

Introduction
As cianobactérias são um filo antiga e diversificada evolutivamente de bactérias encontradas em quase todos ambiente natural na Terra. Em ecossistemas marinhos são particularmente abundantes e desempenham um papel fundamental em muitos ciclos de nutrientes, sendo responsável por cerca de metade da fixação de carbono 1, a maioria da fixação de nitrogênio 2 e centenas de milhões de toneladas de produção de hidrocarbonetos 3 nos oceanos anualmente. Cloroplastos, a organela responsável pela fotossíntese em algas e plantas eucariótica, são susceptíveis de ter evoluído de uma cianobactéria que foi engolida por um organismo hospedeiro 4. As cianobactérias provaram organismos modelo útil para o estudo da fotossíntese, transporte de electrões 5 e vias bioquímicas, muitos dos quais são conservados em plantas. Em adição cianobactérias são cada vez mais utilizados para a produção de alimentos, biocombustíveis 6, 7 e electricidade industriais compostos 8, devido à sua oiconversão ghly eficiente de água e CO 2 a biomassa usando a energia solar 9. Muitas espécies podem ser cultivadas em terras não-aráveis com nutrientes mínimos e água do mar, o que sugere que as cianobactérias potencialmente poderiam ser cultivadas em grande escala, sem afetar a produção agrícola. Certas espécies também são fontes de produtos naturais, incluindo antifúngicos, antibacterianos e anti-câncer compostos 10,11.
A capacidade de gerar mutantes é a chave para a compreensão de cianobactérias fotossíntese, bioquímica e fisiologia, e essencial para o desenvolvimento de cepas para fins industriais. A maioria dos trabalhos publicados produzir estirpes geneticamente modificados por inserção de uma cassete de resistência a antibiótico no local de interesse. Isto limita o número de mutações que podem ser introduzidas numa estirpe, como apenas algumas cassetes de resistência aos antibióticos estão disponíveis para uso em cianobactérias. Estirpes que contêm genes que conferem re antibióticosistance não pode ser usado para a produção industrial em lagoas abertas, o que é susceptível de ser o único meio de baixo custo para a produção de biocombustíveis e outros produtos de baixo valor 12. A geração de mutantes sem marcação supera essas limitações. mutantes não marcadas não contêm DNA estranho, a não ser intencionalmente incluídos, e pode ser manipulado diversas vezes. Por isso, é possível gerar o maior número de alterações em uma cepa como desejado. Além disso, os efeitos polares de genes a jusante do local de modificação pode ser minimizado, permitindo a modificação mais preciso do organismo 13.
Para gerar estirpes mutantes, plasmídeos suicidas contendo dois fragmentos de ADN idênticos às regiões no cromossoma de cianobactérias que flanqueiam o gene a ser suprimido (denominado a 5 'e 3' que flanqueiam regiões) são construídos em primeiro lugar. Dois genes são então inserida entre estas regiões flanqueadoras. Uma delas codifica uma proteína de resistência a antibiótico; o segundo codifica SacB, que produces levanossacarase, um composto que confere sensibilidade à sacarose. Na primeira fase do processo, os mutantes marcadas, as estirpes ou seja, contendo algum ADN estranho, são gerados. A construção de plasmídeo é misturado com as células de cianobactérias e o ADN é tomado naturalmente pelo organismo. Os transformantes são seleccionados por crescimento em placas de agar contendo os antibióticos apropriados e o genótipo mutante verificada por PCR. plasmídeos suicidas não pode replicar dentro da estirpe de interesse. Portanto, quaisquer colónias resistentes ao antibiótico irá resultar a partir de um evento de recombinação em que o gene de interesse é inserida no cromossoma. Para gerar mutantes não marcados, o mutante marcada é, em seguida, misturado com um segundo plasmídeo suicida contendo apenas as regiões f lanqueadoras 5 'e 3'. No entanto, se for necessária a inserção de ADN estranho, num plasmídeo que consiste em 5 'e 3' com regiões de uma cassete que contém os genes de interesse inserido entre estes fragmentos de ADN de flanqueamento, pode ser utilizado. Selecção é através de crescimento em placas de agar contendo sucrose. Como a sacarose é letal para as células quando o produto do gene sacB é expresso, as únicas células que sobrevivem são aquelas nas quais ocorreu um segundo evento de recombinação, em que o gene de sensibilidade de sacarose, em adição ao gene de resistência aos antibióticos, foi recombinado para fora do cromossoma e para o plasmídeo. Como consequência da permuta de recombinação, as regiões f lanqueadoras e qualquer ADN entre eles são inseridos no cromossoma.
