



Overview
Source: Alexander S. Gold1, Tonya M. Colpitts1
1 Département de microbiologie, Boston University School of Medicine, National Emerging Infections Diseases Laboratories, Boston, MA
La transduction est une forme d'échange génétique entre les bactéries qui utilise des bactériophages, ou phages, une classe de virus qui infecte exclusivement les organismes procaryotes. Cette forme de transfert d'ADN, d'une bactérie à l'autre par le phage, a été découverte en 1951 par Norton Zinder et Joshua Ledererg, qui ont appelé le processus de « transduction » (1). Les bactériophages ont été découverts pour la première fois en 1915 par le bactériologiste britannique Frederick Twort, puis découverts de façon indépendante en 1917 par le microbiologiste canadien Français Felix d'Herelle (2). Depuis lors, la structure et la fonction de ces phages ont été largement caractérisées (3), divisant ces phages en deux classes. Les premiers de ces cours sont les phages lytiques qui, lors de l'infection, se multiplient dans la bactérie hôte, perturbant le métabolisme bactérien, lysant la cellule et libérant des phages de descendance (4). En raison de cette activité antibactérienne et de la prédominance croissante des bactéries résistantes aux antibiotiques, ces phages lytiques se sont récemment avérés utiles comme traitement de remplacement pour des antibiotiques. La deuxième de ces classes sont les phages lysogéniques qui peuvent soit se multiplier dans l'hôte par le cycle lytique ou entrer dans un état de quiescent dans lequel leur génome est intégré dans celui de l'hôte (figure 1), un processus connu sous le nom de lysogénie, avec la capacité de phage production à être induite dans plusieurs générations ultérieures (4).
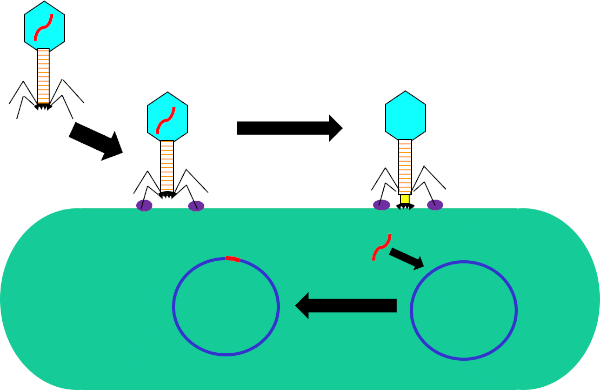
Figure 1 : Infection de la cellule hôte par bactériophage. Adsorption par le phage à la paroi cellulaire bactérienne par des interactions entre les fibres de la queue et le récepteur (violet). Une fois sur la surface de la cellule, le phage est irréversiblement attaché à la cellule bactérienne à l'aide de la plaque de base (noir) qui est déplacé vers la paroi cellulaire par la gaine contractile (jaune). Le génome phage (rouge) entre alors dans la cellule et s'intègre dans le génome de la cellule hôte.
Bien que la transduction bactérienne soit un processus naturel, l'utilisation de la technologie moderne a été manipulée pour le transfert de gènes en bactéries en laboratoire. En insérant des gènes d'intérêt dans le génome d'un phage lysogène, comme le phage, on est capable de transférer ces gènes dans les génomes des bactéries et, par conséquent, de les exprimer à l'intérieur de ces cellules. Alors que d'autres méthodes de transfert de gènes, telles que la transformation, utilisent un plasmide pour le transfert et l'expression des gènes, l'insertion du génome phage dans celui de la bactérie recevable a non seulement le potentiel de conférer de nouveaux traits à cette bactérie, mais permet également mutations naturelles et d'autres facteurs de l'environnement cellulaire pour modifier la fonction du gène transféré.
