

Summary
We describe herein an assay by coupling DNA adenine methyltransferase identification (DamID) to high throughput sequencing (DamID-seq). This improved method provides a higher resolution and a wider dynamic range, and allows analyzing DamID-seq data in conjunction with other high throughput sequencing data such as ChIP-seq, RNA-seq, etc.
Abstract
The DNA adenine methyltransferase identification (DamID) assay is a powerful method to detect protein-DNA interactions both locally and genome-wide. It is an alternative approach to chromatin immunoprecipitation (ChIP). An expressed fusion protein consisting of the protein of interest and the E. coli DNA adenine methyltransferase can methylate the adenine base in GATC motifs near the sites of protein-DNA interactions. Adenine-methylated DNA fragments can then be specifically amplified and detected. The original DamID assay detects the genomic locations of methylated DNA fragments by hybridization to DNA microarrays, which is limited by the availability of microarrays and the density of predetermined probes. In this paper, we report the detailed protocol of integrating high throughput DNA sequencing into DamID (DamID-seq). The large number of short reads generated from DamID-seq enables detecting and localizing protein-DNA interactions genome-wide with high precision and sensitivity. We have used the DamID-seq assay to study genome-nuclear lamina (NL) interactions in mammalian cells, and have noticed that DamID-seq provides a high resolution and a wide dynamic range in detecting genome-NL interactions. The DamID-seq approach enables probing NL associations within gene structures and allows comparing genome-NL interaction maps with other functional genomic data, such as ChIP-seq and RNA-seq.
Introduction
DNA adenine methyltransferase identification (DamID) 1,2 is a method to detect protein-DNA interactions in vivo and is an alternative approach to chromatin immunoprecipitation (ChIP) 3. It uses a relatively low amount of cells and does not require chemical cross-linking of protein with DNA or a highly specific antibody. The latter is particularly helpful when the target protein is loosely or indirectly associated with DNA. DamID has been successfully used to map the binding sites of a variety of proteins including nuclear envelope proteins 4-10, chromatin associated proteins 11-13, chromatin modifying enzymes 14, transcription factors and co-factors15-18 and RNAi machineries 19. The method is applicable in multiple organisms including S. cerevisiae 13, S. pombe 7, C. elegans 9,17, D. melanogaster 5,11,18,20, A. thaliana 21,22 as well as mouse and human cell lines 6,8,10,23,24.
The development of the DamID assay was based on the specific detection of adenine-methylated DNA fragments in eukaryotic cells that lack endogenous adenine methylation 2. An expressed fusion protein, consisting of the DNA-binding protein of interest and E. coli DNA adenine methyltransferase (Dam), can methylate the adenine base in GATC sequences that are in spatial proximity (most significantly within 1 kb and up to roughly 5 kb) to the binding sites of the protein in the genome 2. The modified DNA fragments can be specifically amplified and hybridized to microarrays to detect the genomic binding sites of the protein of interest 1,25,26. This original DamID method was limited by the availability of microarrays and the density of predetermined probes. We have therefore integrated high throughput sequencing into DamID 10 and designated the method as DamID-seq. The large number of short reads generated from DamID-seq enables precise localization of protein-DNA interactions genome-wide. We found that DamID-seq provided a higher resolution and a wider dynamic range than DamID by microarray for studying genome-nuclear lamina (NL) associations 10. This improved method allows probing NL associations within gene structures 10 and facilitates comparisons with other high throughput sequencing data, such as ChIP-seq and RNA-seq.
The DamID-seq protocol described here was initially developed for mapping genome-NL associations 10. We generated a fusion protein by tethering mouse or human Lamin B1 to E. coli DNA adenine methyltransferase and tested the protocol in 3T3 mouse embryonic fibroblasts, C2C12 mouse myoblasts 10 and IMR90 human fetal lung fibroblasts (data not published). In this protocol, we start with constructing vectors and expressing Dam-tethered fusion proteins by lentiviral infection in mammalian cells 24. Next, we describe the detailed protocols of amplifying adenine-methylated DNA fragments and preparing sequencing libraries that should be applicable in other organisms.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Generation and Expression of Fusion Proteins and Free Dam Proteins
- Clone protein of interest into the DamID vector.
- Amplify cDNA of protein of interest (POI) using the desired high fidelity DNA polymerase and appropriate primers according to the manufacturer's protocol. Experimentally determine optimal amplification conditions to ensure proper amplification of inserts.
- Run an agarose gel and purify amplified cDNA of POI by the gel extraction kit according to the manufacturer's protocol.
- Clone cDNA of POI into the pDONR201 vector using BP Clonase II according to the manufacturer's protocol.
- Clone the cDNA of POI from the donor vector into the pLGW-RFC1-V5-EcoDam destination vector or the pLGW-EcoDam-V5-RFC1 destination vector 27 using LR Clonase II according to the manufacturer's protocol, depending on the desired direction of fusing POI to the N-terminus or the C-terminus of the E. coli DNA adenine methyltransferase (EcoDam) 27.
- Verify by sequencing that the cloned cDNA has a correct sequence and forms in-frame fusion to EcoDam.
- Generate lentiviral stocks.
- Generate lentiviral stocks expressing Dam-V5-POI and V5-Dam (from the pLGW-V5-EcoDam vector 27) using lentiviral expression systems. Use the forward transfection procedure according to the manufacturer's protocol.
- Use 293T cells and lipofection to generate lentiviral stocks according to the manufacturer's protocol for transfection.
- Include the 0.45 µm PVDF filter step.
