen auftreten können, die alle sowohl läsin als auch zeitlich stark reguliert sind. Eine der anfälligsten Perioden des Zellzyklus ist die S-Phase, in der die DNA-Synthese stattfindet. Während die Proliferation für das Leben von grundlegender Bedeutung ist, stellt sie auch eine große Herausforderung dar. Zellen müssen eine originalgetreue Replikation ihres Genoms sicherstellen, um zu vermeiden, dass Mutationen an zukünftige Generationen weitergegeben werden. Folglich bietet die Proliferation einen therapeutischen Interventionspunkt, der für die Entwicklung therapeutischer Ansätze im Bereich der Onkologie eingesetzt wurde.
Alle wichtigen Techniken, die zur Untersuchung der Proteinrekrutierung bei DNA-Läsionen verwendet werden, haben ihre Stärken und Grenzen. Die Mikrobestrahlung hat eine bessere räumliche und zeitliche Auflösung1 als die meisten alternativen Methoden wie immunfluoreszierende Bildgebung von ionisierenden strahlungsinduzierten Herden (IRIF), Chromatin-Immunpräzipitation (ChIP) oder biochemische Fraktionierung. Die Mikrobestrahlung schnürt jedoch die Robustheit der oben genannten Techniken, die eine große Anzahl von Zellen gleichzeitig abtasten können.
Um die DNA-Reparatur in der S-Phase zu untersuchen, muss man in der Lage sein, S-Phasen-Zellen in einer asynchronen Zellkulturpopulation zu unterscheiden. Es gibt viele bekannte Methoden, um dies anzugehen, entweder die Synchronisation von Zellen oder die Visualisierung der verschiedenen Zellzyklusphasen. Beide Ansätze bringen jedoch erhebliche Herausforderungen und mögliche Artefakte mit sich. Chemische Synchronisationsmethoden, die häufig zur Anreicherung von Zellen in der frühen S-Phase verwendet werden (z. B. Doppeltymidinblock, Aphidicolin und Hydroxyharnstoffbehandlung), erreichen eine Synchronisation durch die Induktion von Replikationsstress und schließlich DNA-Schäden selbst. Dies schränkt den Einsatz dieser Methoden zur Untersuchung von DNA-Reparaturprozessen in S-Phase2 ein. Die Synchronisation durch Serummangel und -freisetzung ist nur auf eine begrenzte Anzahl von Zelllinien anwendbar, wobei Krebszelllinien weitgehend ausgeschlossen sind, die im Vergleich zu nicht transformierten Zelllinien weniger auf Wachstumsfaktoren für die Zellzyklusprogression angewiesen sind. Das Fluorescence Ubiquitin Cell Cycle Indicator (FUCCI) System ist ein besonders nützliches Werkzeug zur Untersuchung des Zellzyklus, hat aber eine grundlegende Einschränkung bei der Unterscheidung zwischen S- und G2-Zellzyklusphasen3.
Hier wird gezeigt, dass die Verwendung von fluoreszierend markiertem PCNA als nicht-invasiver Marker für die S-Phase die Nachteile chemischer Zellzyklus-Synchronisationsmethoden begrenzt und gleichzeitig mehr Spezifität und Flexibilität als das FUCCI-System ermöglicht. Als einzelner Marker kann PCNA nicht nur S-Phasen-Zellen in einer asynchronen Population hervorheben, sondern auch die genaue Progression von Zellen innerhalb der S-Phase (d. H. Frühe, Mittlere oder späte S-Phase)4anzeigen. Niedrige Expressionsniveaus von exogener, markierter PCNA sorgen für minimale Interferenzen sowohl bei der Zellzyklusprogression als auch bei DNA-Reparaturprozessen. Wichtig ist, dass PCNA auch als interne Kontrolle für die ordnungsgemäße DNA-Schadensinduktion dient, da es an der Reparatur mehrerer DNA-Läsionen beteiligt ist und an lokal induzierten DNA-Schadensstellen rekrutiert wird1,4.
Die hier vorgestellten Experimente zeigen, wie die Rekrutierungsdynamik von EXO1b in der S-Phase gemessen werden kann und wie diese durch den etablierten PARP-Inhibitor Olaparib beeinflusst wird. Die EXO1b-Nukleaseaktivität ist für eine Vielzahl von DNA-Reparaturwegen relevant, einschließlich Mismatch Repair (MMR), Nukleotid-Exzisionsreparatur (NER) und Doppelsträngenbruchreparatur (DSB). In der S-Phase spielt EXO1b eine wichtige Rolle bei der homologen Rekombination (HR) durch die Bildung von 3' ssDNA-Überhängen während der DNA-Resektion5. EXO1b wurde weiter in die DNA-Replikation involviert, mit Rollen bei der Checkpoint-Aktivierung, um blockierte DNA-Gabeln neu zu starten, sowie die Primerentfernung und okazaki-Fragmentreifung am verzögerten Strang während der Strangverschiebung in der Replikation5. Die EXO1b-Rekrutierung an beschädigten DNA-Stellen wird durch die direkte Interaktion mit Poly (ADP-Ribose) (PAR)6,7reguliert. Aufgrund der zahlreichen zellzyklusspezifischen Implikationen von EXO1b ist es eine ausgezeichnete Wahl für S-Phasen-spezifische Rekrutierungsstudien mit PCNA.
Protocol
1. Kultivierung von humanen Osteosarkom-abgeleiteten Zellen (U-2 OS)
HINWEIS: U-2 OS-Zellen sind ideal für diese Studien, da sie eine flache Morphologie, einen großen Kern und eine starke Bindung an mehrere Oberflächen, einschließlich Glas, aufweisen. Andere Zelllinien mit ähnlichen Eigenschaften könnten ebenfalls verwendet werden.
- Für die Kultivierung von U-2 OS-Zelllinien verwenden Sie McCoys 5A-Medium, das mit 10% fetalem Rinderserum (FBS) und Antibiotika (100 U / ml Penicillin und 100 μg / ml Streptomycin) ergänzt wird. Inkubieren Sie Zellen bei 37 °C in einer befeuchteten Atmosphäre mit 5% CO2. Für mikroskopische Untersuchungen die Zellkultur in einer 10-cm-Schüssel aufrechterhalten, um eine ausreichende Zellzahl zu gewährleisten.
- Wenn sich die Zellen einer 90% igen Konfluenz nähern (~ 7 x10 6 Zellen / 10 cm Schüssel), teilen Sie die Zellen.
- Spülen Sie die Zellen mit PBS aus, um die im Serum enthaltenen Trypsinhemmer wegzuwaschen.
- Fügen Sie 1 ml Trypsin-EDTA hinzu und stellen Sie sicher, dass die Zellschicht gleichmäßig bedeckt ist.
- Bei 37 °C inkubieren, bis die Zellschicht von der Platte abgehoben ist (ca. 6 min).
- Resuspendieren Sie die trypsinisierten Zellen in Serum, das Medien enthält, um das Trypsin zu inaktivieren, und fügen Sie 1/10des Volumens (~ 0,7 x10 6 Zellen) in eine neue 10 cm Platte mit 10 ml ergänztem Wachstumsmedium hinzu.
- Testen Sie die Zellen vor dem Experimentieren routinemäßig auf Mykoplasmenkontamination mit dem Universal Mycoplasma Detection Kit gemäß der Empfehlung des Herstellers.
2. Retrovirale Infektion
HINWEIS: Für BSL-2-Sicherheitsmaßnahmen und bei der Arbeit mit rekombinanten Viren lesen Sie bitte: NIH-Richtlinien, Abschnitt III-D-3: Rekombinante Viren in Gewebekulturen.
- Samen Sie 4 x 106 HEK293T-Zellen, um ~ 60% Konfluenz innerhalb von 24 h nach dem Plattieren in eine 10 cm Kulturschale zu erreichen.
- Für die Kultivierung von HEK293T befolgen Sie bitte die in 1.1-1.3 dieses Protokolls beschriebenen Kultivierungsschritte von U-2 OS. Für HEK293T ersetzen McCoy's 5A Medium für DMEM. Achten Sie darauf, HEK293T-Zellen immer sanft zu waschen, da sie schwach an Gewebekulturplatten anhaften.
- Transfizieren Sie HEK293T-Zellen mit einem lipidbasierten Transfektionsreagenz für die virale Verpackung von Plasmiden.
- Für retrovirale Vektoren kombinieren Sie 1,5 μg VSV-G (Addgene #8454) und 1,5 μg pUMVC (Addgene #8449) Verpackungsvektoren zusammen mit 3 μg des Vektors, der das interessierende Gen enthält (in einem retroviralen Vektorrückgrat mit Puromycinresistenz), zu 250 μL Opti-MEM-reduziertem Serummedium in einem Mikrozentrifugenröhrchen. Für jedes μg DNA, das in das Opti-MEM/DNA-Gemisch (in diesem Fall 6 μL) zugeführt wird, wird 1 μL P3000-Reagenz zugegeben und durch Klopfen vorsichtig vermischt. Wirbeln oder Pipette nicht auf und ab piepsen.
