

Summary
The complete construction of a custom, real-time confocal scanning imaging system is described. This system, which can be readily used for video-rate microscopy and microendoscopy, allows for an array of imaging geometries and applications not accessible using standard commercial confocal systems, at a fraction of the cost.
Abstract
Confocal microscopy has become an invaluable tool in biology and the biomedical sciences, enabling rapid, high-sensitivity, and high-resolution optical sectioning of complex systems. Confocal microscopy is routinely used, for example, to study specific cellular targets1, monitor dynamics in living cells2-4, and visualize the three dimensional evolution of entire organisms5,6. Extensions of confocal imaging systems, such as confocal microendoscopes, allow for high-resolution imaging in vivo7 and are currently being applied to disease imaging and diagnosis in clinical settings8,9.
Confocal microscopy provides three-dimensional resolution by creating so-called "optical sections" using straightforward geometrical optics. In a standard wide-field microscope, fluorescence generated from a sample is collected by an objective lens and relayed directly to a detector. While acceptable for imaging thin samples, thick samples become blurred by fluorescence generated above and below the objective focal plane. In contrast, confocal microscopy enables virtual, optical sectioning of samples, rejecting out-of-focus light to build high resolution three-dimensional representations of samples.
Confocal microscopes achieve this feat by using a confocal aperture in the detection beam path. The fluorescence collected from a sample by the objective is relayed back through the scanning mirrors and through the primary dichroic mirror, a mirror carefully selected to reflect shorter wavelengths such as the laser excitation beam while passing the longer, Stokes-shifted fluorescence emission. This long-wavelength fluorescence signal is then passed to a pair of lenses on either side of a pinhole that is positioned at a plane exactly conjugate with the focal plane of the objective lens. Photons collected from the focal volume of the object are collimated by the objective lens and are focused by the confocal lenses through the pinhole. Fluorescence generated above or below the focal plane will therefore not be collimated properly, and will not pass through the confocal pinhole1, creating an optical section in which only light from the microscope focus is visible. (Fig 1). Thus the pinhole effectively acts as a virtual aperture in the focal plane, confining the detected emission to only one limited spatial location.
Modern commercial confocal microscopes offer users fully automated operation, making formerly complex imaging procedures relatively straightforward and accessible. Despite the flexibility and power of these systems, commercial confocal microscopes are not well suited for all confocal imaging tasks, such as many in vivo imaging applications. Without the ability to create customized imaging systems to meet their needs, important experiments can remain out of reach to many scientists.
In this article, we provide a step-by-step method for the complete construction of a custom, video-rate confocal imaging system from basic components. The upright microscope will be constructed using a resonant galvanometric mirror to provide the fast scanning axis, while a standard speed resonant galvanometric mirror will scan the slow axis. To create a precise scanned beam in the objective lens focus, these mirrors will be positioned at the so-called telecentric planes using four relay lenses. Confocal detection will be accomplished using a standard, off-the-shelf photomultiplier tube (PMT), and the images will be captured and displayed using a Matrox framegrabber card and the included software.
Protocol
The choice of laser wavelength, dichroic mirror, and optical filters should be determined based on the specific dyes being used in the experiment. For example, confocal imaging of a sample stained with Alexa Fluor 488 is best accomplished using a 488 nm laser, a 500 nm long-pass dichroic mirror, and a 30 nm bandwidth bandpass mirror centered at 515 nm. In contrast, confocal imaging of the red dye Alexa Fluor 647 would require a different set of components. The microscope in this protocol was built to visualize any dye that absorbs strongly at 400 nm and emits beyond 450 nm. We therefore chose a 406 nm excitation laser and a 425 nm long-pass dichroic to reflect the laser beam. Excited fluorophores can be selectively imagined by selecting the appropriate emission filters. It is important to use proper optical mounting hardware throughout the protocol where indicated; improper or makeshift hardware will not hold alignment as well and can be a safety hazard.
1. Setting up the resonant galvanometric mirror and relay optics
An important concept in building any kind of confocal scanning system is telecentricity. In a telecentric optical system, lenses are spaced from each other by the sum of their focal lengths, such that the magnification of the system is simply defined by the ratio of the focal lengths1. This enables the construction of an optical relay system where the magnifications, and thus the system properties, are easily defined by the choice of lenses. Another important concept involves so-called "stationary" optical planes, also referred to as "aperture planes". An aperture plane is a position along the optical path where the light beam does not undergo any kind of lateral movement. In this microscope design, there are three important aperture planes: the first and second scanning mirrors, and the back-aperture of the objective lens. In order to achieve optimal beam scanning at the focal plane of the objective, the beam entering the back aperture of the objective lens must be stationary, sweeping only in angle. In order to create this stationary, angle-swept plane, we need to place the first and second scanning mirrors at conjugate, telecentric planes to the objective back-aperture. Lenses placed between the mirrors and the objective lens serve to relay the angle-scanned beam between these stationary planes (Fig 2). The scanning mirrors are mounted on two scanning galvos, each of which is responsible for scanning a given direction of the imaging plane (X and Y). To obtain the required line scan rate for video-rate imaging, a high-frequency resonant galvo is required to scan the x-axis (also known as the "fast" axis). These galvos utilize a sensitive, closed-loop feedback circuitry to create a sinusoidal scan pattern and are capable of operating at very high frequencies; we selected an 8 kHz galvo for this build.