Par rapport à d'autres méthodes de transfert horizontal de gènes, comme la conjugaison, la transduction est assez souple dans les critères requis pour les cellules du donneur et du receveur. Tout élément génétique qui peut s'insérer dans le génome du phage utilisé peut être transféré de n'importe quelle souche de bactéries donneuses à n'importe quelle souche de bactéries receveurs tant que les deux sont permissifs au phage, nécessitant l'expression des récepteurs phages nécessaires sur le surfaces cellulaires. Une fois que ce gène est déplacé hors du génome du donneur et emballé dans le phage, il peut être transféré au receveur. Après la transduction, il est nécessaire de sélectionner pour les bactéries récepteurs qui contiennent le gène d'intérêt doivent être sélectionnés pour. Cela pourrait être fait par l'utilisation d'un marqueur génétique, comme un FLAG-tag ou polyhistidine-tag, pour marquer le gène d'intérêt, ou la fonction intrinsèque du gène, dans le cas des gènes qui codent pour la résistance aux antibiotiques. En outre, PCR pourrait être utilisé pour confirmer davantage la transduction réussie. En utilisant des amorces pour une région dans le gène d'intérêt et en comparant le signal à un contrôle positif, les bactéries qui ont le gène d'intérêt, et un contrôle négatif, les bactéries qui ont subi les mêmes étapes que la réaction de transduction sans phage. Bien que la transduction bactérienne soit un outil utile en biologie moléculaire, elle a joué et continue de jouer un rôle important dans l'évolution des bactéries, en particulier en ce qui concerne l'augmentation récente de la résistance aux antibiotiques.
Dans cette expérience, la transduction bactérienne a été utilisée pour transférer l'encodage génétique pour la résistance à l'ampicilline antibiotique de la souche W3110 d'E. coli à la souche J53 via le bactériophage P1 (5). Cette expérience se composait de deux étapes principales. Tout d'abord, la préparation du phage P1 contenant le gène de résistance à l'ampicilline de la souche du donneur. Deuxièmement, le transfert de ce gène à la souche receveuse par transduction avec le phage P1 (figure 1). Une fois effectué, le transfert réussi du gène de résistance à l'ampicilline pourrait être déterminé par qPCR (figure 2). Si la transduction était réussie, la souche J53 de E. coli serait résistante à l'ampicilline, et le gène conférant cette résistance détectable par qPCR. En cas d'échec, il n'y aurait aucune détection du gène de résistance à l'ampicilline et l'ampicilline fonctionnerait toujours comme un antibiotique efficace contre la souche J53.
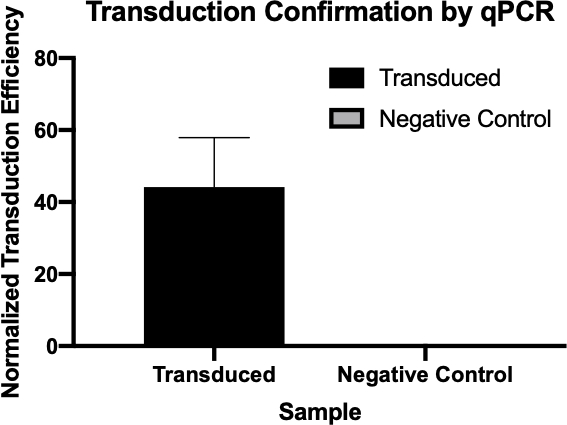
Figure 2 : Confirmation d'une transduction réussie par qPCR. En comparant les valeurs Cq détectées pour le gène d'intérêt de la réaction de transduction et de la réaction de contrôle négative, et en normalisant ces valeurs par rapport à un gène d'entretien ménager, on a pu confirmer que la transduction bactérienne a été réussie.
Procedure
1. Mise en place
- Avant de commencer tout travail impliquant des microbes, stériliser l'espace de travail à l'aide de 70% d'éthanol. Utilisez toujours le PPE nécessaire (manteau de laboratoire et gants).
- Assurez-vous que les plaques d'agar LB avec 1x ampicilline, solution de lysate phage P1 disponible dans le commerce, chloroforme, 1 M de citrate de sodium, le glycérol, et une boîte de pointes de pipette en plastique pré-stérilisé et les épandeurs de cellules sont à portée de main.
- Préparer la LB stérile en autoclamantisant et l'utiliser pour faire trois aliquots 1 mL de solution de sel LB.
- mM MgCl2 (952,11-2,3803 g), 5 mM CaCl2 (11,098 mg), 0,1-0,2 % de glucose (100-20 g)
- mM MgSO4 (12,0366 mg), 5 mM CaCl2 (11,098 mg)
- citrate de sodium mM (25,806 mg)
- Une fois terminé, stériliser toutes les surfaces ainsi que les gants à 70 % d'éthanol et se laver les mains.