- Generate lentiviral stocks expressing Dam-V5-POI and V5-Dam (from the pLGW-V5-EcoDam vector 27) using lentiviral expression systems. Use the forward transfection procedure according to the manufacturer's protocol.
- Infect cells with lentivirus.
- The day before infection (Day 0), pass adherent cells cultured in appropriate growth media for this cell type to a 6-well tissue culture plate using the same growth media without antibiotics to achieve 50% confluence on the day of infection. Place cells in a 37 °C incubator.
- On the day of infection (Day 1), remove 2 cryovials of both Dam-V5-POI and V5-Dam lentiviral supernatants from a -80 °C freezer and place in a 37 °C water bath to thaw.
- Remove growth media from cells and replace with 0.5 ml of fresh growth media without antibiotics.
- Add 1 ml of the thawed lentivirus to each well (2 wells with V5-Dam and 2 wells with Dam-V5-POI). Add 1 ml of growth media without antibiotics to the remaining 2 wells-this will serve as a negative control. Gently shake the 6-well plate to mix and place back in a 37 °C incubator O/N.
- The day after infection (Day 2), remove viral suspensions from cells and replace with 2 ml growth media without antibiotics. Place cells back in a 37 °C incubator for 48 hr.
- Isolate gDNA.
- Aspirate media from each well and detach cells using 250 µl 0.05% trypsin-EDTA. Incubate at 37 °C for 2 min.
- Wash cells off the plate with 1 ml growth media and pipette cells from each well into a 1.5 ml microcentrifuge tube. Pellet cells by centrifuging at 200 x g for 5 min at RT.
- Wash pelleted cells with 500 µl PBS and centrifuge at 200 x g for 2 min at RT.
- Resuspend pelleted cells in 200 µl PBS.
- Isolate gDNA by blood and tissue kit according to the manufacturer's protocol. Elute gDNA in 200 µl buffer AE and determine the concentration by measuring OD260 using a spectrophotometer.
Note: gDNA from uninfected or mock infected cells can be isolated as negative controls. Precipitate gDNA to higher concentrations for long-term storage.- Add 3 volumes of 100% ethanol and 0.1 volume of 3 M sodium acetate (pH 5.5), and mix by inverting tubes 4-6 times.
- Store at -20 °C O/N.
- Centrifuge at 16,000 x g for 15 min at 4 °C.
- Carefully remove the supernatant. Wash pellets with 70% (volume/volume) ethanol and centrifuge at 16,000 x g for 5 min at 4 °C.
- Carefully remove ethanol and allow pellets to air dry for 3 min at RT.
- Dissolve gDNA in T10E0.1 (pH 7.5) to approximately 1 µg/µl. Pool gDNA from each experimental sample or the negative control and measure the concentration. Store at -20 °C.
2. Amplify Adenine-methylated DNA Fragments
- Digest gDNA with DpnI that only cuts adenine-methylated GATCs.
- Set up a reaction on ice with 2.5 µg gDNA, 1 µl 10x buffer, 0.5 µl DpnI (20 U/µl) and fill with H2O to a total volume of 10 µl. For each gDNA sample, prepare three reactions — one without DpnI ("no DpnI", replace DpnI with 0.5 µl H2O) and two with DpnI ("with DpnI").
- Digest O/N at 37 °C and inactivate DpnI at 80 °C for 20 min.
- Ligate DamID adaptors.
- Prepare DamID adaptors.
- Resuspend each of the two DamID adaptor oligos 24 in H2O to a final concentration of 100 µM.
- Combine equal volumes of the two DamID adaptor oligos, mix and place in a tightly closed tube. Seal the tube with Parafilm, sit in a rack and place in a beaker filled with water at 90 °C. Keep the water level below the cap of the tube (to avoid water getting into the tube) but above the surface of the oligo mix.
- Let the water cool to RT so the adaptors anneal slowly.
- Aliquot the annealed adaptors (50 µM) and store at -20 °C.
- Set up a reaction on ice. In each tube from 2.1.2, add 6.2 µl H2O, 2 µl 10x ligation buffer, 0.8 µl 50 µM DamID adaptors (thawed on ice) and 1 µl T4 DNA ligase (5 U/µl). The total volume is 20 µl. In one of the two "with DpnI" tubes, replace ligase with 1 µl H2O ("no ligase"). Note that each gDNA sample has two negative controls — " no DpnI " and " no ligase".
- Ligate O/N at 16 °C and inactivate ligase at 65 °C for 10 min.
- Prepare DamID adaptors.
- Digest DNA with DpnII to destroy fragments that contain unmethylated GATCs.
- Set up a reaction on ice. In each tube from 2.2.3, add 24 µl H2O, 5 µl 10x DpnII buffer and 1 µl DpnII (10 U/µl). The total volume is 50 µl.
- Digest at 37 °C for 2-3 hr and inactivate DpnII at 65 °C for 20 min.
- Amplify adenine-methylated DNA fragments.
- Set up a reaction on ice with 5 µl DpnII digest from 2.3.2, 5 µl 10x PCR buffer, 12.5 µl 5 µM DamID PCR primer 24, 4 µl 10 mM dNTP mix, 1 µl 50x polymerase mix and 22.5 µl H2O. The total volume is 50 µl.
- Run the PCR as follows: 68 °C for 10 min; 94 °C for 1 min, 65 °C for 5 min, 68 °C for 15 min; 4 cycles of 94 °C for 1 min, 65 °C for 1 min, 68 °C for 10 min; 17 cycles of 94 °C for 1 min, 65 °C for 1 min, 68 °C for 2 min.