Temos utilizado com sucesso destes métodos para gerar múltiplas mutações cromossómicas na mesma estirpe de Synechocystis sp. PCC6803 (doravante referida como Synechocystis) 13,14, para introduzir mutações pontuais em um gene de interesse 13 e para a expressão de cassetes de genes. Enquanto a geração de nocautes não marcado foi demonstrada antes de nosso trabalho em Synechocystis 15,16, um método detalhado, auxiliado poruma apresentação visual dos passos críticos, não está disponível publicamente. Nós também aplicaram o mesmo método para geração de nocautes marcados em outro cianobactéria modelo, Synechococcus sp. PCC7002 (doravante referida como Synechococcus). Este protocolo fornece um método claro, simples para a geração de mutantes e um protocolo rápido para validar e armazenar estas estirpes.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Preparação de Meios de Cultura
- Preparar o meio de acordo com a BG11 Castenholz, 1988 17.
- Preparar soluções estoque de BG11 100x, oligoelementos e estoque de ferro (Tabela 1).
- Prepare soluções separadas de partida de fosfato, Na 2 CO 3 estoque, N - [Tris (hidroximetil) metil] -2-aminoetanossulfónico (TES) e tampão de NaHCO 3 (Tabela 1).
- Autoclave o fosfato e Na 2 CO 3 stocks. Filtrar-esterilizar tampão TES e NaHCO3 com 0,2 mm filtros.
- Prepare BG11 através da combinação de 976 ml de água, 10 ml de 100x BG11, 1 ml de elementos vestigiais e 1 ml de estoque de ferro e autoclave a solução. Após esta solução ter arrefecido até à temperatura ambiente, adiciona-se 1 ml de partida de fosfato, 1 ml de Na 2 CO 3 estoque e 10 ml de NaHCO3.
- Para BG11 meio sólido, adicione 15 g de ágar e 700 ml de água a um flask. Para o segundo balão, adicionar 3 g de Na 2 S 2 O 3, 226 ml de água, 10 ml de 100x BG11, 1 ml de elementos vestigiais e 1 ml de estoque de ferro. Autoclave ambas as soluções. Após estas soluções têm arrefecida até à temperatura ambiente, combiná-los e adicionar 1 ml de partida de fosfato, 1 ml de Na 2 CO 3 estoque, 10 ml de tampão TES, e 10 ml de NaHCO3.
NOTA: As soluções são preparadas separadamente para evitar a precipitação de alguns sais.
- Para a selecção em sacarose, preparar um 50% (w / v) de solução de sacarose. Filtro de esterilizar a solução com 0,2 uM filtros e adicionar ao BG11 (100 ml de sacarose a 50% para 900 mL de BG11) para produzir BG11 / 5% de sacarose placas.
Nota: Não adicionar NaHCO 3 a BG11 / 5% placas de agar de sacarose. Adicionar Na 2 CO 3 como normal. - Para a cultura de Synechococcus adicionam-se 10 ml de 1 M de 4- (2-hidroxietil) piperazina-1-ácido etanossulf único, N - (2-hidroxietil) piperazina-NR42; - (2-etanossulfónico) (HEPES) e 1 ml de vitamina B 12 (Tabela 1) a 1 L de meio de BG11.
Nota: Transformação de estirpes cultivadas em meios BG11 disponível comercialmente é significativamente menos eficiente do que nas receitas media BG11 aqui descritos e, portanto, não é recomendado.