- Kombinieren Sie in einem anderen Mikrozentrifugenröhrchen 2 μL pro μg DNA (in diesem Fall 12 μL) Transfektionsreagenz mit 250 μL Opti-MEM-reduzierten Serummedien.
- Die beiden Mischungen (500 μL kombiniert, nicht wirbeln, nur durch sanftes Klopfen mischen) vermischen und 15 min bei Raumtemperatur inkubieren lassen.
- Geben Sie die Mischung vorsichtig tropfenweise zu den ausgesäten HEK293T-Zellen, ohne die Zellen zu lösen. Schwenken Sie die Platten vorsichtig.
- Virusinfektion, um stabile Zelllinien zu erzeugen.
- Entfernen Sie das Virus, das überstandt, 72 h nach der Transfektion aus den HEK293T-Zellen. Filtern Sie die Lösung vorsichtig mit einem 0,45 μm-Filter, um Zelltrümmer und abgelöste Zellen zu entfernen. Fügen Sie dem viralen Überstand optional 8 μg/ml Polybren hinzu, um eine Virusinfektion zu erleichtern.
- Fügen Sie Virus, das Überstände enthält, zu U-2 OS-Zellen bei ~ 50% Konfluenz in einer 10 cm großen Schüssel (~ 3 x 106 Zellen) hinzu. Säen Sie die U-2 OS-Zellen am Vortag.
- Infizieren Sie 6-16 h, bevor Sie den virushaltigen Überstand entfernen und verwerfen.
HINWEIS: Um die gewünschte Überexpression für das interessierende Gen zu erreichen, inkubieren Sie eine Reihe von viralen Verdünnungen für eine bestimmte Zeitspange. Überprüfen Sie die Expressionsniveaus des Transgens in jeder neu etablierten Zelllinie mit Western Blot, um es mit endogenen Spiegeln zu vergleichen. - Erlauben Sie den Zellen, in Gegenwart geeigneter Antibiotika auszuwählen (für 3-4 Tage bei Puromycin bei 2 μg / ml Endkonzentration) und überprüfen Sie die Expression des fluoreszierenden Protein-markierten Gens von Interesse unter einem Mikroskop.
- Wiederholen Sie diese Schritte, um doppelt beschriftete Zelllinien zu generieren. In den hier vorgestellten Experimenten wurde mPlum-PCNA aus einem retroviralen Vektor (pBABE) in Kombination mit EXO1B-AcGFP exprimiert, ebenfalls exprimiert aus einem retroviralen Vektor (pRetroQ-AcGFP1-N1).
3. Vorbereitung von Zellen für die Mikrobestrahlung
- Beschichtungszellen: 24 h vor dem Experiment insgesamt 8,0 x 104 Zellen in ein Volumen zwischen 500 μL-1 ml Medien (für ca. 70% Konfluenz) auf einem vierfach kammerigen Deckglas mit einem Borosilikatglasboden Nr. 1,5, das ideale Ergebnisse für die konfokale Mikroskopie mit hoher Vergrößerung und Lasermikrobestrahlung liefert. Eine höhere Zellkonfluenz ermöglicht es, mehr Zellen in einem einzigen Sichtfeld (FOV) zu messen; vollständig konfluente Objektträger führen jedoch zu Unregelmäßigkeiten im Zellzyklus.
- Bildgebende Medien: Tauschen Sie eine Stunde vor der Mikrobestrahlung reguläres Wachstumsmedium gegen Fluorbrit DMEM aus, ergänzt mit 10% FBS, 100 U/ml Penicillin und 100 μg/ml Streptomycin, 15 mM HEPES (pH=7,4) und 1 mM Natrium-Pyruvat. Diese Bildmedien tragen dazu bei, das Signal-Rausch-Verhältnis zu maximieren, was die Erkennung von sehr schwacher Fluoreszenz ermöglicht. Da es HEPES enthält, stabilisiert es auch den pH-Wert in Abwesenheit einer 5% CO2-Atmosphäre.
- Wenden Sie in diesem Schritt eine zusätzliche Behandlung vor der Bildgebung an. In den hier vorgestellten Experimenten wurden die Zellen eine Stunde vor der Bildgebung entweder mit Olaparib (PARP-Inhibitor, bei 1 μM Endkonzentration) oder einer Fahrzeugkontrolle (DMSO) 1 ,8,9vorbehandelt.
4. Vorbereitung des Mikroskops und Auswahl von S-Phasenzellen für die Bildgebung.
- Verwenden Sie ein konfokales System, das die ähnlichen Eigenschaften wie das hier beschriebene System aufweist, um beste Ergebnisse zu erzielen. Die hier vorgestellten Experimente wurden mit einem konfokalen Mikroskop durchgeführt, das auf einem invertierten Mikroskopständer montiert war (siehe Materialtabelle).
HINWEIS: Das hier verwendete Mikroskop war mit einem 50 mW 405 nm FRAP-Lasermodul und einem 60x 1,4 NA Ölplan-Apochromat-Objektiv ausgestattet. Der konfokale Scankopf hatte zwei Scanneroptionen: einen Galvano-Scanner (für hohe Auflösung) und einen Resonanzscanner (für Hochgeschwindigkeitsbilder).- Einführung der Fluoreszenzrückgewinnung nach dem Photobleaching -Laser (FRAP) in die Probe über ein softwaregesteuertes XY-Galvano-Gerät. Verwenden Sie eine 488-nm-Laserlinie, um AcGFP anzuregen, und eine 561-nm- oder 594-nm-Laserlinie, um mPlum anzuregen.
HINWEIS: Die folgende Filterkombination liefert optimale Ergebnisse: Mit einem 560 nm Langpassfilter wurde Emissionslicht mit einer Wellenlänge von weniger als 560 nm durch einen 525/50 nm Emissionsfilter für AcGFP geleitet, während Emissionslicht mit einer Wellenlänge von mehr als 560 nm durch einen 595/50 nm Emissionsfilter für mPlum geleitet wurde. Es kann ein geeigneter Filterset (z. B. FITC/TRITC, GFP/mCherry, FITC/TxRed) verwendet werden, der eine minimale Fluoreszenzdurchblutung gewährleistet.
- Einführung der Fluoreszenzrückgewinnung nach dem Photobleaching -Laser (FRAP) in die Probe über ein softwaregesteuertes XY-Galvano-Gerät. Verwenden Sie eine 488-nm-Laserlinie, um AcGFP anzuregen, und eine 561-nm- oder 594-nm-Laserlinie, um mPlum anzuregen.
- Schalten Sie die Umgebungskammer und die Mikroskopkomponenten ein.
- Schalten Sie die Heizung (Stufe, Objektiv und Umweltkammer, wenn möglich), die CO2-Versorgung und den Feuchtigkeitsregler mindestens 4 h vor Beginn des Experiments ein, um ein thermisches Gleichgewicht für eine stabile Bildaufnahme zu gewährleisten.
- Initialisieren Sie Lichtquellen zusammen mit den Laserlinien mindestens 1 h vor dem Transfer der Zellen zum Mikroskop.
- Wählen Sie S-Phasen-Zellen in einer asynchronen Population mit fluoreszierend markiertem PCNA als Marker aus. Führen Sie dazu die folgenden Schritte aus.
- Suchen Sie nach dem einzigartigen Lokalisierungsmuster der mPlum-markierten PCNA in der S-Phase, das die Identifizierung dieser Zellzyklusphase ermöglicht. PCNA hat eine völlig homogene Verteilung im Kern in den G1- und G2-Phasen des Zellzyklus, während es von den Nukleoli ausgeschlossen ist. In der S-Phase bildet PCNA Herde an der Stelle von Replisomen im Zellkern. Abbildung 1 zeigt die unterschiedlichen Muster der PCNA-Herde während der S-Phase, wodurch sogar die frühe, mittlere und späte S-Phase unterschieden werden kann.
- Schauen Sie durch das Okular, um ein FOV auszuwählen, das genügend S-Phasen-Zellen für die Mikrobestrahlung hat. Asynchrone U-2 OS-Zellen haben normalerweise 30-40% ihrer Population in der S-Phase.
- Versuchen Sie, Extreme in den Expressionsniveaus (helle und dunkle Zellen gleichermaßen) sowohl für PCNA als auch für das Protein of Interest (POI), in diesem Fall EXO1b-AcGFP, zu vermeiden, was zu experimentellen Artefakten führen könnte.
- Wenn Sie ein geeignetes Sichtfeld finden, vermeiden Sie es, das Feld für eine lange Zeit zu scannen, um Photobleaching und unerwünschte DNA-Schäden zu minimieren.