- Analyze 5 µl PCR products from each reaction on a 1% agarose gel. PCR products should appear as a smear ranging from 0.2 to 2 kb (Figure 2). The "no DpnI" and "no ligase" controls should have no or clearly less amplification.
- If the result from step 2.4.3 is satisfactory, repeat steps 2.4.1-2.4.3 with two or three reactions for the experimental sample and one reaction for each of the two negative controls.
- Pool and purify PCR products from the same experimental sample using PCR purification kits or Solid Phase Reversible Immobilization (SPRI) beads according to the manufacturer's protocol. Do not purify "no DpnI" or "no ligase" controls. Elute DNA with buffer EB.
- Measure the concentration of the purified DNA by measuring OD260 using a spectrophotometer, which should be around 0.1 µg/µl or higher. Collect a minimum of 10 µg DNA for each sample. If using PCR purification kits, purify each 50 µl PCR products with one column, elute in 30 µl buffer EB and pool the eluates.
- Digest DNA with DpnII to prevent DamID PCR primers from being sequenced.
- Set up a reaction on ice with 5 µg purified DNA from 2.4.6, 5 µl 10x DpnII buffer, 1 µl DpnII (10 U/µl) and fill with H2O to a total volume of 50 µl. Prepare two or three reactions for each sample.
- Digest at 37 °C for 2-3 hr and inactivate DpnII at 65 °C for 20 min.
- Pool and purify the digests from the same sample with PCR purification kits or SPRI beads according to the manufacturer's protocol. Elute DNA with buffer EB.
- Measure the concentration of the purified DNA which should be around 0.06 µg/µl or higher. Collect a minimum of 6 µg DNA for each sample. If using PCR purification kits, purify each 50 µl digest with one column, elute in 30 µl buffer EB and pool the eluates.
3. Library Preparation for High-throughput Sequencing
- Fragment DNA
- Experimentally determine the appropriate digestion time for each new batch of dsDNA Fragmentase. As the enzyme activity may decrease over time, repeat the test before performing a new experiment. To save DNA from 2.5.4 for the actual fragmentation, use purified methyl PCR products from 2.4.6 or extra DNA from previous experiments at this step.
- Set up a master mix with 6 µg DNA, 12 µl 10x Fragmentase buffer and fill with H2O to a total volume of 114 µl.
- Vortex the Fragmentase stock vial for 3 sec, add 6 µl into the master mix and vortex the master mix for 3 sec. The total volume is 120 µl.
- Aliquot 20 µl of the master mix to each of 5 new tubes. Incubate all 6 tubes at 37 °C for 5 to 55 min at an increment of 10 min. Add 5 µl 0.5 M EDTA to stop the reaction.
- Analyze 12.5 µl digest (0.5 µg DNA) of each reaction as well as 0.5 µg undigested DNA on an agarose gel (Figure 3). Determine the minimal time (T0.2kb) needed to digest the majority of the smear to around 0.2 kb. Select 6 time durations between 5 min and T0.2kb (including 5 min and T0.2kb) with equal increments for the actual fragmentation.
- Set up the actual fragmentation as described in 3.1.1.1-3.1.1.3. Incubate reactions at 37 °C for the time durations determined in 3.1.1.4.
- Pool 6 reactions and purify the digests with PCR purification kits or SPRI beads according to the manufacturer's protocol. Elute DNA in 51 µl buffer EB. The final eluate is ~50 µl.
- Experimentally determine the appropriate digestion time for each new batch of dsDNA Fragmentase. As the enzyme activity may decrease over time, repeat the test before performing a new experiment. To save DNA from 2.5.4 for the actual fragmentation, use purified methyl PCR products from 2.4.6 or extra DNA from previous experiments at this step.
- Repair ends of the fragmented DNA
- Set up a reaction on ice with the eluate from 3.1.3, 25 µl H2O, 10 µl 10x T4 DNA ligase buffer with 10 mM ATP, 4 µl 10 mM dNTP mix, 5 µl T4 DNA polymerase (3 U/µl), 1 µl Klenow DNA polymerase (5 U/µl), and 5 µl T4 polynucleotide kinase. The total volume is 100 µl. Mix well with pipette. Avoid foam and bubbles.
- Incubate at 20 °C for 30 min.
- Purify the DNA with PCR purification kits or SPRI beads according to the manufacturer's protocol. Elute DNA in 33 µl buffer EB. The final eluate is ~32 µl.
- Add "A" overhangs
- Set up a reaction on ice with the eluate from 3.2.3, 5 µl 10x Klenow buffer, 10 µl 1 mM dNTP, and 3 µl Klenow (3'→5' exo-) (5 U/µl). The total volume is 50 µl. Mix well with pipette. Avoid foam and bubbles.
- Incubate at 37 °C for 30 min.
- Purify the DNA with PCR purification kits or SPRI beads according to the manufacturer's protocol. Elute DNA in 22 µl buffer EB. The final eluate is ~21 µl.
- Ligate sequencing adaptors
- Prepare sequencing adaptors 28.
- Resuspend each adaptor oligo to a concentration of 100 µM in 10 mM Tris-Cl (pH 7.8), 0.1 mM EDTA (pH 8.0) and 50 mM NaCl.
- Mix equal volumes of the universal adaptor 28 and an indexed adaptor 28.
- Anneal the adaptors in a thermal cycler with the following program: 95 °C for 2 min; 140 cycles for 30 sec starting at 95 °C and decreasing by 0.5 °C every cycle; hold at 4 °C.