2. Protocole
- Préparation de la lysate phage de donneur
- Préparer une culture de 1 ml de la souche E3110 du donneur dans LB avec 1x ampicilline cultivée pendant la nuit à 37 oC avec aération et secousses à 220 tr/min.
- Diluer cette culture de nuit 1:100 dans 1 ml de LB frais complété avec 10-25 mM MgCl2, 5 mM CaCl2, et 0,1-0,2% de glucose.
- Cultivez cette dilution bactérienne à 37 oC pendant 2 heures avec l'aération et en secouant à 220 tr/min.
- Une fois que ces cellules ont atteint la phase de croissance logarithmique précoce (croissance notable et légère turbidité), ajoutez 40 l de phage P1 disponible dans le commerce et laissez à 37 oC avec l'aération et secouant à 220 tr/min.
- Avant l'ajout de phage, la densité optique mesurée à 600 nm de ces cellules devrait être approximativement 0.4 (6).
- Surveillez les cellules pendant 1-3 heures jusqu'à ce que la culture se lysed.
- La lyse entraînera une augmentation des débris cellulaires ainsi qu'une diminution notable de la turbidité (c.-à-d. que les cellules seront considérées comme lysées une fois que l'on sera en mesure de voir à travers la culture).
- Ajouter plusieurs gouttes (50-100 l) de chloroforme au lysate et mélanger par vortex.
- Chloroforme stérilise le lysate phage en tuant les cellules donneuses restantes, ne laissant que du phage et augmentant le titer de ce lysate.
- Centrifugeuse lysate à 14 000 tr/min pendant 2 minutes pour enlever les débris et transférer le supernatant dans un tube frais.
- Ajouter quelques gouttes de chloroforme et conserver à 4 oC pendant au plus un jour.
- Transduction
- Préparer une culture de 1 ml de la souche E. coli du receveur J53 cultivée pendant la nuit en LB à 37 oC avec l'aération et en secouant à 220 tr/min.
- Transférer 100 ll de lysate phage donneur (2,1) dans un tube de microfuge de 1,5 ml et couver avec un bouchon ouvert à 37 oC pendant 30 minutes.
- Cette incubation permet à tout chloroforme restant dans la solution de lysate P1 de s'évaporer avant d'être ajouté aux cellules receveuses.
- Cellules de souche de receveur de granules par centrifugation à 6.000 tr/min pendant 5 minutes.
- Resuspendre ces cellules dans 300 l de LB frais contenant 100 mM MgSO4 et 5 mM CaCl2. (Le phage P1 exige que le calcium soit infectieux).
- Mettre en place deux réactions à l'aide des cellules bactériennes réceptrices et du lysate phage du donneur préparé : 1) réaction de transduction combinant 100 'L receveur De la souche E. coli et 100 l de phage de phage de donneur, et 2) contrôle négatif combinant 100 souche J53 du receveur de l'Homme et 2) un contrôle négatif combinant 100 souches J53 du receveur de l'Homme E. coli et 100 l de LB contenant 100 mM MgSO4 et 5 mM CaCl2.
- Incuber à 37 oC pendant 30 minutes en secouant à 220 tr/min.
- Ajouter 1 ml lb et 200 citrate de sodium de 1 ML (pH-5,5) et couver pendant 1 heure à 37 oC en secouant à 220 tr/min.
- Citrate est utilisé pour réduire l'infectiosité de P1 en chélant avec du calcium, empêchant la lyse des bactéries receveurs.
- L'incubation de cette solution permet l'expression du marqueur de résistance à l'ampicilline.
- Pellet cellules par centrifugation à 6000 tr/min pendant 5 minutes.
- Resuspendre les granulés de cellules dans 100 L de LB avec 100 mM de citrate de sodium (pH 5,5). Vortex et plaque solution entière pour les deux réactions sur deux plaques d'agar LB.
- La plaque LB devrait avoir 1x Amp pour l'échantillon transdû et aucun ampli pour le contrôle négatif.