- Legen Sie den gewünschten Interessenbereich (ROI) für die Mikrobestrahlung fest. Mit der zugehörigen Software (siehe Materialtabelle)legen Sie den gewünschten ROI fest, indem Sie zuerst binäre Linien einfügen (die gewünschte Anzahl von Zeilen und Abständen einstellen). Klicken Sie auf Binär, und klicken Sie dann auf Zeile einfügen | Kreis | Ellipse, um die gewünschte Anzahl von Linien zu zeichnen.
- Konvertieren Sie diese binären Linien in ROIs und konvertieren Sie diese ROIs schließlich in Stimulations-ROIs. Klicken Sie dazu zuerst auf ROI, dann auf Binärdatei in ROI verschieben, klicken Sie dann mit der rechten Maustaste auf einen der ROIs und wählen Sie Als Stimulations-ROI verwenden: S1. Legen Sie diese Linien in das Sichtfeld, um den Zellkern zu passieren. ROIs mit einer Länge von 1024 Pixeln, die sich über das gesamte Sichtfeld erstreckten, wurden im gesamten Protokoll verwendet.
5. Mikrobestrahlung zur Immunfluoreszenzfärbung oder Zeitrafferbildgebung.
- Bestimmung der optimalen Mikrobestrahlungseinstellungen.
- Nehmen Sie vor der Mikrobestrahlung der Zellen ein höher aufgelöstes Bild des FOV auf, um PCNA-Herde für die spätere Analyse zu identifizieren. Anstatt sequenzielles Scannen, zeichnen Sie gleichzeitig beide verwendeten optischen Kanäle (grün und rot) auf, um Zellbewegungen zwischen dem Scannen bei den beiden Wellenlängen zu vermeiden. Für die richtige Auflösung der Foci verwenden Sie mindestens 1024 x 1024 Pixel/Feldauflösung mit 1x Zoom (0,29 μm Pixelgröße auf dem hier verwendeten Bildgebungssystem), mit 1/8 Frame/s Scangeschwindigkeit (4,85 μs/Pixel) mit 2x Mittelwertung. Sobald diese Parameter in der A1 LFOV Compact GUI und den A1 LFOV Scan Area Fenstern eingestellt sind, klicken Sie auf die Schaltfläche Capture, um das FOV aufzuzeichnen.
HINWEIS: Es ist wichtig, während der Experimente die gleiche Pixelgröße beizubehalten, um vergleichbare Ergebnisse zu gewährleisten. - Um die Mikrobestrahlung einzurichten, öffnen Sie die Registerkarte ND-Stimulation in der Bildgebungssoftware, um auf das Fenster Zeitplan (A1 LFOV / Galvano Device) zuzugreifen. Dieser verwendet die Galvano-Scanner, um eine Reihe von Vorstimulationsbildern zu erfassen, zu stimulieren (mit dem LUN-F 50 mW 405 nm FRAP-Laser) und dann eine Reihe von Post-Stimulationsbildern erneut mit den Galvano-Scannern zu erfassen. Richten Sie zunächst drei Phasen im Zeitplanfenster ein. Wählen Sie in der Spalte Acq/Stim die Option Acquisition | Bleich | Akquisition für die jeweils drei Phasen. Legen Sie für die Bleichphase S1 als ROI fest.
HINWEIS: In dem hier vorgestellten Experiment wurden während der Stimulationsphase keine Bilder aufgenommen. - Richten Sie im Galvano XY-Fensterdie Schlüsselfaktoren für die Mikrobestrahlung ein: 405 nm Laserleistung, Verweilzeit (Iteration ist standardmäßig 1 auf diesem System). In den hier vorgestellten Experimenten wurden Zellen mit dem 405 nm FRAP-Laser (50 mW an der Faserspitze) bei 100% Ausgangsleistung und einer Verweilzeit von 1000-3000 μs bestrahlt.
HINWEIS: Da die Laserverweilzeit pro Pixel erfolgt, ist die Beziehung zwischen der Verweilzeit und der Leistungsdichte zwischen verschiedenen FOVs vergleichbar, solange die Pixelgröße gleich bleibt. Abbildung 2A zeigt die Verwendung von DNA-Damage-Response-spezifischen Proteinen (DDR) (FBXL10 für DSBs und NTHL1 für oxidative Basenschäden), um die Laserleistungseinstellungen für die spezifische Schadensinduktion zu optimieren. Diese stabilen Zelllinien wurden mit einer Virusinfektion nach Abschnitt 2 des Protokolls erzeugt.
- Nehmen Sie vor der Mikrobestrahlung der Zellen ein höher aufgelöstes Bild des FOV auf, um PCNA-Herde für die spätere Analyse zu identifizieren. Anstatt sequenzielles Scannen, zeichnen Sie gleichzeitig beide verwendeten optischen Kanäle (grün und rot) auf, um Zellbewegungen zwischen dem Scannen bei den beiden Wellenlängen zu vermeiden. Für die richtige Auflösung der Foci verwenden Sie mindestens 1024 x 1024 Pixel/Feldauflösung mit 1x Zoom (0,29 μm Pixelgröße auf dem hier verwendeten Bildgebungssystem), mit 1/8 Frame/s Scangeschwindigkeit (4,85 μs/Pixel) mit 2x Mittelwertung. Sobald diese Parameter in der A1 LFOV Compact GUI und den A1 LFOV Scan Area Fenstern eingestellt sind, klicken Sie auf die Schaltfläche Capture, um das FOV aufzuzeichnen.
- Zeitraffer-Bildgebung.
- Richten Sie zeitraffer-Imaging für das gewünschte Zeitfenster und die gewünschten Intervalle mithilfe des Zeitplans, der A1 LFOV Compact GUI und der A1 LFOV Scan Area Fenster ein. In den hier vorgestellten Experimenten wurde die Rekrutierung von EXO1b und PCNA für 12 Minuten abgebildet, wobei das Sichtfeld alle 5 Sekunden mit 1024 x 1024 Pixeln / Feld gescannt wurde, wobei 1x Zoom (was zu 0,29 μm Pixelgröße auf dem hier verwendeten Bildgebungssystem führte) mit 0,35 Bildern / s Scangeschwindigkeit (1,45 μs / Pixel) ohne Mittelwert zur Reduzierung der Fotobleiche verwendet wurde.
- Optimieren Sie die Einstellungen für Laserleistungsprozent, Verstärkung und Offset, um das Photobleichen während der Bildgebung im A1 LFOV Compact GUI-Fenster zu reduzieren. Wenn man sowohl POI als auch PCNA messen möchte, verwenden Sie gleichzeitiges Scannen anstelle von sequenziellem Scannen, um Zellbewegungen zwischen dem Scannen des Feldes für die beiden separaten Fluorophore zu vermeiden.
- Das Imaging-System wurde mit den folgenden Einstellungen verwendet. Für die 488 nm Laserlinie (20 mW): 7% Laserleistung, Verstärkung: 45 (GaAsP-Detektor) mit und Offset von 2, für die 561 nm Laserlinie (20 mW): 4% Laserleistung, Gain 40 (GaAsP-Detektor) mit und Offset von 2.
- Je nach Kinetik des Proteins verlängern oder verkürzen Sie das Intervall zwischen den Bildern oder die Dauer des gesamten Zeitraffers. Legen Sie im Fenster Zeitplan das gewünschte Intervall und die gewünschte Dauer für die Zeile erfassung der dritten Phase fest.
- Drücken Sie jetzt ausführen, um die Mikrobestrahlung und die anschließende Zeitrafferbildgebung auszuführen.
- Speichern Sie am Ende der Zeitraffer-Bildgebung die Stimulations-ROIs als separate Bilder, die eine nützliche Hilfe sind, um die Koordinaten der Mikrobestrahlung in jeder nachgeschalteten Software zu identifizieren, die für die Analyse verwendet wird.
- Immunfluoreszenzfärbung.
HINWEIS: Schritt 5.1.3 und Abbildung 2A zeigen die Verwendung bekannter DNA-Reparaturproteine zur Beurteilung der Arten von DNA-Läsionen, die durch Mikrobestrahlung eingeführt werden. Bestimmte DNA-Läsionen können auch durch die Verwendung spezifischer Antikörper nach der Fixierung der Zellen nachgewiesen werden. Es ist auch möglich, die Rekrutierung des POI durch Antikörpernachweis des endogenen Proteins nachzuweisen. Die Visualisierung von γH2A.X zur Überprüfung auf DSBs wird unten gezeigt (Abbildung 2B). Abbildung 3 zeigt die Konsistenz der PCNA-Lokalisierung und -Rekrutierung während des gesamten Zellzyklus sowohl für endogene als auch für exogene markierte PCNA.- Nehmen Sie nach Schritt 5.1.3 nur ein Bild nach der Mikrobestrahlung auf, um ein ordnungsgemäßes FRAP-Ereignis basierend auf der Rekrutierung von mPlum-PCNA sicherzustellen. Notieren Sie sich die genauen Koordinaten des FOV, um das Feld später nach der immunfluoreszierenden Markierung zu finden.