- Aliquot adaptors and store at -20 °C.
- Set up a reaction on ice with the eluate from 3.3.3, 25 µl 2x ligation buffer, 1.5 µl 50 µM annealed sequencing adaptors (thawed on ice) from 3.4.1.4 and 2.5 µl T4 DNA ligase. Mix well with pipette. Avoid foam and bubbles. If multiplex sequencing is desired, use a different indexed adaptor for each sample.
- Incubate at RT for 1 hr.
- Purify the DNA with PCR purification kits or SPRI beads according to the manufacturer's protocol. Elute DNA in 24 µl buffer EB. The final eluate is ~23 µl.
- Prepare sequencing adaptors 28.
- Convert Y-shaped adaptors to dsDNA to enable accurately determining DNA fragment sizes 28
- Set up a reaction on ice with the eluate from 3.4.4, 12.5 µl H2O, 1 µl 25 µM primer 1 28, 1 µl 25 µM primer 2 28, 1.5 µl 10mM dNTP, 10 µl 5x PCR Buffer and 1 µl DNA polymerase (1 U/µl).
- Run the PCR as follows: 95 °C for 3 min; 5 cycles of 98 °C for 15 sec, 63 °C for 30 sec, 72 °C for 30 sec; 72 °C for 1 min; 4 °C on hold.
- Purify the DNA with PCR purification kits or SPRI beads according to the manufacturer's protocol. Elute DNA in 11 µl buffer EB. The final eluate is ~10 µl.
- Size select the library
- Prepare a 2% agarose gel with 1x TAE buffer. Add ethidium bromide (CAUTION!) to a final concentration of 500 ng/ml when the melted TAE-agarose solution has cooled to avoid the inhalation of ethidium bromide. Make sure to have enough lanes for all the samples, DNA ladder and empty lanes.
- Add 8 µl 6x loading dye to the eluate from 3.5.3.
- Prepare DNA ladder by mixing 1 kb plus DNA ladder (1.0 µg/µl), 6x loading dye and H2O in a ratio of 1:1:4.
- Load 6 µl DNA ladder, the samples from 3.6.2 and another 6 µl DNA ladder, each in a separate lane and with at least one empty lane from the adjacent samples/ladders.
- Run the gel at 120 V for 60 min.
- View the gel on a UV transilluminator (minimize the exposure time to UV). Wear safety glasses and a face shield. Make sure at least one of the DNA ladders run well with appropriate spacing (enough for excising 3 gel slices) between 300 bp and 400 bp bands. Narrow spacing increases the difficulties in excising multiple gel slices, while wide spacing increases the volume of excised gel slices.
- Use a new scalpel or a razor blade for each lane. Excise three thin gel slices between 300 and 400 bp from each lane (Figure 4) and place them each in a microcentrifuge tube. Keep the volume of each gel slice as low as possible (< 100 µl).
- Measure the volume of each gel slice (1 µl gel weighs approximately 1 mg), add 6x volumes of QG buffer and incubate at 50 °C.
- Vortex the QG-gel mixture every 2-3 min until the gel slice has completely dissolved. Add 2x gel volumes of isopropanol and mix.
- From this step, follow the protocol from gel extraction kits. Elute DNA in 51 µl buffer EB. The final eluate is ~50 µl.
- Enrich sequencing adaptor modified DNA fragments
- Optimize the number of PCR cycles 29.
- Set up a reaction on ice with 1 µl eluate from 3.6.10, 1 µl 25 µM primer 1 28, 1 µl 25 µM primer 2 28, 7 µl H2O and 10 µl SYBR Green Supermix. The total volume is 20 µl.
- Run the qPCR as follows: 95 °C for 3 min; 20 cycles of 95 °C for 30 sec, 63 °C for 30 sec, 72 °C for 30 sec, plate read.
- Analyze data using a qPCR analysis software and determine the Quantification Cycle (Cq) or Threshold Cycle (Ct) for each sample using the manufacturer's protocol. Continue with the samples that have Cq/Ct ≤ 14. Use the maximal Cq (Cq0) minus 1 (rounded up to the next higher integer) as the final PCR cycle number (NPCR).
- Adjust the quantities of DNA templates so that different samples will be amplified to approximately equal amounts after running the same number of PCR cycles. Use 8 µl template for the sample with the highest Cq (Cq0), and calculate the template volume of other samples with the following formula:
Voli = 8 x 1.8Cqi-Cq0
- Set up PCR reactions on ice with the template volumes calculated from 3.7.1.4: 1 µl 25 µM primer 1 28, 1 µl 25 µM primer 2 28, 1.5 µl 10 mM dNTP, 10 µl 5x buffer, 1 µl DNA polymerase (1 U/µl) and fill with H2O to a total volume of 50 µl.
- Run the PCR as follows: 95 °C for 45 sec; NPCR cycles (determined in 3.7.1.3) of 98 °C for 15 sec, 63 °C for 30 sec, 72 °C for 30 sec; 72 °C for 1 min; 4 °C on hold.
- Analyze 5 µl PCR products in a 2% agarose gel (Figure 5A). A clear "single" band indicates that the amplified DNA fragments are within a narrow length range and that the DNA library can be subject to further analysis.
- Repeat one reaction for selected samples and pool the amplified DNA libraries from the same sample.
- Purify selected DNA libraries for sequencing.