- Set up the fiber collimator in the optical mount and roughly steer the beam using the adjustment screws such that it travels in a straight line both horizontally and vertically. Now, take an iris and place it in front of the fiber collimator, adjusting the iris’s vertical height such that the beam passes cleanly through the iris center. Next, move the iris away from the collimator along the beam path and observe if the beam still travels through the iris center. If not, adjust the beam position on the iris using the two adjustment screws.
- Place the mounted dichroic mirror in the beam path with the laser beam positioned at approximately the center of the mirror. Before clamping the mirror to the table, rotate the mirror holder to reflect the beam at approximately 90 degrees and roughly adjust the reflection so that the reflected laser beam’s vertical height does not change.
- Place a mounted resonant galvanometric mirror into the laser beam path, taking care to ensure that the laser beam is positioned at the exact horizontal center of the mirror surface. In this protocol, the resonant galvo mirror was expoxied directly to a mirror mount. Rotate the mirror mount to reflect the laser beam at a 90-degree angle. Roughly adjust the reflection off the mirror to maintain the same laser beam vertical height.
- In order to direct any ray of light in a given direction, one must by definition define two points in space through which the ray will travel. This is typically accomplished by placing two irises along the desired horizontal and vertical path and manipulating the laser beam to pass through the center of each iris. Four degrees of freedom are required to adjust the beam; two horizontal and vertical degrees of freedom for each iris. The most common and straightforward way to achieve these degrees of freedom is to use two mirrors to steer, or “walk,” a laser beam.
Take two irises, and set their vertical height as in step 1.1, using the laser beam reflected off the resonant galvo mirror as a reference. Now, using the screw holes on the optical breadboard as a guide to the eye, clamp the two irises down in a straight line. - Adjust the dichroic mirror and resonant galvo mirror to steer the laser beam through the center of the two irises. Use the first mirror in the path (the dichroic mirror) to center the beam on the first iris, then use the second mirror in the path (the resonant galvo mirror) to center the beam on the second iris. Iteratively adjust these two mirrors until the beam is aligned through both irises, ensuring throughout that the laser beam reflected from the resonant galvo mirror is still reflected from the approximate mirror center. If the beam has deviated, adjust the fiber collimator mount and repeat the iterative steps above.
- With the beam centered on both irises, we will now place the two relay lenses that will image our first stationary, telecentric plane (i.e., the resonant galvo mirror) onto our second stationary, telecentric plane (i.e., the standard speed galvo mirror). For this particular microscope, the selected lenses in the first relay have the same focal length, “f”, so the distance between the two mirrors in our telecentric system is simply 4f. To ensure that the lenses are precisely centered in the beam path, use the lens alignment trick. Place the first lens in the beam path and look at the laser beam spot on the iris next in the beam path following the lens. Next, adjust the lens height vertically so that the vertical center of the beam is at the iris center. Finally, adjust the horizontal beam position to center the beam on the iris. Carry out this same procedure for the second lens.
2. Setting up the second scanning mirror and rotating the microscope
- To find the exact position of the second telecentric plane, hook up the resonant galvo to its scanning unit and turn it on. Use a white business card to track the scanning beam through the two lenses. You will find the telecentric plane at the approximate distance of 4f from the resonant galvo, where the laser beam will appear completely stationary. Mark this position on the breadboard.
- Position the standard scanning galvo mirror at this exact telecentric plane location, and adjust the mirror height and position such that the beam at the telecentric plane strikes the exact center of the scanning mirror. It is crucial to power up the mirror control hardware and put a voltage of 0 volts on the scanning mirror input so that the mirror settles to its neutral position during this process. Carefully adjust the mirror angle to direct the beam vertically, and gently tighten the mirror in position.
- As we are building an upright microscope, we will now attach the second breadboard at a 90 degree angle using 90 degree mounting brackets. Make sure to turn off the laser and scanning electronics, disconnect the fiber, and disconnect the scanning mirrors during this process. To make the rest of the alignment easier, once the brackets are bolted in place, carefully rotate the entire microscope so that the new breadboard is now lying flat. Use a clamp to fix the breadboard to the working surface. Now the remainder of the formerly vertical setup can be easily carried out on the flat breadboard.