- Nehmen Sie die Zellkulturkammer aus dem Mikroskop und inkubieren Sie die Zellen bei 37 °C in einer befeuchteten Atmosphäre mit 5% CO2 für 5-10 min.
HINWEIS: Paraformaldehyd (PFA) ist giftig und die Arbeit sollte in einem gut belüfteten Bereich oder einem Abzug durchgeführt werden. Alle nachfolgenden Wasch- und Inkubationen erfolgen mit 0,5 mL Volumen im 4-Well-Kammer-Schlitten. Nach der Inkubationszeit die Zellen mit 0,5 mL PBS (137 mM NaCl, 2,7 mM KCl, 8 mMNa2HPO4und 2 mM KH2PO4)waschen und mit 0,5 mL 4% PFA in PBS für 10 min bei Raumtemperatur (RT) fixieren. - Waschen Sie die Zellen einmal mit PBS, dann waschen Sie sie mit 50 mM NH4Cl, um Rest-PFA abzuschrecken.
- Permeabilisieren Sie die Zellen für 15 min bei RT mit 0,1% Triton X-100 in PBS.
- Blockieren Sie die Proben für 1 h mit Blockierungspuffer (5% FBS, 3% BSA, 0,05% Triton X-100 in PBS).
- Entfernen Sie die Blockierende Lösung und fügen Sie den verdünnten primären Antikörper (anti-γH2A.X, 1:2000) in einen Sperrpuffer für 1 h bei RT hinzu.
- Waschen Sie die Vertiefungen mit Sperrpuffer 3 x 10 min.
- Verdünnter sekundärer Antikörper (Anti-Maus Alexa 488 Plus Konjugat, 1:2000) in Blockierungspuffer für 1 h bei RT hinzufügen.
- Waschen Sie die Vertiefungen mit Sperrpuffer 3 x 10 min.
- Den Kern mit 1 μg/ml DAPI-Lösung in PBS für 15 min gegenzinnen.
- Waschen Sie die Zellen einmal mit PBS. Die Bildgebung kann direkt in PBS oder einer PBS-Lösung mit Antifade-Reagenzien (z. B. AFR3) durchgeführt werden, um Photobleaching zu reduzieren.
6. Rekrutierungsanalyse
HINWEIS: Abbildung 4A zeigt repräsentative Bilder der Exo1b- und PCNA-Rekrutierung in Gegenwart von DMSO oder Olaparib. Abbildung 4B zeigt ein repräsentatives Bild zur Datenanalyse. Die mittleren Fluoreszenzwerte wurden berechnet, indem die mittleren AcGFP-Dichten unter Verwendung eines Rechtecks entlang der Laserspur gemessen wurden, das durch die mPlum-PCNA (A, gelbe Rechtecke) über verschiedene Zeitpunkte mit Fidschi hervorgehoben wurde. PCNA kann als interne Kontrolle dienen, um die erfolgreiche Bestrahlung entlang der ROI-Koordinaten hervorzuheben. In ähnlicher Weise wurden mittlere AcGFP-Fluoreszenzwerte auch für unbeschädigte Bereiche des Kerns (B, blaue Rechtecke) berechnet. Die Hintergrundsignalintensität wurde in unbewohnten Gebieten (C, rote Rechtecke) gemessen und von den mittleren Fluoreszenzwerten abgezogen (Abbildung A und B). Somit wurde die relative mittlere Fluoreszenzeinheit (RFU) für jeden Datenerfassungspunkt durch die Gleichung RFU = (A − C)/ (B − C)8,9berechnet. Die resultierenden RFU-Werte des mikrobestrahlten Bereichs werden vor der Mikrobestrahlung auf die RFU-Werte normalisiert.
- Um die Region A der mikrobestrahlten Stelle zu definieren, schließen Sie Nukleolarregionen, Replikationsherde und unregelmäßige Kernregionen der Zelle von der Messung aus. Halten Sie die Umschalttaste zwischen dem Zeichnen von zwei ROIs in Fidschi gedrückt, um zwei separate Regionen als eine zu gruppieren.
HINWEIS: Die Proteinrekrutierung variiert zwischen verschiedenen Genen und Bestrahlungsbedingungen. daher muss die Größe der Region A individuell bestimmt werden. Sobald die Pixelbreite von Region A bestimmt ist, sollte sie für alle vergleichenden Rekrutierungen konstant bleiben. In den hier vorgestellten Experimenten wurden Rechtecke mit einer Breite von 7 Pixeln verwendet. - Schließen Sie Zellen, die sich während der Dauer der aufgezeichneten Videos bewegt haben, von der Analyse aus. Um hochmobile Zellen einzubeziehen, muss die beschriebene Analyse Frame für Frame durchgeführt werden.
- Um das Rekrutierungsprofil zu visualisieren, zeichnen Sie die normalisierten RFU-Werte mithilfe einer Statistiksoftware gegen die Zeit auf.
- Berechnen Sie die Differenz zu einem angegebenen Zeitpunkt zwischen DMSO und Olaparib (n = 31) Behandlung mit einem Mann-Whitney-Test.
Representative Results
Zellen adressieren jede Art von DNA-Läsion auf eine bestimmte Weise, die auch davon abhängt, in welcher Zellzyklusphase sie sich befinden. Beispielsweise werden nach der Mikrobestrahlung doppelsträngige Brüche (DSB) je nach Zellzyklusphase entweder durch nicht-homologe Endfügung (NHEJ) oder HR verarbeitet. Nukleasen, die während der S- und G2-Phasen des Zellzyklus am stärksten wirken, erzeugen DNA-Überhänge, die für die richtige HR entscheidend sind. Um die Bewertung von Zellen in der S-Phase zu fördern, wurde PCNA als einfarbiger Zellzyklusmarker eingesetzt. Abbildung 1A zeigt das Lokalisierungsprofil von mPlum-PCNA während des Zellzyklusverlaufs. PCNA hat eine völlig homogene Verteilung im Kern in der G1- und G2-Phase (während es auch weitgehend von den Nukleoli ausgeschlossen ist). In der S-Phase lokalisiert PCNA an Stellen der DNA-Replikation, die als helle Flecken im Zellkern visualisiert werden können. In frühen S-Phasenzellen sind die Flecken relativ klein und gleichmäßig im Zellkern verteilt. Im Verlauf der mittleren S-Phase verschwimmen die Flecken und lokalisieren sich mehr in Richtung des Umfangs des Kerns und der Nukleoli. In der späten S-Phase verringern sich die Flecken in der Anzahl, werden aber immer größer, da sich PCNA an späten Replikationsstellen konzentriert (Abbildung 1B). Wichtig ist, dass die exogene PCNA-Expression aus dem pBABE-Vektor-Rückgrat geringer war als die endogenen Werte, aber für den Nachweis durch Mikroskopie ausreichte, was potenzielle Artefakte in der Zellzyklusprogression und DDR minimiert. Abbildung 1C zeigt das Ausmaß der PCNA-Überexpression im Vergleich zu endogenen Werten. Bitte beachten Sie, dass das Band, das mPlum-PCNA entspricht, aufgrund seiner größeren Größe langsamer migriert.
Unser Ziel war es, DSBs während der Mikrobestrahlung einzuführen, um die PARP1/2-abhängige Rekrutierung von EXO1b für diese Läsionen in der S-Phase zu untersuchen. Abbildung 2A zeigt, dass niedrige Energiedosen (1000 μs Verweilzeit) nicht die Rekrutierung von EGFP-FBXL10, einem DSB-Responder (Bestandteil des FRUCC-Komplexes 8),induzieren, während es ausreichte, um die Rekrutierung von NTHL1-mCherry, einem Base Excision Repair (BER) Pathway Protein, zu induzieren, das an Stellen oxidativer DNA-Schädenrekrutiert 10,11,12. Bei einer Verweilzeit von 3000 μs rekrutieren sowohl EGFP-FBXL10 als auch NTHL1-mCherry und demonstrieren eine Laserleistung, die sowohl oxidative Läsionen als auch DSBs erzeugt. Um diese Ergebnisse zu verstärken, zeigt Abbildung 2B eine Immunfluoreszenzfärbung gegen γH2A.X (DSB-Marker), die bei Verwendung höherer Energiedosen deutlich deutlicher wird. PCNA dient sowohl als Zellzyklusmarker als auch als Marker für eine erfolgreiche Mikrobestrahlung, da es mit beiden Laserverweilzeiteinstellungen adäquat rekrutiert. Wichtig ist, dass sowohl exogene als auch /oder endogene fluoreszierende proteinmarkierte PCNA für diese Reporterfunktion verwendet werden können, da sie sich ähnlich verhalten (Abbildung 3). Endogen markierte PCNA wurde entwickelt, indem mRuby im Rahmen mit dem ersten Exon in ein Allel des PCNA-Locus13 eingefügt wurde (die Zelllinie war ein freundliches Geschenk von Jörg Mansfeld).