2. Crescimento das Estirpes cianobactérias
- Cultura estirpes em 100 ml garrafas cónicas com um volume máximo de 50 ml e agitar a 120 rpm. Selar as placas com parafilme e BG11 punção três pequenos orifícios no lado da placa para permitir a troca de gás. Incubar todas as estirpes a 30 ° C sob lâmpadas fluorescentes em um fotobiorreactor com uma intensidade de luz entre 20-40 umol fotões m-2 s-1.
- Usar as melhores técnicas estéreis. Lidar com todas as linhagens de cianobactérias em uma capela de fluxo laminar.
Nota: Isto é especialmente importante quando as estirpes são cultivadas com meio contendo sacarose, o qual pode ser facilmente contaminated.
3. Geração do plasmídeo Constructos
- Desenho conjuntos de iniciadores, incluindo os locais necessários de enzimas de restrição, utilizando software de desenho de iniciadores, tais como Primer3 (http://frodo.wi.mit.edu/primer3/), para amplificar dois ~ 1 kb regiões 5 'e 3' do gene de interesse. Consulte a sequência do genoma das espécies de cianobactérias via Cyanobase (http://genome.kazusa.or.jp/cyanobase). Ver Tabela 2 para todos os iniciadores utilizados aqui. Ao projetar primers considerar os seguintes fatores:
- Certifique-se de que as regiões amplificadas incluir regiões 5 'e 3' do gene que vai ser mutada, por exemplo, Figura 1.
- Não se transformar regiões intergênicas para evitar a mutação não intencional de antisense e RNAs não-codificantes. Para a geração dos mutantes em Synechocystis, referem-se a lista de sítios de início da transcrição documentados em Mitschke et al., 2011 18, a fim de evitar a mutação de anti-sentidoou RNAs não-codificantes.
- Ao escolher as regiões flanqueadoras não incluir a totalidade da grelha de leitura aberta de genes adjacentes como expressão destes genes em Escherichia coli pode interferir com a clonagem.
- Amplificar produtos por PCR utilizando ADN-polimerase de alta fidelidade de acordo com as instruções do fabricante.
Nota: Em nossa experiência esta enzima produz alguns erros.- Defina-se reacções de PCR de 50 ul contendo tampão de HF e de 0, 1,5 ou 3 ul de DMSO. Use 100 ng de ADN genómico por reacção. Utilizar um programa que consiste em um passo de desnaturação inicial de 98 ° C durante 30 seg, 35 ciclos de 98 ° C durante 10 seg, 67 ° C durante 30 seg, 72 ° C durante 30 seg, seguido por um passo final de extensão de 72 ° C durante 5 min. Isto dá tipicamente produtos consistentes.
- Os produtos de PCR e verificar amostras digerido com as enzimas de endonuclease para o tamanho correcto através de electroforese em gel. Corra 1% (w / v) de geles de agarose contendo 0,02%(V / v) de brometo de etídio durante 45 min a 100 V.
CUIDADO: O brometo de etídio é um agente mutagênico potencial e deve ser manuseado com proteção adequada. - Purifica-se os produtos de PCR utilizando um kit de purificação de ADN de acordo com as instruções do fabricante. Também usar este kit para a purificação de fragmentos de plasmídeos, incluindo peças cortadas de géis de agarose. Elui-se o ADN purificado em 14 uL de água.
- Para os passos de clonagem, incubar as misturas reaccionais para endonucleases de restrição a 37 ° C durante> 1 hora num volume total de 30 ul de acordo com as instruções do fabricante.
- Para passos de ligação, os fragmentos de ADN ligadura à temperatura ambiente durante> 1 hora num volume total de 20 uL, contendo 5 ul de plasmídeo digerido purificado, 12 ul de inserção digerido purificado, 2 ul de tampão e 1 ul de ligase.
- Preparar células transformantes de Escherichia coli DH5a de acordo com o método seguinte.
- Crescer um dia para o outro E. coli
- Inocular 400 ml de LB num frasco cónico de 1 L contendo 6 ml de 1 M de MgCl2 (Tabela 1) com 1 ml da cultura durante a noite.