3. Setting up the scan, tube, and objective lenses
Next we will set up the second set of relay lenses, formally referred to as the “scan lens” and “tube lens”. It is important to choose the right combination of lenses so as to achieve the correct magnification at the objective focus and optimize the final image resolution. First, to achieve the maximum numerical aperture (NA) of any given objective lens, the laser beam striking the back of the objective must fill the back aperture completely; only then will the objective lens be able to create the tightest focus. Objective lenses have a range of back aperture sizes; chose a lens magnification ratio to slightly overfill the back aperture of the selected objective. Second, in order to achieve the right magnification, the objective lens must be matched with the tube lens focal length for which it was designed. Unfortunately, different microscope objective manufacturers have chosen to use different tube lens focal lengths, so it is important to build a microscope with the correct tube lens for the specific objective lens employed. Furthermore, certain manufacturers, such as Zeiss, design their tube lenses to compensate for the specific chromatic aberrations of their matched objective, such that using an improper objective-tube lens pair will in fact introduce new aberrations that would not otherwise be present. We typically prefer Olympus objectives, as all chromatic compensation is performed in the objective itself, making the objective/tube lens pairing easier. Although the microscope will still work if the objective and tube lens do not match, the actual microscope magnification will likely not match the magnification listed on the objective lens. For this particular microscope build, the optimal back aperture size was determined to be 4 mm, requiring a 1:4 magnification ratio between the scan lens and tube lens. For this custom microscope build, we will use a scan lens length of 75 mm and a tube lens length of 300 mm.
- As the total distance between the second scan mirror and objective focus is large, this segment of the microscope build will first layout the mirrors needed to steer the beam to the objective lens. Place the first large, 2” (50 mm) diameter mirror close to the edge of the breadboard, and rotate the mirror mount to reflect the laser beam approximately 90 degrees. Roughly adjust the reflection off the mirror to maintain the same vertical beam height. Place the other 2” mirror at the opposite edge of the breadboard at an orientation that directs the beam downward at a 90-degree angle. Use the adjustment screws to ensure the beam’s vertical height does not change. Set up two irises, as in step 1.4, and adjust the two mirrors as directed in step 1.5 to center the beam on the irises.
- With the irises still in place, place the scan lens in the beam path and adjust its horizontal and vertical position to center the laser spot on the first iris. At a distance of 75 mm + 300 mm from the lens (between the two mirrors), carefully place the large 2” tube lens and adjust its horizontal and vertical position to center the beam on the first iris. For purposes of maintaining alignment in the future, it is useful to leave these irises in place; for this application, a business card with an appropriately-sized hole could be glued to a stand and inserted in the beam path.
- With all mirrors and lenses now in place, begin scanning the resonant galvo mirror and the standard scanning mirror. In this build, the standard scanning mirror will ultimately be synced to the scan rate of the resonant mirror through a custom-built control circuit, such as that described in Figure 3; this provides superior vertical and horizontal synchronization. However, for alignment purposes and many imaging applications, the mirror can easily be scanned using a sawtooth pattern from a function generator. Using a business card, locate the laser beam at a position 300 mm after the tube lens. Though the beam is scanning elsewhere in the microscope in both the vertical and horizontal directions, the beam should be perfectly stationary near this location. This is where the back aperture of the objective lens will be placed. If the horizontal and vertical stationary planes do not coincide at the same plane along the beam path, carefully unclamp and translate the tube lens along the optical path to ensure that both planes overlap as closely as possible. Re-center the vertical and horizontal position of the tube lens and clamp it securely in position.
- Place the objective lens in the beam path, making sure to position the objective lens back aperture as close to the stationary plane as possible. The true objective back aperture may not actually be always located at the physical back opening of the objective due to varying manufacturer design choices. It is therefore always best the check with the manufacturer to determine the true back aperture position.
- Set up the sample stage, making sure that the translation mount that will allow for z-axis motion can move over its full range without running into the objective lens mount.
4. Setting up and aligning the confocal pinhole and detector
- Disconnect all power supplies and fiber optics, and rotate the microscope assembly such that it again rests on the breadboard holding the resonant scanning mirror. Clamp the breadboard securely in place, then re-connect the fiber to the collimator, and re-connect both galvos and their control cables. As before, place 0 volts on the control voltage driving the standard scanning galvo.
- On the sample stage, place a business card or a small volume of a bright dye in the objective focus, sandwiched between two coverslips. The choice of dye will depend on the laser and dichroic selected; in this case we will use the fluorescence emission from a white business card to align the confocal detection system. Quantum dots can also be useful for alignment purposes, as they are bright and do not photobleach. Other alternatives include fluorescent beads and/or fabric samples exposed to color/laundry brightener, both of which fluoresce brightly. Turn on the laser source and bring the sample into the microscope focus using the translation stage. Once in focus, the fluorescence generated from the sample should be visible behind the dichroic mirror, as described in the next step. Maximize the laser power to make the fluorescence as bright as possible.
- Using a business card, trace the fluorescence emission from the sample through the objective lens and back through the scanning system to the dichroic mirror. The dichroic mirror will transmit the fluorescence emission while reflecting the laser beam; find this fluorescence signal on the other side of the dichroic mirror. Now, place a mirror behind the dichroic mirror and use it to reflect the emission at a 90-degree angle. Take an iris, as was done in step 1.1, and use it along with the mirror to direct the fluorescence beam as straight and parallel to the breadboard as possible. This step may be best carried out in dim light.