- If primer/adaptor dimers are not visible in 3.7.4, purify DNA libraries with PCR purification kits or SPRI beads according to the manufacturer's protocol. Elute DNA in 21 µl buffer EB. The final eluate is ~20 µl. If the user's sequencing facility has specific instructions on the elution buffer, the final volume, etc., prepare samples accordingly.
- If primer/adaptor dimers are clearly visible in 3.7.4, purify libraries as follows.
- Load DNA from 3.7.5 and DNA Ladder in a 2% precast agarose gel. Samples can be purified as described in 3.7.6.1 to reduce the volume or can be loaded into multiple wells. Place the agarose gel in an appropriate power system. Allow the gel to run for 15 min.
- Using an appropriate transilluminator, cut out the desired band with a clean scalpel/razor for each sample and place the gel slice in a 1.5 ml microcentrifuge tube.
- Isolate DNA using gel extraction kits according to the manufacturer's protocol with two modifications: incubate buffer QG-gel mixture in a thermo-mixer at 37 °C and 1,000 rpm for 30 min, and add 2x gel volumes of isopropanol.
- Optimize the number of PCR cycles 29.
- Submit purified DNA libraries to a sequencing facility. Follow all facility guidelines.
Note: A quality analysis by a Bioanalyzer (Figure 5B) should be performed prior to sequencing in order to determine the exact size range and the concentration of a DNA library.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
The Dam-V5-LmnB1 fusion protein was verified to be co-localized with the endogenous Lamin B protein by immunofluorescence staining (Figure 1).
The successful PCR amplification of adenine-methylated DNA fragments is a key step for DamID-seq. The experimental samples should amplify a smear of 0.2 - 2 kb while the negative controls (without DpnI, without ligase or without PCR template) should result in no-or clearly less-amplification (Figure 2).
The methylated DNA fragments are in the range from 0.2 to 2 kb, while the desired insert size for an NGS library is from 200 to 300 bp. Therefore, it is essential to fragment the methyl PCR products into the suitable size range. Nonetheless, it was found to be impractical to simultaneously break larger DNA fragments down to suitable sizes and keep the majority of smaller DNA fragments intact in a single fragmentation duration. Therefore, time course experiments were performed to determine the minimal time (T0.2kb) needed to fragment 1 µg DNA to a smear centered at 200 bp (Figure 3). Then 6 time durations in equal increments were selected between 5 min and T0.2kb for the actual fragmentation. The enzymatic activity of double strand DNA Fragmentase may vary from batch to batch and may decrease over time, so it is recommended to repeat this step for a new batch of Fragmentase or after storage for a period of time.
The desired insert size is between 200 and 300 bp corresponding to DNA fragments between 300 and 400 bp (including 121 bp sequencing adaptors) on the agarose gel. Three thin slices within this range were excised from each experimental sample to narrow the size range of a library and increase the possibility of obtaining at least one qualified sequencing library (Figure 4).
An aliquot of 5 µl of each amplified DNA library was analyzed on the agarose gel to determine which library may qualify for sequencing. As shown in Figure 5A, a clear single band of the same size as the excised gel slice should be visible on the agarose gel (step 3.7.4). Next, selected libraries were examined by a Bioanalyzer (Figure 5B) to determine the exact size range and concentrations prior to sequencing. If desired, amplified DNA libraries can be directly examined by a Bioanalyzer without gel analysis. When multiple libraries are of good quality, it is recommended to sequence libraries of similar size ranges for a pair of experimental (cells expressing Dam-V5-POI) and control (cells expressing V5-Dam) samples.
The short reads generated by sequencing systems were first mapped back to the corresponding genome. Uniquely aligned reads were then passed to subsequent analyses. A pipeline to process short reads, construct a genome-NL interaction map and analyze gene-NL associations were described in detail in our previous work 10. Representative results are shown in Figure 6.

Figure 1. Validating subnuclear localizations of fusion proteins by immunofluorescence staining. IMR90 human lung fibroblast cells were transiently transfected with a plasmid expressing Dam-V5-LmnB1 and were stained by anti-Lamin B (A, red) and anti-V5 (B, green). (C) Merge of images in A and B. Please click here to view a larger version of this figure.

Figure 2. PCR amplifying adenine-methylated DNA fragments. An aliquot of 5 µl from each PCR reaction was analyzed on a 1% agarose gel. A smear ranging from 0.2 to 2 kb was amplified from each experimental sample, but no amplification was observed in negative controls (no DpnI in step 2.1, no Ligase in step 2.2, or no PCR template). Please click here to view a larger version of this figure.

Figure 3. Determining optimal fragmentation durations. Purified methyl PCR products were subject to fragmentation for time durations from 5 to 55 min at an increment of 10 min (undigested DNA, labeled as "0 min"). 1 µg of DNA ladder and 0.5 µg of each fragmented DNA sample were analyzed on an agarose gel. The minimal time to digest the majority of the DNA smear to around 200 bp was determined to be around 35 min, therefore six evenly spaced time durations between 5 and 35 min (5 and 35 min included) were selected to perform the actual fragmentation. Please click here to view a larger version of this figure.

Figure 4. Size selecting the DNA libraries. DNA samples of Dam-V5-LmnB1 and V5-Dam from step 3.6 were run on a 2% agarose gel, and three equally sized gel slices (L, M and H corresponding to low, medium and high in size) were excised between 300 and 400 bp as shown by the yellow lines. Please click here to view a larger version of this figure.

Figure 5. Amplified DNA libraries for high throughput sequencing. (A) Amplified DNA libraries analyzed on an agarose gel. PCR templates were purified from gel slices shown on Figure 4. (B) Bioanalyzer results of the V5-Dam (L) sample and the Dam-V5-LmnB1 (L) sample that were underlined in (A). These two libraries had the similar narrow size ranges and thus qualified for high throughput sequencing. Please click here to view a larger version of this figure.