- Crescer a cultura a 37 ° C a 220 rpm durante cerca de 4 h ou até que OD 600 nm alcança 0,4-0,6.
- Coloque as células em gelo durante 1 hora.
- Centrifugar a 2.800 xg durante 10 min para sedimentar as células a 4 ° C.
- Remover o sobrenadante e ressuspender em 160 ml de solução A (Tabela 1) e incubar em gelo durante 20 min.
- Centrifugar a 2.800 xg durante 10 min para sedimentar as células a 4 ° C.
- Remover o sobrenadante e ressuspender em 4 ml de solução A + glicerol (Tabela 1).
- Prepare 50 mL alíquotas, congelar no N2 líquido, armazenar a -80 ° C.
- Misturar 5 ul de mistura de ligação com 50 ul de células competentes e incubar durante 1 hora em gelo.
- Choque térmico às células a 42 ° C durante 90 seg, seguinconduzido por incubação em gelo durante 2 min.
- Adicionar 950 ul de meio LB (Tabela 1) e incuba-se a 37 ° C durante 1 h.
- Alíquota de 50 e 200 ul em placas com o antibiótico adequado, quer de ampicilina (100 ug / ml) e / ou canamicina (30 ug / ml).
ATENÇÃO: Tanto canamicina e ampicilina são tóxicos e devem ser manuseados com proteção adequada. - Escolha e incubar colónias individuais em 2 ml de meio LB inoculados com o antibiótico apropriado.
- Purifica-se todos os plasmídeos utilizando um kit de purificação de plasmídeo miniprep de acordo com as instruções do fabricante.
- Gerar plasmídeos, neste exemplo específico de bater para fora os genes cpcC1C2, de acordo com os seguintes passos.
- Amplificar a região flanqueadora 1012 pb 5 '(fragmento esquerdo) utilizando iniciadores cpcC1C2leftfor e cpcC1C2leftrev (Ver passo 3.2, Tabela 2). Retirar uma pequena quantidade da reacção de PCR e confirmar se aproduto tamanho correto foi amplificado através de eletroforese em gel (passo 3.3). Digerir este fragmento e pUC19 com Xba I e Bam Hl (passo 3.5).
- Purificar ambas as preparações (passo 3.4), ligadura (passo 3.6), transformar (passo 3.7) e criou quatro 2 ml culturas líquidas LB com ampicilina (100 ug / ml) a partir de colônias separadas para purificação de plasmídeo via minipreps (passo 3.8).
- Entrada para a inserção do fragmento em pUC19 através de digestão com Xba I / Bam HI e electroforese em gel (passo 3.3). Bandas de 2.660 pb e 1.012 pb indicam introdução correcta da inserção no plasmídeo.
- Amplificar a região adjacente 1.016 bp 3 '(fragmento direita) usando primers cpcC1C2rightfor e cpcC1C2rightrev (Veja o passo 3.2, Tabela 2). Retirar uma pequena quantidade da reacção de PCR e confirmar se o produto de tamanho correcto foi amplificado por meio de electroforese em gel (passo 3.3). Digerir este fragmento e pUC19 com Sac I e Eco RI (step 3.5).
- Purificar ambas as preparações (passo 3.4), ligadura (passo 3.6), transformar (passo 3.7) e criou quatro 2 ml culturas líquidas LB com ampicilina (100 ug / ml) a partir de colônias separadas para purificação de plasmídeo via minipreps (passo 3.8).
- Entrada para a inserção do fragmento em pUC19 através de digestão com Sac I / Eco RI (passo 3.5) e electroforese em gel (passo 3.3). Bandas de 2.660 pb e 1.016 pb indicam introdução correcta da inserção no plasmídeo.