- Set up the confocal pinhole unit as described in Fig 2. We have found that the spatial filter cage mount assembly from ThorLabs is ideal for this task. It is important to select an appropriate pinhole size to ensure that the confocal system reaches its optimal resolution without sacrificing too much signal. For this custom microscope, a pinhole size of 100 μm was selected. Place the spatial filter unit in line with the fluorescence beam path, taking care to center the first focusing lens mount on the fluorescence emission beam. After mounting a short focal length lens in the unit (a microscope objective can also be used), slide the z-translation mount until a clear focus can be observed on the pinhole surface. Make sure the entire unit is oriented along the exact straight line set by the fluorescence beam. Clamp the unit to the breadboard.
The emission from most samples is weak compared to ambient light levels in even dark rooms. It is therefore crucial that adequate shielding/light baffling be used along the emission path to protect from stray light contamination. Furthermore, high ambient light levels will overload and damage many PMTs, particularly those with no current protection. Readers are therefore strongly urged to use lens tubes to enclose the emission beam path; a properly shielded system, like the one demonstrated here, is capable of operation in room light with little to no stray light contamination. - Now, using the adjustment knobs on the translation stage, systematically move the confocal pinhole to find the point where the fluorescence signal through the pinhole is maximized. This position is most easily identified through iterative adjustment of the two axes to perform a 2D search over the pinhole mount surface. Once the signal maximizing position is found, place the collimating lens on the cage mount after the pinhole. Find the fluorescence emission that goes through the confocal unit using a business card, and slide the collimating lens along the posts until the emitted fluorescence signal is as collimated as possible. Once the beam is collimated, be sure to place the appropriate filter in the beam path in a lens tube.
- Set up the photomultiplier tube (PMT) assembly. Place a 50 mm focal length lens in the fluorescence emission beam path and find its focal point using a business card. Mark this position on the breadboard. Now, turn off the laser completely – this is important, as stray or unattenuated laser light can permanently damage most PMTs. Position the PMT so that its active area is located as close to the marked focal point as possible. Connect the PMT assembly to the focusing lens using adjustable lens tubes, and carefully wrap dark tape around all exposed beam paths following the pinhole.
- Turn on the laser, but be sure to keep its power extremely low such that the fluorescence emission is barely visible. Turn on the PMT, carefully reading out its voltage on an oscilloscope as the control voltage is increased. A PMT generates signal through a series of electron multiplying stages; if the photocurrent is too high for the incident light level, the tube can be irreversibly damaged. PMTs with current limiting circuitry are therefore highly recommended, especially for users who have not worked with such detectors before.
Increase the PMT control voltage until a spike-like readout and/or a DC offset can be seen on the oscilloscope screen; for most PMTs, this signal will be negative relative to ground. Confirm that this signal indeed arises from the fluorescence by turning the laser power off to observe a loss of signal. - Finally, iteratively align the pinhole for maximum signal on the oscilloscope by first manipulating the focusing lens z-position, and then adjusting the yz translation stage.
- The video-rate microscope hardware is complete! Now hook up the mirrors, custom control boards, and computer as diagrammed in Fig 3. As above, it is recommended to use the imaging system to first visualize a known size standard to find the optimal resolution of the microscope and calculate the pixel-to-resolution constant for the imaging system. There are many size standards that can be used, such as white business cards with well-known letter sizes, fluorescence or reflective air force targets, and fluorescent microspheres.
5. Preparing the system for confocal scanning microendoscopy
In this build we use a coherent image fiber, which consists of a bundle of many thousands of fiber cores; such an arrangement allows an image to be transmitted through the fiber and easily reconstructed and/or expanded at the other end (Fig 4). The coherent fiber bundle used in the construction of this endoscope is polished at both ends, making it a so-called “contact-mode” microendoscope. An in-focus image will therefore only be formed when the microendoscope tip is brought in close contact with an object. In this pseudo-confocal arrangement, the microscope’s scanning action focuses the laser on one fiber core at a time, while the confocal pinhole ensures that no out-of-focus light from the surrounding fibers is allowed to pass through to the detector. For different imaging applications, a set of lenses can be added on the distal tip to allow for forward-facing, long-distance fluorescence imaging. Microoptic lenses, as well as gradient refractive index (GRIN) lenses can easily be adapted for this use, and can be affixed to the distal fiber tip using optical quality glues.
- To set up the imaging system for microendoscopy, carefully remove the sample stage and replace it with a fiber holding stage (Fig 5). Dip one end of the fiber bundle in a weak solution of dye so that fluorescence emission is generated evenly across all of the fiber cores. Turn on the scanning system and adjust the fiber holder to bring the other end of the fiber bundle (the proximal end, or the end nearest to the microscope optics) in focus. First, use translation adjustment screws to center the fiber in the scanned field. Now, look at the fluorescence emission image from the proximal fiber tip while scanning. When the complete microendoscope surface is in the focal plane of the objective, the fluorescence emission across all fiber cores will be as uniform as possible. Use the angle adjustment knobs to adjust the fiber face to make all fiber cores evenly bright. During this adjustment, it will likely be necessary to re-adjust the translation position to re-center the fiber in the scanned field. Iterate through these adjustments until the entire fiber tip is correctly in focus.