- La contamination par le phage P1 sur cette plaque nécessite un re-streaking avant que les stocks de congélateur puissent être préparés.
- Si le phage n'est pas enlevé, les cultures cultivées à partir de ces colonies ne se développeront que si en présence d'un chélateur de calcium.
- Choisissez environ 3-4 colonies des deux plaques et strie zons à nouveau sur deux plaques d'agar LB étalées avec 100 l de citrate de sodium de 1 M (pH-5,5).
- La plaque LB devrait avoir 1x Amp pour l'échantillon transdû et aucun ampli pour le contrôle négatif.
- Incuber les plaques à 37 oC pendant la nuit pour permettre aux colonies exemptes de phage de se développer.
- Choisissez les colonies des deux plaques et utilisez-les pour faire pousser des cultures de nuit dans 5 ml de LB à 37 oC avec l'aération et les secousses à 220 tr/min.
- Isoler l'ADN de ces cultures par miniprep d'ADN en utilisant 4,5 ml du volume total de culture.
- Adn d'Elute utilisant 35 l d'eau nucléane-
- Mesurez la concentration résultante par Nanodrop. L'ADN pur générera un rapport d'absorption (A260/280) d'environ 1,8.
- Utilisez les 0,5 ml restants de chaque culture pour préparer 1 ml de glycérol en faisant un mélange 1:1 de 100% de glycérol et de culture bactérienne.
- Entreposer les stocks bactériens de glycérol à -80 oC.
3. Analyse des données et résultats
- Confirmation de la transduction par qPCR
- Préparer deux mélanges de maître qPCR pour six réactions qPCR, trois à l'aide d'amorces qPCR pour le gène de résistance à l'ampicilline, et les trois autres à l'aide d'amorces qPCR pour un gène d'entretien ménager (14,5 L par réaction) : 12,5 l qPCR buffer mix - 1 'L amorce avant '1 'L' amorce inversée.
- Dans cette expérience, nous avons utilisé SYBR Green master mix.
- Les amorces de gène d'entretien ont été conçues pour amplifier un segment de l'ADN dans le gène bactérien codant pour le gyrase B d'ADN (7).
- Pour chaque réaction qPCR, combiner 100 g d'ADN de chaque réaction (10,5 l) avec 14,5 L de mix principal qPCR.
- À l'aide d'une machine qPCR et du protocole thermocycling énuméré séditié dans le tableau 1, l'amplification a été mesurée pour la résistance à l'ampicilline et les gènes d'entretien ménager pour les six réactions.
- Les valeurs cq générées par qPCR ont été utilisées pour calculer l'efficacité de transduction normalisée du gène de résistance à l'ampicilline (figure 3), confirmant la transduction réussie du gène de résistance à l'ampicilline.
- La valeur Cq, ou valeur de quantification du cycle, d'un échantillon est le plus ancien numéro de cycle PCR auquel un signal dépassant le seuil d'arrière-plan est détecté. Les faibles valeurs cqées correspondent à une séquence cible plus nombreuse, vice versa.
- L'efficacité de transduction normalisée d'un gène dans un échantillon peut être calculée à l'aide de ces valeurs cq en soustrayant la valeur du gène d'entretien ménager de celle du gène cible, générant une valeur cq, qui peut être utilisée pour calculer la transduction normalisée l'efficacité de 2(-Cq).
- Préparer deux mélanges de maître qPCR pour six réactions qPCR, trois à l'aide d'amorces qPCR pour le gène de résistance à l'ampicilline, et les trois autres à l'aide d'amorces qPCR pour un gène d'entretien ménager (14,5 L par réaction) : 12,5 l qPCR buffer mix - 1 'L amorce avant '1 'L' amorce inversée.