Nota: Xba I / Bam HI de locais para a clonagem da região 5 'e Sac I / Eco RI para a clonagem da região 3' em pUC19 são usados sempre que possível. Se possível, sempre incluir um local BamHI no iniciador inverso para "região ou o iniciador directo para a extremidade 3 'da região 5 para garantir que as etapas de clonagem mais tarde são mais fáceis de executar. - Sequência de ambos os insertos para determinar se a sequência estiver correcta utilizando iniciadores que abrangem o sítio de inserção, por exemplo, M13 para a frente e reverso M13 (Tabela 2). A sequência deve ser correta para garantir que não haja erros são introduzidos em regiões de flanqueamento.
- Excisar o fragmento esquerdo de pUC19 através de Xba I / Bam HI digestão. Digerir o pUC19 + fragmento incorrectamente com Xba I / Bam HI (passo 3.5).
- Purificar o fragmento deixou 1.012 pb e 3.676 pb pUC19 + fragmento de direito a partir de um gel de agarose (passo 3.3) através de excisão do DNA usando uma lâmina de bisturi.
- Purificar ambas as preparações (passo 3.4), ligadura (passo 3.6), transformar (passo 3.7) e criou quatro 2 ml culturas líquidas LB com ampicilina (100 ug / ml) a partir de colônias separadas para purificação de plasmídeo via minipreps (passo 3.8).
- Entrada para a inserção do fragmento em pUC19 + fragmento direito através de Xba I / Bam Hl de digestão (passo 3.5) e electroforese em gel (passo 3.3). Bandas de 3.676 pb e 1.012 pb indicam a inserção correta da inserção no plasmídeo (referem a isso como plasmídeo B).
Nota: A cassete NPT1 / sacB não tem de ser purificada a partir de géis de agarose, uma vez pUM24cm codifica uma proteína que confere resistência ao cloranfenicol. Portanto, se as colónias são cultivados em LB / ampicilina / canamicina placas de agar a única combinação possível que levará a colónias resistentes é a incorporação do NPT1 / cassete sacB no plasmídeo B. - Purifica-se ambas as preparações (passo 3.4), ligadura (passo 3.6), transformar (passo 3.7) e definir-se quatro 2 ml culturas líquidas de LB com ampicilina (100 ug / ml) e canamicina (30 ug / ml) a partir de colónias independentes para a purificação de plasmídeo através de mini-preparações (passo 3.8).
- Entrada para a inserção do NPT1 / cassete de sacB no plasmídeo B através da digestão com BamHI (passo 3.5) e electroforese em gel (passo 3.3). Bandas de 4688 pb e 3.894 pb indicam a inserção correta de the inserir no plasmídeo (referem a isso como plasmídeo A).
- Em alternativa, a extremidade cega / cassete NPT1 sacB e o clone em um local de endonuclease de restrição diferente entre os fragmentos esquerdo e direito no plasmídeo B. A / cassete de sacB NPT1 deve ser clonado entre os fragmentos esquerdo e direito.
Nota: Se a expressão de uma cassete externa é necessária, então esta deve ser inserido entre os fragmentos esquerdo e direito do plasmídeo B. Este plasmídeo é então utilizado nos passos knockout não marcados.
4. Geração de Marcadas Mutantes Synechocystis e Synechococcus
- Configurar uma cultura fresco inoculando um ciclo completo de células em 30-50 ml de meio BG11. Crescer a cultura durante 2-3 dias a 750nm OD = 0,2 a 0,6.
Nota: Normalmente as colónias individuais são demasiado pequenos para usar-se para a inoculação e a exposição das células individuais, mesmo para níveis baixos de luz irá resultar em fotoinibição e selecçãopara os mutantes resistentes luz. - Centrifugar 1-2 ml da cultura a 2.300 x g durante 5 min e desprezar o sobrenadante. Não centrifugar quaisquer culturas de cianobactérias em> 2.300 xg, pois isso pode danificar as células. Lava-se a pelete uma vez com meio BG11.
Nota: Não ressuspender as células em vortex como isto pode resultar em perda de pili que são essenciais para a absorção de ADN. Ressuspender as células por pipetagem suave. - Adicionar meio BG11 para um volume final de 100 ul. Transferência de células para um tubo de 14 ml de fundo redondo.