- Before using the microendoscope, gently clean the distal tip using lens cleaning paper slightly wetted with HPLC-grade methanol. As before, use a known size standard to measure and calculate the resolution of the microendoscope imaging system.
6. Representative Results:
Figure 6 shows an example of a finished upright confocal scanning microscope configured for microendoscopy. The laser and emission beams have been drawn as a guide to the eye. A fiber mount holds the image fiber in place during microendoscopy operation. This fiber mount can be readily replaced with a xy or xyz translation stage for use as an upright microscope platform. ThorLabs parts PT3 (XYZ translation) or two stacked PT1 stages (XY translation) work well for this application, along with a right-angle bracket such as ThorLabs part AP90.
A video-rate framegrabber card is used to generate images from the incoming signal. Figure 7 shows a representative test image taken of a lower-case "m" printed on a white business card using the video-rate microscope scanning system. Bleached white paper contains fluorophores that are excited by UV and blue light, resulting in the bright background behind dark letter "m". An emission filter centered at 515 nm was chosen to collect this fluorescent emission. A minor distortion of the image can be observed, especially near the lateral edges of the image frame. This distortion results from the sinusoidal scanning pattern of the 8kHz gavlo mirror, and will be discussed in detail below.

Figure 1. Diagram demonstrating the operating principle of a confocal microscope. Rays originating from the objective focus are relayed back through the system and focused through the confocal pinhole (red). Rays originating either above (blue) or below (green) the objective focus do not emerge from the objective collimated, and therefore are not efficiently transmitted through the confocal pinhole.

Figure 2. Diagram showing all light paths through the beam scanning system. The scanning mirrors sit at planes telecentric with the stationary, objective back aperture plane. Pairs of lenses between the stationary planes act to relay the scanned beams. The first two relay lenses have equal focal lengths, forming a 1:1 telescope. The second pair of lenses, known formally as the scan lens and tube lens, do not need to be equal in focal length, and often serve as a beam expanding telescope to ensure the objective back-aperture is overfilled. Light emitted from the sample travels back through the scanning system and is passed through the dichroic mirror. A short focus lens focuses the emission light through the confocal pinhole, which is then collimated by a lens. A final lens focuses the confocal-filtered emission onto a photomultiplier tube. Click here to view a full-sized version of this image.


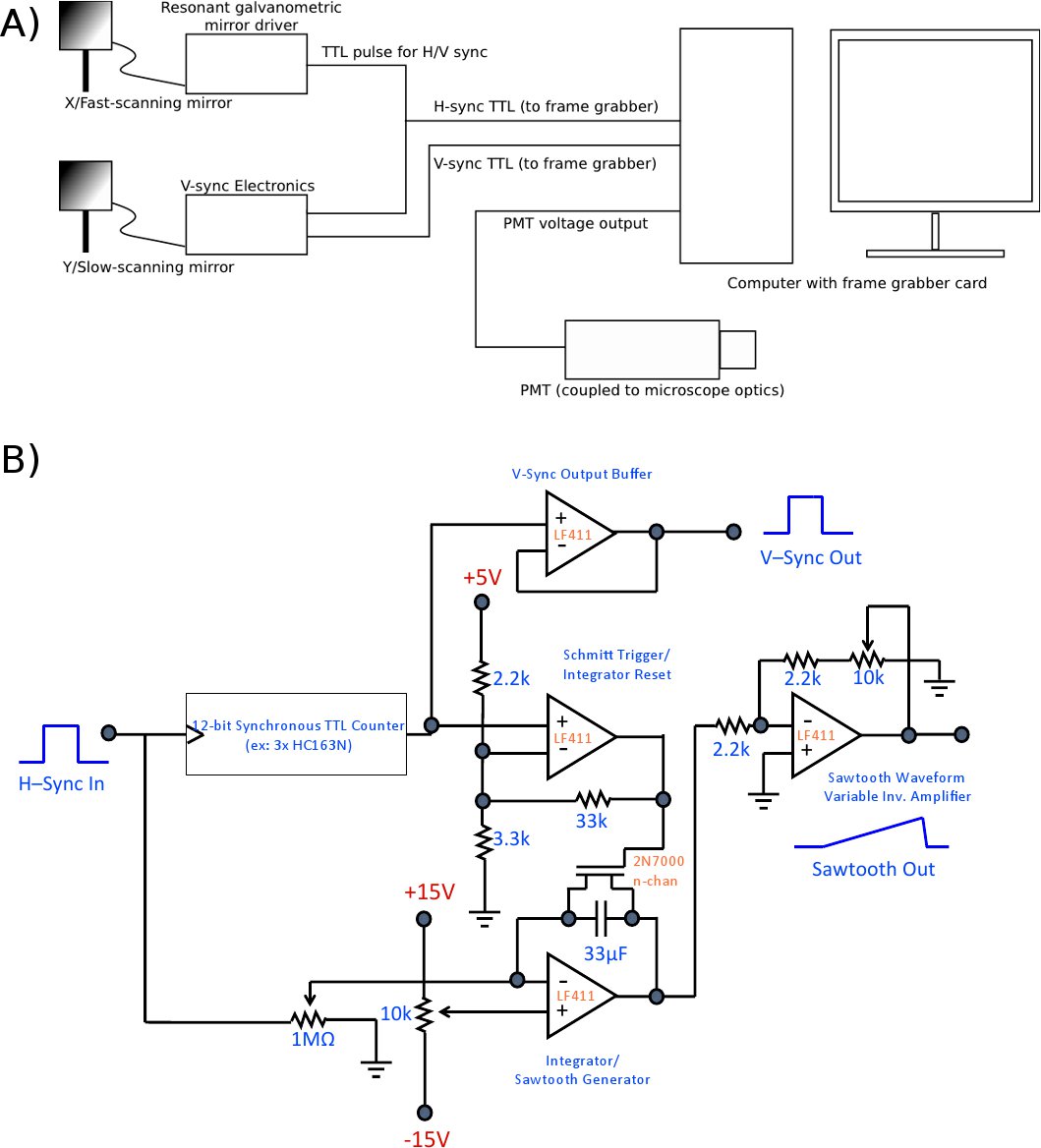
Figure 3. (a) Overview diagram of the scanning electronics setup. The microscope’s overall reference signal and timebase is the “sync” TTL output of the fast axis resonant galvo mirror, which generates a TTL pulse at the end of each line scanned (i.e., when the galvo has completed a scan cycle). This provides the H-sync signal to the framegrabber card. The galvo’s sync output is also connected to the V-sync control board, which incrementally increases its output voltage in response to each H-sync pulse to generate the sawtooth waveform that drives the slow scanning axis. Once all lines have been scanned, the V-sync board resets the sawtooth waveform and generates a TTL pulse that serves as the framegrabber’s V-sync signal. The final input to the framegrabber card is the analog