Abbildung 4A und Abbildung 4C zeigen die Rekrutierung von AcGFP-markiertem EXO1b in S-Phasenzellen. EXO1b erreicht etwa 1 Minute lang ein maximales Akkumulationsniveau an Mikrobestrahlungsstellen und beginnt sich danach langsam von den DNA-Läsionen zu löst. Anreicherungen an Mikrobestrahlungsstellen werden durch eine relative Fluoreszenzeinheit > 1 auf der Grafik gekennzeichnet. In Gegenwart von Olaparib ist die Akkumulation von EXO1b am Laserstreifen nach 1 Minute im Vergleich zur Fahrzeugsteuerung signifikant geringer. Diese Ergebnisse stehen im Einvernehmen mit der Literatur6,7. Abbildung 4B zeigt repräsentative Regionen für die Quantifizierung (Gebiete A, B und C), wie in Nummer 6 des Protokolls beschrieben. Abbildung 4D zeigt die vergleichbaren Expressionsniveaus von endogenem EXO1b und exogenem EXO1b-AcGFP in Zellen, die für die Mikrobestrahlung verwendet werden.
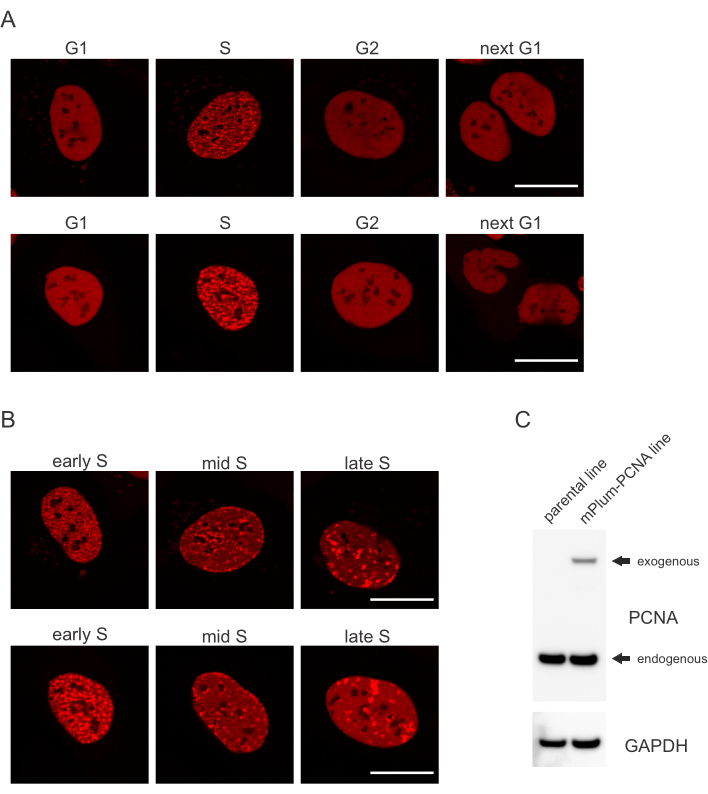
Abbildung 1:Lokalisierungsmuster von PCNA. (A) Bilder zeigen ein Lokalisierungsmuster stabil integrierter, exogener PCNA während des gesamten Zellzyklus in U-2 OS-Zellen. (B) Bilder zeigen PCNA-Herdemuster in verschiedenen Stadien der S-Phase (früh, mittel und spät) in U-2 OS-Zellen. (C) Western Blot, der endogene und exogene PCNA-Spiegel in den für die Bildgebung verwendeten U-2-OS-Zellen zeigt. Der Maßstabsbalken entspricht 20 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 2: Induktion von DSBs durch optimierte Laserleistung. (A) Lasereinstellungen können optimiert werden, um verschiedene Formen von DNA-Schäden zu induzieren. U-2 OS-Zellen, die sowohl EGFP-FBXL10 als auch NTHL1-mCherry stabil exprimieren, wurden verwendet, um DSBs bzw. Stellen oxidativer Läsionen zu identifizieren. Die Mikrobestrahlung mit einer 405 nm Laserlinie wurde auf asynchronen U-2 OS-Zellen mit entweder 1000 μs oder 3000 μs Verweilzeit durchgeführt. Der Schuppenbalken stellt 20 μm dar. (B) Immunfluoreszierende Färbung gegen γH2A.X wurde an menschlichen retinalen Pigmentepithelzellen (hTERT RPE-1) mit mRuby-markierter endogener PCNA durchgeführt. Die Zellen wurden 5 Minuten nach der Mikrobestrahlung mit entweder 1000 μs oder 3000 μs Verweilzeit fixiert und verarbeitet. Der Maßstabsbalken entspricht 20 μm. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 3: Vergleichbare Rekrutierung von endogener mRuby-PCNA und exogener mPlum-PCNA an Mikrobestrahlungsstellen bei 1000 μs oder 3000 μs Laserverweilzeit. Sowohl endogene als auch exogene markierte PCNA bilden während der S-Phase Replikationsherde. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.

Abbildung 4: PARP1/2-abhängige Rekrutierung von EXO1b in der S-Phase. U-2 OS-Zellen, die stabil EXO1b-AcGFP und mPlum-PCNA exprimierten, wurden mit einer 405 nm FRAP-Laserlinie unter Verwendung einer Verweilzeit von 3000 μs mikrobestrahlt. (A) Repräsentative Bilder von mikrobestrahlten Zellen zu den angegebenen Zeitpunkten nach der Vorbehandlung mit Fahrzeugkontrolle (DMSO) oder Olaparib (1 μM). Der Maßstabsbalken stellt 20 μm dar. (B) Repräsentative Bilder definierter Regionen von A-, B- und C-Bereichen für die Rekrutierungsanalyse. Der Maßstabsbalken stellt 20 μm dar. (C) Die Rekrutierungsdynamik von DNA-Schäden wurde durch Live-Zell-Bildgebung erfasst. Relative mittlere Fluoreszenzwerte und Bilder wurden alle 5 s für 12 min aufgenommen. Für jede Bedingung wurden ≥30 Zellen ausgewertet. Mittlere relative Fluoreszenzwerte (durchgezogene schwarze Linien) und Standardfehler (Bereich visualisiert durch einen schattierten Bereich) wurden gegen die Zeit aufgezeichnet. Die gestrichelte Linie zeigt Rekrutierungswerte bei 1 minute nach Mikrobestrahlung an. Der Unterschied zwischen DMSO (n=32) und Olaparib (n=31) Behandlung wurde mit einem Mann-Whitney-Test berechnet. Asterix bezeichnet S<0,0001. (D) Western Blot vergleicht die Expressionsniveaus von endogenem EXO1b und exogenem EXO1b-AcGFP in Zellen, die für die Mikrobestrahlung verwendet werden. Bitte klicken Sie hier, um eine größere Version dieser Abbildung anzuzeigen.
Discussion
Kritische Schritte und mögliche Protokollfehlerbehebung/-änderungen
Das richtige Gewebekulturgefäß für die Mikrobestrahlung ist entscheidend für den Erfolg. Die meisten hochauflösenden Bildgebungssysteme sind für eine Deckglasstärke von 0,17 mm optimiert. Die Verwendung von Bildgebungskammern mit höherer oder geringerer Dicke oder aus Kunststoffpolymeren (nicht für die 405-nm-Bildgebung optimiert) kann die Bildqualität erheblich reduzieren. Wenn Sie Glasoberflächen verwenden, stellen Sie sicher, dass sie gewebekulturbehandelt sind, um die Zelladhäsion zu verbessern. Wenn sie nicht mit Gewebekultur behandelt werden, müssen diese Kammern beispielsweise mit Poly-D-Lysin beschichtet werden, bevor die Zellen ausgesät werden. Beim Plattieren von Zellen in das kammerbewältigte Deckglas ist eine ideale Zelldichte von größter Bedeutung, um Unregelmäßigkeiten im Zellzyklus und zusätzliche Belastung der Zellen zu vermeiden. Die richtige thermische Gleichgewichtierung der Mikroskopkomponenten vor dem Experimentieren, um eine stabile Temperatur aufrechtzuerhalten, ist entscheidend für die Aufrechterhaltung des Fokus während der gesamten Zeitrafferaufnahme und ist auch notwendig, um eine homogene DDR über Zeit und Proben hinweg zu gewährleisten.