| température | temps | |
| Dénaturation | 94 oC | 2 min |
| 40 cycles: | ||
| Dénaturation | 94 oC | 15 sec |
| Annealing, extension, et fluorescence lire | 60 oC ou 5 oC au-dessous de l'apprêt le plus bas Tm | 1 min |
Tableau 1 : Protocole de thermocyclisme qPCR
Les bactéries peuvent s'adapter rapidement à un environnement en évolution rapide en échangeant du matériel génétique et l'une des façons de le faire est par la transduction, l'échange de matériel génétique médié par des virus bactériens. Un bactériophage, souvent abrégé en phage, est un type de virus qui infecte les bactéries en se fixant d'abord à la surface de l'hôte, puis en injectant son ADN dans la cellule bactérienne. Il dégrade ensuite l'ADN de la cellule hôte et reproduit son génome viral, tout en détournant la machinerie de la cellule pour synthétiser de nombreuses copies de ses protéines. Ces protéines phages s'auto-assemblent et emballent les génomes de phage pour former la progéniture multiple. Cependant, en raison de la faible fidélité du mécanisme d'emballage de l'ADN, de temps en temps, le phage emballe des fragments d'ADN bactérien dans la capside phage. Après avoir induit la lyse de l'hôte, la progéniture phage sont libérés et, une fois qu'un tel phage infecte une autre cellule hôte, il transfère le fragment d'ADN de son hôte précédent. Cela peut ensuite se recombiner et s'intégrer en permanence dans le chromosome du nouvel hôte, ce qui permet de transférer des gènes entre les deux bactéries.
Pour effectuer la transduction de phage en laboratoire nécessite une souche de donneur qui contient un gène d'intérêt, une souche receveur qui n'en a pas, un phage qui peut infecter les souches, et une méthode pour sélectionner les bactéries transductées. Dans la plupart des cas, il s'agira d'un média sélectif de croissance solide qui soutient la croissance des bactéries cédées, mais inhibe la croissance des bactéries non cédées. Pour commencer, la souche de donneur qui contient le gène d'intérêt est cultivée dans un milieu de croissance liquide. Lorsque toutes les bactéries se divisent activement dans la phase de notation de leur croissance, la culture est inoculée avec le phage cible. Après trois à quatre heures d'incubation, lorsque presque toutes les bactéries ont lysé et libéré les particules de phage, le lysate phage donneur est inoculé dans une culture fraîchement cultivée de la souche bactérienne bénéficiaire. Après une brève incubation d'une heure, la culture devrait maintenant contenir un mélange de cellules bactériennes transductées et non transducées et cela est examiné pour les cellules transductées en répandant une fraction de la suspension sur un support de croissance solide sélectif approprié. Lors d'une incubation ultérieure, les cellules transducées devraient se développer et se multiplier pour produire des colonies visibles. Ces colonies peuvent ensuite être sélectionnées pour une analyse plus approfondie à l'aide d'une variété de méthodes pour confirmer davantage la transduction réussie, comme la pcR de la colonie, le séquençage de l'ADN ou le PCR quantitatif.
Avant de commencer la procédure, mettez tout équipement de protection individuelle approprié, y compris une blouse de laboratoire et des gants. Ensuite, stérilisez l'espace de travail avec 70% d'éthanol et essuyez la surface.
Après cela, préparer trois aliquots d'un millilitre de solution de sel LB. Maintenant, préparez une culture de souche de donneur en ajoutant 100 microlitres d'E. coli à un flacon conique de 15 millilitres contenant cinq millilitres de milieu de croissance LB avec 500 microgrammes d'ampicilline. Ensuite, cultivez la culture du jour au lendemain à 37 degrés Celsius avec l'aération et les secousses à 220 tr/min. Le lendemain, essuyez le dessus du banc avec 70% d'éthanol avant de retirer la culture de l'incubateur secouant. Ensuite, diluer la culture du jour au lendemain de 1 à 100 en ajoutant 10 microlitres de souche de donneur à 990 microlitres de LB frais complété s'est accompagné d'une solution de sel.
Laisser la dilution bactérienne se développer à 37 degrés Celsius pendant deux heures avec l'aération et secouer à 220 tr/min. Une fois que les cellules ont atteint la phase de journal précoce, retirer la culture de l'incubateur, ajouter 40 microlitres de phage P1 à la culture et incuber à nouveau. Continuer à surveiller les cellules pendant une à trois heures jusqu'à ce que la culture a lysed. Ensuite, ajouter 50 à 100 microlitres de chloroforme au lysate et mélanger par vortex. Ensuite, centrifuger le lysate pour enlever les débris et transférer le supernatant dans un tube frais. Ajoutez quelques gouttes de chloroforme au supernatant et conservez-le à quatre degrés Celsius pendant au plus un jour.