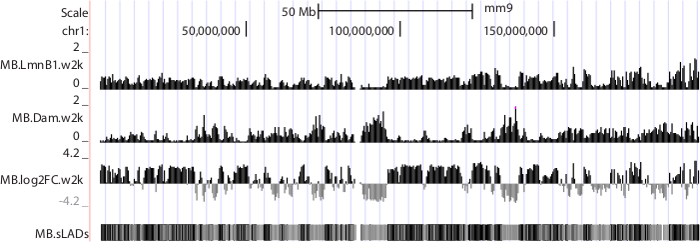
Figure 6. NGS data displayed in UCSC genome browser. Mouse chromosome 1 is shown as an example. Sequence data were produced from mouse C2C12 myoblasts 10. Tracks "MB.LmnB1.w2k" and "MB.Dam.w2k", corresponding to data from cells expressing Dam-V5-LmnB1 and V5-Dam respectively, plot normalized read densities (reads per kilobase per million uniquely mapped reads, or RPKM) in non-overlapping consecutive 2 kb windows along the chromosome. Track "MB.log2FC.w2k" plots genome-NL associations, i.e. log2 RPKM ratios of LmnB1 over Dam, at 2 kb resolution. Track "MB.sLADs" paints sequencing-based Lamina Associated Domains (sLADs, i.e. genomic regions that have higher read densities of LmnB1 over Dam with statistical significance) in black, non-sLADs in gray and undetermined regions in white. Please click here to download this file.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Whether Dam-tagged proteins retain the functions of endogenous proteins should be examined before a DamID-seq experiment. The subcellular localization of Dam-tagged nuclear envelope proteins should always be determined and compared with that of the endogenous proteins. For studying transcription factors, it is suggested to examine whether the Dam-fusion protein can rescue the functions of the endogenous protein in regulating gene expression. This functional test can be performed in organisms in which knockout mutants of endogenous DNA-binding proteins are available. Because advances in genome engineering have potentially allowed knocking out any endogenous gene of interest, functions of Dam-tagged DNA-binding proteins can be examined in cultured mammalian cells.
The critical step in this protocol is to successfully fragment the DpnII-digested DamID PCR products to around 200 bp. This step is designed to render the amplified adenine-methylated fragments to a narrow size range for sequencing and to randomize the starting nucleotides of the DNA fragments in a sequencing library. Inefficient fragmentation will leave the majority of the DNA fragments starting with GATC (the 5'-overhang from the second DpnII digestion), and will result in a much lower performance and yield or even a failure in Illumina sequencing. Other DNA fragmentation methods may be used as an alternative approach.
The resolution of DamID (and DamID-seq described here) is limited by the frequency of GATCs in the genome to be studied. Moreover, even with high throughput sequencing, the genomic localizations of a DNA-binding protein can only be mapped within two consecutive GATCs rather than to the actual DNA-binding sites.
Despite its limitation, the DamID assay has important advantages. Because DamID does not require highly-specific antibodies, it can be used to detect a subset of nuclear proteins that could be difficult to assay by ChIP (such as the nuclear envelope proteins). To study how these proteins regulate genome functions, it is important to integrate and cross-analyze their genome-wide localization data with the current epigenomic mapping data (such as data from the ENCODE and NIH Roadmap Epigenomics Projects 30,31). The DamID-seq approach provides both higher resolution and higher sensitivity than DamID by microarray and enables detecting differential NL-associations within gene structures 10. A combinatorial analysis of DamID-seq data, ChIP-seq data 32 and gene expression data has identified a class of NL-associated genes with distinct epigenetic and transcriptional features (data not published).
Another advantage of DamID is that it only requires a small number of cells. In recent years, there has been an explosion in single cell analysis of gene regulation 33,34. Although genome sequence 35, genome-wide gene expression 36 and chromatin conformation 37 can be assayed in a single cell, there has not been an available approach for detecting protein-DNA interactions genome-wide in a single cell. DamID-seq is a highly promising approach for this goal, and may complement the single cell imaging approach in detecting the dynamics of genome-NL interactions 38. One complication is that because the Dam-fusion protein is expressed at a much lower level than the endogenous protein in the DamID assay, it is possible that the Dam-fusion protein may only occupy a subset of genomic binding sites as compared to the endogenous protein.
DamID assay has mostly been used in cultured animal cells to detect protein-DNA interactions. Notably, developmental biologists have applied this assay in detecting protein-DNA interactions in specific cell types in vivo. For example, Dam-tagged RNA polymerase II was expressed specifically in Drosophila neural stem cells to detect their genome-wide occupancy without cell isolation 39. DamID-seq will be highly useful to study the genome-wide localizations of nuclear envelope proteins, transcription factors and chromatin regulators during development in animal models.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Dr. Bas van Steensel for providing the DamID mammalian expression vectors. We thank Yale Center for Genome Analysis and the Genomics Core in Yale Stem Cell Center for advice on preparing NGS libraries and implementing high throughput DNA sequencing. This work was supported by the startup funding from Yale School of Medicine, a Scientist Development Grant from American Heart Association (12SDG11630031) and a Seed Grant from Connecticut Innovations, Inc. (13-SCA-YALE-15).