Es ist wichtig, dass sich die Zellen vor der Mikrobestrahlung in einem gesunden Zustand befinden, um die artefaktischen Daten zu reduzieren. Wenn Zellen nach der Infektion/ Selektion eine unregelmäßige Morphologie aufweisen, lassen Sie die Zellen durch mehrere Passagen fortschreiten, bis sich die Morphologie wieder normalisiert. Achten Sie immer darauf, dass die verwendeten Zelllinien frei von Mykoplasmenkontamination sind. Unter den vielen Nebenwirkungen der Mykoplasmeninfektion verursacht es auch DNA-Schäden an den Wirtszellen und könnte ihre DDR-Signalwege beeinflussen14,15. Der empfindlichste Weg, Mykoplasmen in der Zellkultur nachzuweisen, ist die PCR (versus. Nachweis mit DAPI oder Hoechst).
Eine optimale Überexpression des interessierenden Reparaturproteins sollte mit endogenen Werten vergleichbar sein, jedoch hoch genug für den Nachweis. Der auf den viralen Vektoren verwendete Promotor, der virale Titer während der Infektion und die Länge der Infektionszeit können alle für ideale Expressionsniveaus angepasst werden. Für konsistente Ergebnisse isolieren Sie einzelne Zellklone, um homogene Expressionsniveaus und eine normale Zellmorphologie zu gewährleisten. Es wird empfohlen, Vektorkonstrukte zu verwenden, die markierte PCNA nicht in höheren als endogenen Konzentrationen überexprimieren, um den richtigen Zellzyklus und die DNA-Reparaturmarkerfunktion zu erreichen. Selbst geringe PCNA-Überexpressionsraten reichen aus, um S-Phasen-Zellen zu unterscheiden. Zu diesem Zweck wurden erfolgreich retrovirale pBABE-Vektoren eingesetzt (Addgene #1764, #1765, #1766, #1767). PCNA kann mit beliebigen monomeren roten(z. B. mPlum, mCherry, mRuby usw.) oder monomeren grün fluoreszierenden Proteinen (z. B. mEGFP, AcGFP, mWasabi, mNeonGreen, mEmerald usw.) markiert werden, die dann mit einem abwechselnd markierten POI kombiniert werden können. Die Überexpressierung eines fluoreszierend markierten POI hat einige Einschränkungen und Überlegungen. Fluoreszierende Tags können die normale Proteinfunktion und -lokalisation stören. Somit muss die Position des Tags (N- oder C-Terminal) berücksichtigt werden. Verwenden Sie immer monomere fluoreszierende Proteine, da die Oligomerisierung nicht-monomerer Varianten die Funktion des POI beeinflussen kann.
Die Lasereinstellungen müssen für jedes Bildgebungssystem bestimmt werden, da viele Komponenten des optischen Pfades die tatsächliche Leistung der Zellen beeinflussen. Die Lasermikrobestrahlung kann verschiedene Arten von DNA-Läsionen verursachen, abhängig von der Anregungswellenlänge, der Leistungsabgabe des FRAP-Lasers und ob vorsensibilisierende Mittel (wie Bromodeoxyuridin oder Hoechst) verwendet wurden. 405 nm Laser können oxidative DNA-Schäden verursachen, einzel- und doppelsträngigeBrüche 16,17. Durch die Verwendung höherer Laserausgabeeinstellungen erhöht sich die Anzahl der DSBs. In diesem Protokoll wurden keine Vorsensibilisierungsmethoden verwendet, aber diese Techniken werden in der Literatur ausführlich behandelt und in der folgenden Diskussion erneut behandelt. Unserer Meinung nach ist der beste Weg, um zu testen, ob die gewünschte Läsion erzeugt wird, die Rekrutierung bekannter DNA-Schadensweg-spezifischer Gene. Die Rekrutierung von NTHL1 oder OGG1, Komponenten des BER-Signalwegs, deutet auf die Induktion von oxidierten DNA-Basen10,11,17,18,19hin, während FBXL10 oder XRCC5 das Vorhandensein von DSBs8,20,21anzeigen . Die Rekrutierung von XRCC1 kann sowohl auf das Vorhandensein von oxidierten DNA-Basen als auch auf einzelsträngige Brüche (SSB)hinweisen 22,23. XPC (d.h. RAD4) ist ein guter Indikator für NER, der die sperrigen DNA-Addukte entfernt, die durch ultraviolettes Licht (UV) erzeugt werden17,24. Da die Rekrutierung exogener Proteine zu bestimmten Unregelmäßigkeiten führen kann, kann die immunfluoreszierende Färbung von endogenen DNA-Reparaturproteinen oder Markern (wie γH2A.X für doppelsträngige Brüche) das Vorhandensein spezifischer DNA-Läsionen bestätigen. Alternativ könnten auch Antikörper gegen bestimmte Arten von DNA-Läsionen verwendet werden. Um die gelieferte Laserleistung einzustellen, können sowohl die Verweilzeit als auch die Laserleistung verändert werden.
Mit Hilfe der mathematischen Modellierung konnte eine detaillierte kinetische Analyse durchgeführt werden, die wertvolle Einblicke in die Rekrutierungseigenschaften des POI liefern kann (z. B. Beitrag mehrerer DNA-Bindungsdomänen, Empfindlichkeit gegenüber verschiedenen Signalereignissen usw.). Automatisierte Rekrutierungsauswertung und Zellverfolgung könnten kombiniert werden, um robuste Workflows zu schaffen 1,25.
Vorteile und Grenzen der DNA-Vorsensibilisierung
Die Vorsensibilisierung der DNA vor der Mikrobestrahlung ist ein häufig verwendetes Werkzeug zur Rekrutierung von DNA-Reparaturproteinen16,17. Die Sensibilisierung der DNA vor der Mikrobestrahlung macht sie anfälliger für DSBs. Die beiden gebräuchlichsten Methoden zur DNA-Vorsensibilisierung sind die Vorbehandlung von Zellen mit Bromodeoxyuridin (BrdU) oder Hoechst-Farbstoff. Für Systeme, die bei hohen Laserleistungen nicht mikrobestrahlen können, können diese Methoden notwendig sein, um DNA-Läsionen wie DSBs zu induzieren. Darüber hinaus fungiert Hoechst in Ermangelung eines Durchlichtdetektors oder eines fluoreszierenden Signals, das den Zellkern hervorhebt (z. B. bei der Untersuchung der Rekrutierung von nicht markierten endogenen DNA-Reparaturproteinen), sowohl als vorsensibilisierendes Werkzeug als auch als fluoreszierender Kernfleck. Eine DNA-Vorsensibilisierung kann jedoch zu erheblichen Komplikationen führen. BrdU (verwendet in einer Endkonzentration von 10 μM) muss den Zellen 24 Stunden (oder einer Zeit, die einem vollständigen Zellzyklus in der verwendeten Zelllinie entspricht) zugesetzt werden, um sie ordnungsgemäß in die DNA einzubauen, und kann Zellzyklusinterferenz verursachen26. Hoechst 33342 (verwendet in einer Endkonzentration von 1 μg/ml) ist nach langen Inkubationszeitzeiten zytotoxisch, benötigt aber ausreichend Zeit, um den Zellkern mit dem Farbstoff zu sättigen. Daher sollte es nur 15-20 Minuten vor der Mikrobestrahlung angewendet werden; Andernfalls sind die Rekrutierungsdaten nicht konsistent. Die auf diese Weise gefärbten Zellen können nicht länger als ein paar Stunden in Kultur gehalten werden27,28. Achten Sie darauf, Hoechst 33358 nicht zu verwenden, das nicht so zelldurchlässig ist wie der Farbstoff Hoechst 33342. Die Vorsensibilisierung kann auch zu unnötigen Abweichungen zwischen den Experimenten führen und macht das Experiment noch empfindlicher gegenüber Unterschieden in der Zelldichte (da dies die Menge des eingearbeiteten Farbstoffs / der Zelle beeinflusst).
Vorteile und Grenzen der konfokalen Mikroskopie
Die Bildgebungsgeschwindigkeit der konfokalen Mikroskopie kann im Vergleich zur Weitfeldmikrokopie einschränkend sein. Ein konfokales Mikroskop, das mit einem Resonanzscanner ausgestattet ist, kann jedoch die Bildgeschwindigkeit (auf Kosten der Auflösung) enorm verbessern und den Geschwindigkeiten der Spinnscheibenmikroskopie nahe kommen. Drei Eigenschaften machen das konfokale System A1R HD25 zu einer ausgezeichneten Wahl für das hier vorgestellte Protokoll. Erstens ermöglicht das 25-mm-Sichtfeld des Systems die Abbildung von 15-20 Zellen in einem einzigen gescannten Feld (im Vergleich zu 5-10 Zellen in normalen Setups), wodurch die Anzahl der Erfassungen begrenzt wird, die erforderlich sind, um genügend Zellen für die statistische Analyse zu erhalten. Zweitens ermöglichen das FRAP-Modul und zwei Scanheads die gleichzeitige Abbildung und Mikrobestrahlung der Zellen, nicht nur nacheinander. Schließlich bietet die Flexibilität, sowohl den Resonanz- als auch den Galvano-Scanner zu haben, die Möglichkeit, einfach zwischen hochauflösender Bildgebung mit außergewöhnlicher Geschwindigkeit, die das Abschrecken von Fluorophoren minimiert, und hochauflösender Bildgebung, die langsamere Scangeschwindigkeiten verwendet, um Bilder mit einem höheren Signal-Rausch-Verhältnis zu erzeugen, zu wechseln. Während das verwendete System die oben genannte Flexibilität ermöglichte, um weiter verbreiteten konfokalen Mikroskopkonfigurationen zu ähneln, wurde in den vorgestellten Experimenten nur der Galvano-Scanner verwendet (sowohl für die Mikrobestrahlung als auch für die anschließende Bildgebung).