signal from the photomultiplier tube (Note that many PMTs generate negative output voltage; be sure to design your circuit and choose your hardware accordingly). The video-rate images are generated and displayed in the Matrox framegrabber software. (b) Example control circuit. In this design, the voltage of each H-Sync pulse is “added”/integrated at the op amp integrator to generate the sawtooth waveform ramp; pulses are concomitantly counted at the TTL counter stage. When the desired number of lines has been reached (i.e., when the raster scan is complete), the counter generates an active-low “carry out” pulse, which drives the Schmitt trigger to generate a reset pulse for the integrator. This resets both the counter and the op amp integrator, readying the circuit for the next cycle. Appropriate component choice makes this circuit widely applicable to a variety of raster sizes. This is only one implementation; numerous other implementations are possible and may be preferred under certain circumstances. Also, this circuit is designed for use with Matrox framegrabber cards, which detect and correct image phase automatically. If the circuit is to be used with other framegrabbers, phase correction circuitry or software may be required. Click here to view a full-sized version of this image.

Figure 4. Image transmission through a coherent fiber bundle. In this schematic, the lenses on either side of the bundle are in place to scale both the image projected onto the fiber bundle input as well as expand the image on the fiber bundle output.

Figure 5. Example of a fiber bundle mounted in a 5-axis mount. A small 1” diameter aluminum block was bored so that the image fiber bundle could be inserted. The fiber was epoxied inside the aluminum block at both the top and bottom of the block for stability.

Figure 6. Image of the completed microscopy system with the microendoscope attached. To better visualize the light paths, the excitation beam path is drawn in blue, while the emission beam path after the dichroic mirror is drawn as a red line.

Figure 7. Example image generated by the video-rate confocal scanning microscopy system. A dark lower case letter "m" appears on the bright fluorescence background of a white business card.
Discussion
This video-rate imaging system makes use of a resonant galvanometric mirror operating at about 8 kHz. Resonant mirrors can be quite loud when run at full power, and their high pitch can be bothersome or even dangerous at sufficient exposure times. Though not demonstrated here, it is recommended to shield the resonant galvanometric mirror inside a transparent case to significantly reduce the system volume and/or to wear appropriate hearing protective gear, such as earplugs.
The resonant galvanometric mirror scans in a sinusoidal pattern. However, framegrabber cards read in signal assuming a completely linear sweep rate in both the horizontal and vertical directions. Since a sinusoidal sweep slows down at the edges of the scan, image compression artifacts can be observed along the fast (horizontal) image axis. One way to minimize this problem is to purposely drive the resonant galvo mirror scanning range significantly larger than the relay lens diameter. In doing this, only the nearly linear central sweep of the sinusoidal scan pattern will traverse the sample, minimizing image distortions. Another approach would be to post-process collected images to linearize the fast axis. This can be accomplished by imaging a known fluorescent pattern (such as a grid) and using the known pattern dimensions to create a processing script that unwarps the collected images.