Materials
| Name | Company | Catalog Number | Comments |
| ViraPower Lentiviral Expression Systems | Life Technologies | K4950-00, K4960-00, K4970-00, K4975-00, K4980-00, K4985-00, K4990-00, K367-20, K370-20, and K371-20 | |
| Gateway BP Clonase II Enzyme Mix | Life Technologies | 11789-020 | |
| Gateway LR Clonase II Enzyme Mix | Life Technologies | 11791-020 | |
| DNeasy Blood & Tissue Kit (250) | QIAGEN | 69506 or 69504 | |
| Gateway pDONR 201 | Life Technologies | 11798-014 | |
| 293T cells | American Type Culture Collection | CRL-11268 | |
| Trypsin-EDTA (0.05%), phenol red | Life Technologies | 25300-054 | |
| DMEM, high glucose, pyruvate | Life Technologies | 11995-065 | |
| Fetal Bovine Serum | Sigma | F4135 | |
| Tris | brand not critical | ||
| EDTA | brand not critical | ||
| 200 Proof EtOH | brand not critical | ||
| Isopropanol | brand not critical | ||
| Sodium Acetate | brand not critical | ||
| DpnI | New England Biolabs | R0176 | supplied with buffer |
| DamID adaptors "AdRt" and "AdRb" | Integrated DNA Technologies | sequences available in ref. 24; no phosphorylation of the 5' or 3' end to prevent self-ligation. | |
| T4 DNA Ligase | Roche Life Science | 10481220001 | supplied with buffer |
| DpnII | New England Biolabs | R0543 | supplied with buffer |
| DamID PCR primer "AdR_PCR" | Integrated DNA Technologies | sequences available in ref. 24 | |
| Deoxynucleotide (dNTP) Solution Set | New England Biolabs | N0446 | 100 mM each of dATP, dCTP, dGTP and dTTP |
| Advantage 2 Polymerase Mix | Clontech | 639201 | supplied with buffer |
| 1Kb Plus DNA Ladder | Life Technologies | 10787018 | 1.0 µg/µl |
| QIAquick PCR Purification Kit | QIAGEN | 28104 or 28106 | |
| MinElute PCR Purification Kit | QIAGEN | 28004 or 28006 | for an elution volume of less than 30 µl |
| SPRI beads / Agencourt AMPure XP | Beckman Coulter | A63880 | apply extra mixing and more elution time if less than 40 µl elution buffer is used |
| Buffer EB | QIAGEN | 19086 | |
| NEBNext dsDNA Fragmentase | New England Biolabs | M0348 | supplied with buffer |
| T4 DNA Ligase Reaction Buffer | New England Biolabs | B0202 | |
| T4 DNA Polymerase | New England Biolabs | M0203 | |
| DNA Polymerase I, Large (Klenow) Fragment | New England Biolabs | M0210 | |
| T4 Polynuleotide Kinase | New England Biolabs | M0201 | |
| Klenow Fragment (3’ → 5’ exo-) | New England Biolabs | M0212 | supplied with buffer |
| sequencing adaptors | Integrated DNA Technologies | sequences available in ref. 28 | |
| Quick Ligation Kit | New England Biolabs | M2200 | used in 3.4.3; supplied with Quick Ligation Reaction Buffer and Quick T4 DNA Ligase |
| sequencing primer 1 and 2 | Integrated DNA Technologies | sequences available in ref. 28 | |
| KAPA HiFi PCR Kit | Kapa Biosystems | KK2101 or KK2102 | supplied with KAPA HiFi DNA Polymerase, 5x KAPA HiFi Fidelity Buffer and 10 mM dNTP mix |
| agarose | Sigma Aldrich | A4679 | |
| ethidium bromide | Sigma Aldrich | E1510-10ML | 10 mg/ml |
| QIAquick Gel Extraction Kit | QIAGEN | 28704 or 28706 | |
| iTaq Universal SYBR Green Supermix | Bio-Rad Laboratories | 1725121 or 1725122 | |
| Spectrophotometer | brand not critical | ||
| 0.45 μm PVDF Filter | brand not critical | ||
| 25 ml Seringe | brand not critical | ||
| 10 cm Tissue Culture Plates | brand not critical | ||
| 6-well Tissue Culture Plates | brand not critical | ||
| S1000 Thermal Cycler | Bio-Rad Laboratories | ||
| C1000 Touch Thermal Cycler | Bio-Rad Laboratories | for qPCR | |
| Vortex Mixer | brand not critical | ||
| Dry Block Heater or Thermomixer | brand not critical | ||
| Microcentrifuge | brand not critical | ||
| Gel electrophoresis system with power supply | brand not critical | ||
| Magnet stand | for purification of DNA with SPRI beads; should hold 1.5-2 ml tubes; brand not critical | ||
| UV transilluminator | brand not critical | ||
| E-gel electrophoresis system | Life Technologies | G6400, G6500, G6512ST |
References
- van Steensel, B., Delrow, J., Henikoff, S. Chromatin profiling using targeted DNA adenine methyltransferase. Nat Genet. 27, 304-308 (2001).
- van Steensel, B., Henikoff, S. Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nat Biotechnol. 18, 424-428 (2000).
- Fu, A. Q., Adryan, B. Scoring overlapping and adjacent signals from genome-wide ChIP and DamID assays. Mol Biosyst. 5, 1429-1438 (2009).
- Guelen, L. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 453, 948-951 (2008).
- Kalverda, B., Pickersgill, H., Shloma, V. V., Fornerod, M. Nucleoporins directly stimulate expression of developmental and cell-cycle genes inside the nucleoplasm. Cell. 140, 360-371 (2010).