Vorteile und Grenzen der Mikrobestrahlung
Die Mikrobestrahlung bietet zwar eine konkurrenzlose räumliche und zeitliche Auflösung, ist aber nicht ohne Einschränkungen. DNA-Schäden durch Laser-Mikrobestrahlung sind im Vergleich zu natürlich vorkommenden schädlichen Wirkstoffen stark auf bestimmte Teile des Kerns konzentriert. Daher kann sich die Chromatinreaktion aufgrund von Mikrobestrahlung von homogen verteilten Schäden unterscheiden. Darüber hinaus ist die Mikrobestrahlung zeitaufwendig und kann nur an einigen Dutzend Zellen durchgeführt werden, während große populationsbasierte biochemische Methoden (Chromatinfraktionierung, Immunpräzipitation, ChIP) eine erhöhte Robustheit bieten können, indem Tausende von Zellen gleichzeitig untersucht werden. Die Überprüfung von Beobachtungen durch Mikrobestrahlung mit traditionellen biochemischen Techniken ist eine effektive Strategie für zuverlässige Schlussfolgerungen. Obwohl die gleichzeitige Mikrobestrahlung vieler Zellen in einem bestimmten Sichtfeld möglich ist, benötigt das Bildgebungssystem mehr Zeit, um die Aufgabe auszuführen. Daher begrenzt die Messung der Dynamik von Proteinen, die sich sehr schnell zu DNA-Läsionen rekrutieren, die Anzahl der möglichen ROIs für die Mikrobestrahlung, die gleichzeitig verwendet werden. Auf dem für dieses Protokoll verwendeten Bildgebungssystem dauert die Mikrobestrahlung eines einzelnen 1024 Pixel langen ROI 1032 ms mit 1000 μs Verweilzeit und 3088 ms mit 3000 μs Verweilzeit. Die Verwendung mehrerer RoIs-Linien erhöht die Zeit, die für die Mikrobestrahlung benötigt wird, erheblich (z. B. dauert der ROI von 7 x 1024 Pixeln 14402 ms bei 1000 μs Verweilzeit und 21598 ms bei 3000 μs Verweilzeit). Diese Zeit geht durch die Bildaufnahme verloren und muss berücksichtigt werden. Verwenden Sie bei der Abbildung von schnellen Rekrutierungsereignissen den kürzest möglichen ROI und bestrahlen Sie nur eine Zelle gleichzeitig mit Mikrobestrahlung.
Vorteile und Einschränkungen gegenüber Synchronisationsmethoden
Für zellzyklusspezifische Studien beinhalten die bestehenden Methoden entweder die Synchronisation von Zellen in bestimmte Zellzyklusphasen oder die Verwendung von fluoreszierenden Reportern, um die spezifische Zellzyklusphase der Zelle zu identifizieren. Jede dieser Methoden bietet jedoch ihre eigenen Herausforderungen und Grenzen.
Das FUCCI-System3 (basierend auf fluoreszierenden Protein-markierten verkürzten Formen von CDT1 und Geminin) ist ein besonders nützliches Werkzeug für Zellzyklusstudien, hat aber Einschränkungen, wenn es darum geht, zwischen S- und G2-Phasen des Zellzyklus zu unterscheiden. Die Gemininspiegel sind bereits ab der mittleren S-Phase hoch und bleiben bis zur M-Phase hoch, was es schwierig macht, diese Phasen zu trennen. Die Verwendung des FUCCI-Systems bedeutet auch, dass zwei optische Kanäle des Mikroskops nicht für die Abbildung des POI verwendet werden können.
Nicht-Krebszelllinien könnten durch die Entfernung von Wachstumsfaktoren im Serum (Serummangel) in G0 synchronisiert werden, was zu geringen oder keinen DNA-Schäden an den Zellen führt. Die meisten Krebszelllinien werden jedoch teilweise auch ohne ausreichende Mengen an Serum in ihren Medien durch den Zellzyklus fortschreiten. Darüber hinaus beginnen Zellen teilweise die Synchronisation durch die späte G1-, frühe S-Phase zu verlieren. Neben dem Serummangel gibt es zahlreiche chemische Methoden, um eine Zellzyklussynchronisation zu erreichen. Hydroxyharnstoff-, Aphidicolin- und Thymidinblöcke sind Methoden, um die DNA-Replikation zu stoppen, um Zellen in die frühe S-Phase zu synchronisieren. Während diese Methoden billig und einfach sind, führen sie zu Replikationsstress, der zu DNA-Schäden führt. Es wurde gezeigt, dass diese DNA-Replikationsinhibitoren die Phosphorylierung von H2A induzieren. X, ein bekannter Marker von DSBs2,29. Die Methode, Tagged-PCNA als Marker für S-Phasen-Zellen zu verwenden, reduziert das Potenzial für Artefakte, die durch chemische Synchronisation verursacht werden, und kann im Vergleich zum Serummangel auf eine Vielzahl von Zelllinien angewendet werden.
Schlussfolgerung
DNA-Schäden sind eine treibende Kraft für genetische Erkrankungen, bei denen mutagene Läsionen zur bösartigen Transformation von Zellen führen können. Das Targeting der DNA-Synthesemaschinerie ist eine grundlegende therapeutische Strategie bei der Behandlung von hyperproliferativen Erkrankungen wie Krebs. Um diese Krankheiten gezielter behandeln zu können, brauchen wir ein besseres Verständnis der Proteine, die DNA-Läsionen reparieren. Das hier beschriebene Protokoll hilft mikrobestrahlungsbasierten Studien in der S-Phase, indem es die Herausforderungen herkömmlicher Synchronisationsmethoden minimiert, um mögliche Artefakte zu reduzieren und die Reproduzierbarkeit der Experimente zu erhöhen.
Disclosures
Die Autoren geben an, dass die Veröffentlichung der präsentierten Arbeit von der Nikon Corporation gesponsert wurde. Die Autoren erklären, dass keine interessenkonflikte bestehen.
Acknowledgments
Die Autoren danken M. Pagano für seine kontinuierliche Unterstützung sowie D. Simoneschi, A. Marzio und G. Tang für ihre kritische Durchsicht des Manuskripts. B. Miwatani-Minter dankt R. Miwatani und B. Minter für ihre anhaltende Unterstützung. G. Rona dankt K. Ronane Jurasz und G. Rona für ihre anhaltende Unterstützung.