This particular scanning system was designed for the purpose of in vivo imaging, which often requires an upright oriented video-rate microscope. For cellular imaging experiments, inverted microscopes are more typically used. The design presented here can be easily changed to build such an inverted microscope; all that is required is a rotation of the final 2” diameter mirror. Instead of orienting the mirror to direct the scanning beam downward, the mirror can direct the beam upward. Placing the objective lens at the same distance from the mirror along with a sample stage would allow for imaging in an inverted geometry. If the imaging system is being built solely for microendoscopic imaging, there is no reason to “fold” the microscope design vertically at all. Instead, the entire scanning system can be built on a single horizontal breadboard with the objective lens oriented parallel to the optical table.
Note that the microscope in this build uses a fixed pinhole configuration; while this provides for the greatest build simplicity and ease of alignment, users desiring a more versatile system might consider incorporating a variable pinhole, as can be found in most commercial confocal microscopes. By allowing the user to adjust the size of the pinhole to compensate for samples of varying emission intensity, this allows the user to better optimize the tradeoff between signal strength and resolution for a given sample.
The choice of image fiber selected for the microscope is important. We recommend using Sumitomo coherent image fibers due to their close fiber core spacing and low relative autofluorescence. Image fibers manufactured by Fujikura have been found to have high amounts of autofluorescence10, which can overwhelm weak fluorescence signals from a sample and limit the ultimate sensitivity of the microendoscope. Sumitomo manufactured fibers, such as the 8-30N used in this particular setup, have much lower autofluorescence levels than their Fujikura equivalents. While leeched fiber bundles might be considered attractive for microendoscopy, their design typically places individual fiber cores too far apart, meaning that the fiber cores sparsely sample objects, leaving out significant regions of potential interest.
Finally, it should be noted that while the microscope described here will be useful in a variety of in vitro and in vivo applications and can be created for a fraction of the cost of a full-featured commercial system, it does not have features such as transmitted light detection, an eyepiece for viewing, or a beam path for non-confocal widefield epifluorescence. While it is possible to construct a system with these features from scratch, readers desiring such a system may wish to modify an existing commercial system to meet their needs rather than initiate an entirely new build.
Disclosures
Production of this video-this video was sponsored by Thorlabs, Inc.
Acknowledgments
The authors would like to thank ThorLabs for their support of this project. AJN wishes to acknowledge the support of an NSF Graduate Fellowship.
This work was partially funded by the National Institutes of Health through the NIH Director's New Innovator Award Program, grant number 1 DP2 OD007096-01. Information on the New Innovator Award Program is at http://nihroadmap.nih.gov/newinnovator/. The authors would like to thank Tom Hayes for use of the Harvard Electronics lab.
Materials
| Name | Company | Catalog Number | Comments |
| 515 nm Band Pass Filter | Chroma Technology Corp. | HQ515/50M | 46 FWHM |
| Achromatic Doublet Lens 25.4mm Dia. x 50mm FL, MgF2 Coating | Edmund Scientific | NT49-766 | |
| Achromatic Doublet Lens 25.4mm Dia. x 76.2mm FL, MgF2 Coating | Edmund Scientific | NT49-768 | |
| Achromatic Doublet Lens 25.4mm Dia. x 88.9mm FL, MgF2 Coating | Edmund Scientific | NT49-769 | |
| Achromatic Doublet Lens 50mm Dia. x 300mm FL, MgF2 Coating | Edmund Scientific | NT45-179 | |
| 8 kHz R High Frequency Optical Scanner | Electro-Optical Products Corporation (EOPC) | SC-30 | 8 kHz |
| AGC Driver | Electro-Optical Products Corporation (EOPC) | ACG:8K | |
| H7422-PA Photosensor Module | Hamamatsu Corp. | H7422-PA | Current limiting recommended |
| M9012 Power Supply | Hamamatsu Corp. | M9012 | For use with H7422-PA |
| HC PL APO CS Objective | Leica Microsystems | 11506284 | 10x/0.40 |
| Solios eA/XA Framegrabber Card | Matrox | Solios eA/XA | MIL software required; –M interconnects recommended |