Pour commencer la procédure de transduction, obtenir une culture d'un millilitre de la souche du destinataire. Ensuite, transférer 100 microlitres de lysate de phage de donneur dans un tube de microcentrifuge de 1,5 millilitre et l'incuber à 37 degrés Celsius avec le bouchon ouvert pendant 30 minutes pour permettre à tout chloroforme restant de s'évaporer. Pendant que le phage lysate phage du donneur couve, granulez les cellules de la souche du receveur par centrifugation douce. Jetez le supernatant et resuspendre la pastille cellulaire en 300 microlitres de LB frais contenant 100 millimolaires de sulfate de magnésium et cinq chlorures de calcium millimolaires.
Ensuite, configurez la réaction de transduction en combinant 100 microlitres de la souche réceptrice et 100 microlitres du lysate phage du donneur dans un tube de microcentrifuge. Ensuite, configurez le contrôle négatif en combinant 100 microlitres de la souche récepteur et 100 microlitres de la LB avec du sulfate de magnésium et du chlorure de calcium. Après l'incubation, ajouter 200 microlitres d'un citrate de sodium molaire et un millilitre de LB dans les deux tubes, et mélanger en faisant doucement monter et descendre. Puis, après que les tubes ont été incubés pendant une heure, granulez doucement les cellules par centrifugation.
Après centrifuage, jeter le supernatant et resuspendre les cellules granulées dans 100 microlitres de LB avec 100 millimolar de sodium citrate. Vortex les solutions et pipette l'ensemble de l'échantillon transducé sur une plaque d'agar LB avec 1X ampicilline. Enfin, pipette le volume entier du mélange négatif de cellule de contrôle sur une plaque d'agar de LB sans ampicilline. Après avoir incubé les plaques pendant la nuit à 37 degrés Celsius, utilisez une pointe de pipette stérile pour cueillir trois à quatre colonies de la plaque de transduction et les stries sur une nouvelle plaque d'agar LB contenant 1X ampicilline et 100 microlitres d'un citrate de sodium molaire. Répétez cette méthode de placage pour le contrôle négatif sur une autre plaque d'agar LB contenant seulement 100 microlitres d'un citrate de sodium molaire. Ensuite, incuber les plaques à 37 degrés Celsius pendant la nuit pour permettre aux colonies exemptes de phage de se développer.
Le lendemain, essuyez le dessus du banc avec 70 % d'éthanol avant de retirer vos assiettes de l'incubateur. À l'aide d'une pointe de pipette stérile, choisissez trois colonies de la plaque de transduction et ajoutez-les chacune à un tube séparé contenant cinq millilitres de support LB. Ensuite, sélectionnez trois colonies de la plaque de contrôle négative et ajoutez-les à un autre tube contenant cinq millilitres de support LB. Cultivez les cultures pendant la nuit à 37 degrés Celsius avec l'aération et les secousses à 220 tr/min. Après avoir stérilisé le dessus du banc comme nous l'avons déjà démontré, utilisez un kit de miniprep d'ADN pour isoler l'ADN de 4,5 millilitres de chaque culture selon les instructions du fabricant. Ensuite, élichez l'ADN avec 35 microlitres d'eau sans nucléarité et mesurez la concentration résultante par spectrophotomètre de laboratoire. Enfin, préparer les stocks de glycérol en ajoutant les 0,5 millilitres restants des deux solutions bactériennes à 0,5 millilitres de 100% de glycérol.
Pour confirmer la transduction, préparez d'abord deux mélanges de maître qPCR pour 24 réactions qPCR. Pour le premier mix maître, ajouter 150 microlitres de mélange tampon qPCR à un tube microcentrifuge et 12 microlitres chacun d'une amorce avant et inverse conçue pour amplifier le gène de résistance à l'ampicilline. Ensuite, préparez un deuxième mix maître qPCR en ajoutant 150 microlitres de mélange maître qPCR à un tube microcentrifuge, puis en ajoutant 12 microlitres chacun d'une amorce avant et d'une amorce inversée conçue pour amplifier un gène d'entretien ménager.