Materials
| Name | Company | Catalog Number | Comments |
| Ammonium chloride | Sigma-Aldrich | A9434-500G | For quenching formaldehyde |
| Anti-EXO1 Rabbit Polyclonal Antibody | Proteintech | 16253-1-AP | primary antibody |
| Anti-phospho-Histone H2A.X (Ser139) Antibody, clone JBW301 | Millipore | 05-636 | primary antibody |
| Bovine Serum Albumin | Sigma-Aldrich | 3117332001 | BSA for blocking |
| BrdU (5-Bromo-2'-deoxyuridine) | Merck | 19-160 | pre-sensitizing agent |
| Citifluor™ Mountant Solution AFR3 | Electron Microscopy Sciences | 17973-10 | antifade containing PBS solution for imaging |
| DAPI | Sigma-Aldrich | D9542-1MG | nucleic acid stain |
| DMEM Medium | Thermo Fisher Scientific | 10569010 | Cell culture medium for HEK293T cells |
| DMSO | Sigma-Aldrich | D2650-100ML | Vehichle control and dissolution solvent |
| EGFP-FBXL10 | Addgene | #126542 | viral expression vector for EGFP-FBXL10 |
| EXO1b-AcGFP (in pRetroQ) | custom cloning | na | EXO1b cDNA was cloned in the NheI, BamHI sites of pRetroQ-AcGFP1-N1 vector. |
| Fetal Bovine Serum | Gibco | 16140071 | Media supplement |
| FluoroBrite DMEM | Thermo Fisher Scientific | A1896701 | Phenol red free medium for microscopy |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | A32723 | secondary antibody |
| HEK293T cells | ATCC | ATCC CRL-3216 | Cell line for viral packaging |
| HEPES | Sigma-Aldrich | H0887-100ML | Buffering agent to supplement live cell imaging medium |
| Hoechst 33342 | Thermo Fisher Scientific | H3570 | pre-sensitizing agent |
| Lipofectamine 3000 | Thermo Fisher Scientific | L3000015 | Transfection reagent |
| McCoy’s 5A (Modified) Medium | Life Technologies | 16600-108 | Cell culture medium for U-2 OS cells |
| mCherry-PCNA | Addgene | #55117 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA | Addgene | #55994 | non-viral PCNA construct suitable for cell cycle marker |
| mPlum-PCNA (in pBABE) | custom cloning | na | mPlum-PCNA cDNA was cloned from Addgene #55994 in the BamHI, SalI sites of pBABE (puro) |
| Nikon A1R-HD25 Confocal Scanhead and Controller | Nikon | na | confocal imaging system |
| Nikon LUN4 laser unit | Nikon | na | excitation system |
| Nikon LUN-F 50 mW 405 nm FRAP laser unit | Nikon | na | FRAP laser unit |
| Nikon NIS Elements Confocal Controller Software | Nikon | na | Confocal controlling software |
| Nikon Ti2-E Inverted Microscope | Nikon | na | inverted epifluorescent microscope base |
| Nikon Ti2-LAPP Modular Illumination System | Nikon | na | illumination system |
| NTHL1-mCherry (in pRetroQ) | custom cloning | na | NTHL1 cDNA was cloned in the NheI, SalI sites of pRetroQ-mCherry-N1 vector. |
| Nunc Lab-Tek II Chambered Coverglass (4 well) | Thermo Fisher Scientific | 155382PK | Live cell microscopy cell culture chamber |
| Olaparib | Selleck Chemicals | S1060 | PARP inhibitor |
| Opti-MEM reduced serum media | Thermo Fisher Scientific | 31985062 | Dilution medium for transient transfection |
| Paraformaldehyde aqueous solution (32%) | Thermo Fisher Scientific | 50-980-494 | Fixative |
| pBABE (hygro) | Addgene | #1765 | retroviral expression vector (for low expression levels) |
| pBABE (neo) | Addgene | #1767 | retroviral expression vector (for low expression levels) |
| pBABE (puro) | Addgene | #1764 | retroviral expression vector (for low expression levels) |
| pBABE (zeo) | Addgene | #1766 | retroviral expression vector (for low expression levels) |
| PCNA Antibody (PC10) | Santa Cruz | sc-56 | primary antibody |
| Penicillin-Streptomycin-Glutamine (100x) | Gibco | 10378016 | Media supplement |
| polybrene | Sigma-Aldrich | TR-1003 | Increase viral infection efficiency |
| pRetroQ-AcGFP-C1 | Takara | 632506 | retroviral expression vector |
| pRetroQ-AcGFP-N1 | Takara | 632505 | retroviral expression vector |
| pRetroQ-mCherry-C1 | Takara | 632567 | retroviral expression vector |
| pRetroQ-mCherry-N1 | Takara | 632568 | retroviral expression vector |
| pUMVC | Addgene | #8449 | Viral packaging vector |
| Sodium-pyruvate | Thermo Fisher Scientific | 11360070 | Supplement for live cell imaging medium |
| Triton X-100 aqueous solution (10%) | Sigma-Aldrich | 11332481001 | Dilute in PBS for cell permeabilization buffer |
| Trypsin-EDTA Solution 10X | Sigma-Aldrich | 59418C-100ML | Dilute in PBS to split cells |
| U-2 OS Cells | ATCC | HTB-96 | Optimal cell line for microscopy experiments |
| Universal Mycoplasma Detection Kit | ATCC | 30-1012K | PCR based Mycoplasma detection kit |
| VSV-G | Addgene | #8454 | Viral protein envelope vector |
References
- Aleksandrov, R., et al.
- Darzynkiewicz, Z., Halicka, H. D., Zhao, H., Podhorecka, M. Cell synchronization by inhibitors of DNA replication induces replication stress and DNA damage response: Analysis by flow cytometry. Methods in Molecular Biology. 761, 85-96 (2011).
- Sakaue-Sawano, A., et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 132 (3), 487-498 (2008).
- Herce, H. D., Rajan, M., Lattig-Tunnemann, G., Fillies, M., Cardoso, M. C. A novel cell permeable DNA replication and repair marker. Nucleus. 5 (6), 590-600 (2014).
- Keijzers, G., et al. Human exonuclease 1 (EXO1) regulatory functions in dna replication with putative roles in cancer. International Journal of Molecular Sciences. 20 (1), (2018).
- Cheruiyot, A., et al. Poly(ADP-ribose)-binding promotes Exo1 damage recruitment and suppresses its nuclease activities. DNA Repair (Amsterdam). 35, 106-115 (2015).
- Zhang, F., Shi, J., Chen, S. H., Bian, C., Yu, X. The PIN domain of EXO1 recognizes poly(ADP-ribose) in DNA damage response. Nucleic Acids Research. 43 (22), 10782-10794 (2015).
- Rona, G., et al. PARP1-dependent recruitment of the FBXL10-RNF68-RNF2 ubiquitin ligase to sites of DNA damage controls H2A.Z loading. elife. 7, (2018).
- Young, L. M., et al. TIMELESS forms a complex with PARP1 distinct from its complex with TIPIN and plays a role in the dna damage response. Cell Reports. 13 (3), 451-459 (2015).
- Kong, X., et al. Laser microirradiation to study in vivo cellular responses to simple and complex dna damage. Journal of Visualized Experiments. (131), e56213 (2018).
- Kong, X., et al. Condensin I recruitment to base damage-enriched DNA lesions is modulated by PARP1. PLoS One. 6 (8), 23548 (2011).
- Lan, L., et al. Novel method for site-specific induction of oxidative DNA damage reveals differences in recruitment of repair proteins to heterochromatin and euchromatin. Nucleic Acids Research. 42 (4), 2330-2345 (2014).
- Zerjatke, T., et al. Quantitative cell cycle analysis based on an endogenous all-in-one reporter for cell tracking and classification. Cell Reports. 19 (9), 1953-1966 (2017).
- Ji, Y., Karbaschi, M., Cooke, M. S. Mycoplasma infection of cultured cells induces oxidative stress and attenuates cellular base excision repair activity. Mutation Research. 845, 403054 (2019).
- Sun, G., et al. Mycoplasma pneumoniae infection induces reactive oxygen species and DNA damage in A549 human lung carcinoma cells. Infection and Immunity. 76 (10), 4405-4413 (2008).
- Gassman, N. R., Wilson, S. H. Micro-irradiation tools to visualize base excision repair and single-strand break repair. DNA Repair (Amsterdam). 31, 52-63 (2015).
- Muster, B., Rapp, A., Cardoso, M. C. Systematic analysis of DNA damage induction and DNA repair pathway activation by continuous wave visible light laser micro-irradiation. AIMS Genetics. 4 (1), 47-68 (2017).
- Ikeda, S., et al. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. Journal of Biological Chemistry. 273 (34), 21585-21593 (1998).
- Rosenquist, T. A., Zharkov, D. O., Grollman, A. P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proceedings of the National Academy of Science U. S. A. 94 (14), 7429-7434 (1997).
- Reid, D. A., et al. Organization and dynamics of the nonhomologous end-joining machinery during DNA double-strand break repair. Proceedings of the National Academy of Science U. S. A. 112 (20), 2575-2584 (2015).
- Taccioli, G. E., et al. Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science. 265 (5177), 1442-1445 (1994).
- Marsin, S., et al. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. Journal of Biological Chemistry. 278 (45), 44068-44074 (2003).
- Thompson, L. H., Brookman, K. W., Jones, N. J., Allen, S. A., Carrano, A. V. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Molecular and Cell Biology. 10 (12), 6160-6171 (1990).
- Scharer, O. D.
- Oeck, S., et al. High-throughput evaluation of protein migration and localization after laser micro-irradiation. Science Reports. 9 (1), 3148 (2019).
- Mistrik, M., et al. Cells and stripes: A novel quantitative photo-manipulation technique. Science Reports. 6, 19567 (2016).
- Durand, R. E., Olive, P. L. Cytotoxicity, mutagenicity and dna damage by hoechst 33342. Journal of Histochemistry and Cytochemistry. 30 (2), 111-116 (1982).
- Tobey, R. A., Oishi, N., Crissman, H. A. Cell cycle synchronization: reversible induction of G2 synchrony in cultured rodent and human diploid fibroblasts. Proceedings of the National Academy of Science U. S. A. 87 (13), 5104-5108 (1990).
- Podhorecka, M., Skladanowski, A., Bozko, P. H2AX phosphorylation: Its role in DNA damage response and cancer therapy. Journal of Nucleic Acids. 2010, (2010).