| 12V Power Supply | Meanwell | LPV-100-12 | +12V, 8.5A |
| 5x Microscope Objective Lens | Newport Corp. | M-5X | 0.10 NA, 25.4 mm Focal Length |
| Coherent Image Fiber | Sumitomo Bakelite Co., Ltd. | 8-30N | |
| 1/4"-20 Cap Screw and Hardware Kit | Thorlabs Inc. | HW-KIT2 | |
| 100 μm Mounted Pinhole | Thorlabs Inc. | P100S | Ideal for building spatial filters |
| 30 mm Cage Cube Clamp | Thorlabs Inc. | B6C | |
| 30 mm Cage System Cube, 4-Way | Thorlabs Inc. | C4W | |
| 406 nm, 5 mW, B Pin Code, SM Fiber Pigtailed Laser Diode, FC/PC | Thorlabs Inc. | LPS-406-FC | Product obsolete; replaced by LP405-SF10 |
| 5-Minute Epoxy, 1 Ounce | Thorlabs Inc. | G14250 | |
| 6 Axis Kinematic Optic Mount | Thorlabs Inc. | K6X | |
| 8-32 Cap Screw and Hardware Kit | Thorlabs Inc. | HW-KIT1 | |
| 8-32 Setscrew and Hardware Kit | Thorlabs Inc. | HW-KIT3 | |
| Adapter with External RMS Threads and Internal SM1 Threads | Thorlabs Inc. | SM1A4 | |
| Adj. FC/PC and FC/APC Collimator, f = 2.0 mm, ARC: 400-600 nm | Thorlabs Inc. | CFC-2X-A | f = 2.0 mm |
| Adjustable Fiber Collimator Adapter, SM1 Threaded | Thorlabs Inc. | AD9.5F | |
| Aluminum Breadboard, 12" x 18" x 1/2" | Thorlabs Inc. | MB1218 | 1/4"-20 Threaded |
| Benchtop Laser Diode/TEC Controller | Thorlabs Inc. | ITC4001 | 1 A/96 W |
| DMLP 425 nm Long-Pass Dichroic Mirror | Thorlabs Inc. | DMLP425 | |
| Kinematic Mount for 1" Optics | Thorlabs Inc. | KM100 | |
| LD/TEC Mount for ThorLabs Fiber-Pigtailed Laser Diodes | Thorlabs Inc. | LM9LP | |
| Lens Mount for 18 mm Optics | Thorlabs Inc. | LMR18 | One retaining ring included |
| Lens Mounts for 2" Optics | Thorlabs Inc. | LMR2S | With internal and external threading; retainer ring included |
| Mini Series Cage Assembly Rod, 6" Long, 4 mm, Qty. 1 | Thorlabs Inc. | SR6 | |
| 1.0" Pedestal Pillar Post, 8-32 Taps, 1" Long | Thorlabs Inc. | RS1P8E | |
| 1" Pillar Post Extension, Length=0.5 | Thorlabs Inc. | RS05 | |
| 1" Pillar Post Extension, Length=0.75" | Thorlabs Inc. | RS075 | |
| 1" Protected Silver Mirror, 3.2 mm Thick | Thorlabs Inc. | ME1-P01 | |
| 1" SM1 Rotating Adjustable Focusing Element, L = 1" | Thorlabs Inc. | SM1V10 | |
| 2" Protected Silver Mirror, 3.2 mm Thick | Thorlabs Inc. | ME2-P01 | |
| P100S - 100 μm Mounted Pinhole | Thorlabs Inc. | P100S | |
| Polaris Low Drift 1" Kinematic Mirror Mount | Thorlabs Inc. | POLARIS-K1 | Low drift |
| SM1 Lens Tube, L = 1" | Thorlabs Inc. | SM1L-10 | One retaining ring included |
| SM1 Threaded 30 mm Cage Plate, 0.35" Thick | Thorlabs Inc. | CP02 | |
| SM1 to M25 Optical Component Threading Adaptor | Thorlabs Inc. | SM1A24 | External SM1 Threads and Internal M25.5x0.5 Threads |
| Small Beam Diameter Galvo System | Thorlabs Inc. | GVSM001 | |
| Small Clamping Fork | Thorlabs Inc. | CF125 | 1/25" counterbored slot, universal |
| Spatial Filter System | Thorlabs Inc. | KT310 | Pinhole sold separately |
| TE-Cooled Mount for 5.6 & 9 mm Lasers | Thorlabs Inc. | TCLDM9 | |
| Vertical Bracket for Breadboards | Thorlabs Inc. | VB01 | Each |
| Plan-Apochromat | Carl Zeiss, Inc. | 1101-957 | 20x/0.75 NA |
References
- Pawley, J. B. Handbook of biological confocal microscopy. , Springer Verlag. 985-985 (2006).
- Lippincott-Schwartz, J., Snapp, E., Kenworthy, A. Studying protein dynamics in living cells. Nature Reviews Molecular Cell Biology. 2, 444-456 (2001).
- Klonis, N., Rug, M., Harper, I., Wickham, M., Cowman, A., Tilley, L. Fluorescence photobleaching analysis for the study of cellular dynamics. European Biophysics Journal. 31, 36-51 (2002).
- Stephens, D. J. Light Microscopy Techniques for Live Cell Imaging. Science. 300, 82-86 (2003).
- McMahon, A., Supatto, W., Fraser, S. E., Stathopoulos, A. Dynamic Analyses of Drosophila Gastrulation Provide Insights into Collective Cell Migration. Science. 322, 1546-1550 (2008).
- Wallingford, J. B. Dishevelled controls cell polarity during Xenopus gastrulation. Nature. 405, 81-85 (2000).
- Laemmel, E. Fibered Confocal Fluorescence Microscopy (Cell-viZio) Facilitates Extended Imaging in the Field of Microcirculation. Journal of Vascular Research. 41, 400-411 (2004).
- Moussata, D. The confocal laser endomicroscopy. Acta Endoscopica. 39, 448-451 (2010).
- Dunbar, K., Canto, M. Confocal endomicroscopy. Current Opinion in Gastroenterology. 24, 631-637 (2008).
- Udovich, J. A. Spectral background and transmission characteristics of fiber optic imaging bundles. Applied optics. 47, 4560-4568 (2008).

{kind=link}
{kind=link}