Pour chaque réaction qPCR, combinez 100 microgrammes d'ADN expérimental de chaque réaction avec 14,5 microlitres de mix maître qPCR. Maintenant, préparez les réactions restantes comme précédemment démontré. Transférer les réactions à un thermocycleur préchauffé à 94 degrés Celsius, puis lancer le programme. Enfin, utilisez les valeurs de quantification du cycle, ou Cq, générées par qPCR pour calculer l'efficacité de transduction normalisée du gène de résistance à l'ampicilline.
Les valeurs de la quantitation du cycle, ou Cq, pour les gènes d'intérêt ont été compilées pour chacun des témoins négatifs et des échantillons transductés. Les faibles valeurs cqides, généralement inférieures à 29 cycles, comme les échantillons transducés dans cet exemple indiquent des quantités élevées de la séquence cible.
Un gène d'entretien ménager, également compilé ici, est utilisé comme un contrôle de chargement pour normaliser la quantité d'ADN dans chaque réaction et comme un contrôle positif pour s'assurer que le qPCR fonctionne. Si les mêmes quantités du gène d'entretien ménager sont chargées, on le trouve relativement au même rythme dans chaque échantillon.
Ensuite, pour calculer la valeur delta Cq pour chaque échantillon, soustrayez la valeur Cq du gène d'entretien ménager pour chaque échantillon de la valeur Cq de son gène cible correspondant. Par exemple, le delta Cq du premier contrôle négatif est de 13,54. Ensuite, utilisez cette valeur pour calculer l'efficacité de transduction normalisée de chaque échantillon à l'aide de la formule indiquée ici. Enfin, l'efficacité moyenne de transduction normalisée pour chaque groupe d'échantillons peut être calculée.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
Le transfert de gènes vers et depuis les bactéries par bactériophage, bien qu'il s'avère un processus naturel, s'est avéré extrêmement utile pour une multitude de fins de recherche. Tandis que d'autres méthodes de transfert de gène telles que la transformation et la conjugaison sont possibles, la transduction utilise uniquement des bactériophages ; permettant non seulement l'intégration des gènes dans le génome hôte, mais aussi la livraison de gènes à plusieurs bactéries qui ne sont pas sensibles à d'autres méthodes. Ce processus, bien que particulièrement utile en laboratoire, a également été utilisé dans le domaine récemment émergent de la thérapie génique, plus spécifiquement dans la thérapie génique alternative, une stratégie thérapeutique qui utilise des bactéries pour fournir des thérapies pour cibler les tissus, beaucoup d'entre eux ne sont pas sensibles à d'autres méthodes d'administration et ont beaucoup de pertinence clinique (8,9).
Subscription Required. Please recommend JoVE to your librarian.
References
- Lederberg J, Lederberg E.M., Zinder, N.D., et al. Recombination analysis of bacterial heredity. Cold Spring Harbor symposia Quantitative Biol. 1951;16:413-43.
- Duckworth DH. "Who Discovered Bacteriophage?". Bacteriology Reviews. 1976;40:793-802.
- Yap ML, Rossman, M.G. Structure and Function of Bacteriophage T4. Future Microbiol. 2014;9:1319-27.
- Sulakvelidze A, Alavidze, Z., Morris, J. G. Bacteriophage Therapy Antimicrobial Agents and Chemotherapy 2001;45(3):649-59.
- Moore S. Sauer:P1vir phage transduction 2010 [Available from: https://openwetware.org/wiki/Sauer:P1vir_phage_transduction].
- Kobayashi A, et al. Growth Phase-Dependent Expression of Drug Exporters in
- Escherichia coli and Its Contribution to Drug Tolerance. Journal of Bacteriology. 2006;188(16):5693-703.
- Rocha D, Santos, CS, Pacheco LG. Bacterial reference genes for gene expression studies by RT-qPCR: survey and analysis. Antonie Van Leeuwenhoek. 2015;108:685-93.
- Pálffy R. et al. Bacteria in gene therapy: bactofection versus alternative gene therapy. Gene Ther. 2006 13:101-5.
- O'Neill JM, et al. Intestinal delivery of non-viral gene therapeutics: physiological barriers and preclinical models. Drug Discovery Today. 2011;16:203-2018.