- Kubben, N. Mapping of lamin A- and progerin-interacting genome regions. Chromosoma. 121, 447-464 (2012).
- Steglich, B., Filion, G. J., van Steensel, B., Ekwall, K. The inner nuclear membrane proteins Man1 and Ima1 link to two different types of chromatin at the nuclear periphery in S. pombe. Nucleus. 3, 77-87 (2012).
- Harr, J. C. Directed targeting of chromatin to the nuclear lamina is mediated by chromatin state and A-type lamins. J Cell Biol. 208, 33-52 (2015).
- Gonzalez-Aguilera, C. Genome-wide analysis links emerin to neuromuscular junction activity in Caenorhabditis elegans. Genome Biol. 15, R21 (2014).
- Wu, F., Yao, J. Spatial compartmentalization at the nuclear periphery characterized by genome-wide mapping. BMC Genomics. 14, 591 (2013).
- Filion, G. J. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell. 143, 212-224 (2010).
- Vogel, M. J. Human heterochromatin proteins form large domains containing KRAB-ZNF genes. Genome Res. 16, 1493-1504 (2006).
- Venkatasubrahmanyam, S., Hwang, W. W., Meneghini, M. D., Tong, A. H., Madhani, H. D. Genome-wide, as opposed to local, antisilencing is mediated redundantly by the euchromatic factors Set1 and H2A.Z. Proc Natl Acad Sci U S A. 104, 16609-16614 (2007).
- Shimbo, T. MBD3 localizes at promoters, gene bodies and enhancers of active genes. PLoS Genet. 9, e1004028 (2013).
- Orian, A. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 17, 1101-1114 (2003).
- Artegiani, B. Tox: a multifunctional transcription factor and novel regulator of mammalian corticogenesis. EMBO J. , (2014).
- Schuster, E. DamID in C. elegans reveals longevity-associated targets of DAF-16/FoxO. Mol Syst Biol. 6, 399 (2010).
- Bianchi-Frias, D. Hairy transcriptional repression targets and cofactor recruitment in Drosophila. PLoS Biol. 2, e178 (2004).
- Woolcock, K. J., Gaidatzis, D., Punga, T., Buhler, M. Dicer associates with chromatin to repress genome activity in Schizosaccharomyces pombe. Nat Struct Mol Biol. 18, 94-99 (2011).
- Luo, S. D., Shi, G. W., Baker, B. S. Direct targets of the D. melanogaster DSXF protein and the evolution of sexual development. Development. 138, 2761-2771 (2011).
- Germann, S., Gaudin, V. Mapping in vivo protein-DNA interactions in plants by DamID, a DNA adenine methylation-based method. Methods Mol Biol. 754, 307-321 (2011).
- Zhang, X. The Arabidopsis LHP1 protein colocalizes with histone H3 Lys27 trimethylation. Nat Struct Mol Biol. 14, 869-871 (2007).
- Orian, A. Chromatin profiling, DamID and the emerging landscape of gene expression. Curr Opin Genet Dev. 16, 157-164 (2006).
- Vogel, M. J., Peric-Hupkes, D., van Steensel, B. Detection of in vivo protein-DNA interactions using DamID in mammalian cells. Nat Protoc. 2, 1467-1478 (2007).
- Greil, F., Moorman, C., van Steensel, B. DamID: mapping of in vivo protein-genome interactions using tethered DNA adenine methyltransferase. Methods Enzymol. 410, 342-359 (2006).
- de Wit, E., Greil, F., van Steensel, B. Genome-wide HP1 binding in Drosophila: developmental plasticity and genomic targeting signals. Genome Res. 15, 1265-1273 (2005).
- DamID mammalian vectors. , Available from: http://research.nki.nl/vansteensellab/Mammalian_plasmids.htm (2015).
- Illumina TruSeq adaptors & PCR primers. , Available from: https://ethanomics.wordpress.com/chip-seq-library-construction-using-the-illumina-truseq-adapters/ (2015).
- Optimization of PCR cycles for NGS. , Available from: https://ethanomics.wordpress.com/ngs-pcr-cycle-quantitation-protocol/ (2015).
- Bernstein, B. E. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 28, 1045-1048 (2010).
- Encode Project Consortium. A user's guide to the encyclopedia of DNA elements (ENCODE). PLoS Biol. 9, e1001046 (2011).
- Asp, P. Genome-wide remodeling of the epigenetic landscape during myogenic differentiation. Proc Natl Acad Sci U S A. 108, E149-E158 (2011).
- Hoppe, P. S., Coutu, D. L., Schroeder, T. Single-cell technologies sharpen up mammalian stem cell research. Nat Cell Biol. 16, 919-927 (2014).
- Avital, G., Hashimshony, T., Yanai, I. Seeing is believing: new methods for in situ single-cell transcriptomics. Genome Biol. 15, 110 (2014).
- Navin, N. E. Cancer genomics: one cell at a time. Genome Biol. 15, 452 (2014).
- Saliba, A. E., Westermann, A. J., Gorski, S. A., Vogel, J. Single-cell RNA-seq: advances and future challenges. Nucleic Acids Res. 42, 8845-8860 (2014).
- Nagano, T. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 502, 59-64 (2013).
- Kind, J. Single-cell dynamics of genome-nuclear lamina interactions. Cell. 153, 178-192 (2013).
- Southall, T. D. Cell-type-specific profiling of gene expression and chromatin binding without cell isolation: assaying RNA Pol II occupancy in neural stem cells. Dev Cell. 26, 101-112 (2013).