- Adicionar 1 ug de plasmídeo A e para as células Misturar agitando ligeiramente. Adicionar <10 ul de plasmídeo.
Nota: De um modo preferido o plasmídeo deve estar a uma concentração de> 100 ng / ul, mas concentrações mais baixas do que isso são adequados para a transformação bem sucedida. - Colocar os tubos para baixo horizontalmente na incubadora. Incubar as culturas de 4-6 hr.
Nota: As células podem ser misturada brevemente tocando cada 1-2 horas, mas isto não é essencial. As amostras podem ser colocadas em umagitando incubadora embora este não melhorar significativamente a eficiência. - Espalhe-se alíquotas da mistura de ADN de cultura de células / plasmídeo em placas de agar BG11 sem antibióticos. Tipicamente, 20 uL e 80 mL alíquotas são espalhadas em placas separadas.
- ~ 24 horas mais tarde, adicionar 2,5-3 mL de 0,6% solução de agar em água contendo canamicina (por 20 mL: 0,12 g de agar, 100 uL de 100 mg / ml de canamicina) para a placa de agar. Arrefecer a solução a ~ 42 ° C, e adicionar à borda da placa de agar. Inclinar a placa de modo que a solução forma um mesmo 'ágar de topo' camada na superfície.
- Incubar as placas de ágar por um novo período de tempo. Colónias deve ser visível após aproximadamente 7 dias.
Nota: As placas de agar podem ser empilhados 3 elevado em uma incubadora. Tipicamente centenas de colónias são obtidos por transformação. - Streak colónias individuais sobre BG11 + canamicina (30 ug / ml) em placas de agar. Divida a placa de agar em 6 setores e use um palito fim brusco à raia foraas colônias mais de cada sector. A obtenção de colónias isoladas não é importante apenas o crescimento dos transformantes.
- Confirmar nocaute marcada por PCR utilizando polimerase do ADN de Taq de acordo com as instruções do fabricante. Adicionar 2 mL de MgCl2 (25 mM) por reacção.
- Retirar uma pequena proporção das células e transferir para um tubo contendo 50 ul de água e 20 ~ ^ M 425-600 esferas de vidro. Agitar num vibrador durante 5 min a ~ 2000 rpm. Centrifuga-se a 15700 xg durante 5 min e utilizar 5 ul de sobrenadante por 50 ul de reacção de PCR.
Nota: Não ressuspender a solução. Os restos celulares precisa de ficar no fundo do tubo.
- Retirar uma pequena proporção das células e transferir para um tubo contendo 50 ul de água e 20 ~ ^ M 425-600 esferas de vidro. Agitar num vibrador durante 5 min a ~ 2000 rpm. Centrifuga-se a 15700 xg durante 5 min e utilizar 5 ul de sobrenadante por 50 ul de reacção de PCR.
- validar mutantes
- primers de design que abrangem a região do knockout usando software de desenho de primer (como Primer3). iniciadores de desenho a partir de ~ 200 pb de cada lado da região de nocaute.
Nota: Os Primers para verificar o mutante cpcC1C2 são apresentadas na Tabela 2e são denominadas cpcC1C2for e cpcC1C2rev. - Amplificar produtos usando um programa que consiste em um passo de desnaturação inicial de 95 ° C durante 2 min, 35 ciclos de 95 ° C durante 1 min, 60 ° C durante 1 min, 72 ° C durante 1 min por kb de sequência, seguido de um passo de extensão final de 72 ° C durante 5 min. Incluir um controlo de tipo selvagem. Isto dá tipicamente produtos consistentes.
- Verificar o genótipo através de electroforese em gel. Knockout transformantes marcados irão mostrar uma banda de ~ 4 kb (0,2 kb a partir de ambos os fragmentos esquerdo e direito, mais o NPT1 / cassete sacB) e a ausência da banda de tipo selvagem (Figura 2).
Nota: Em certos casos, uma banda de ~ 4 kb não é observada no mutante marcada, devido ao grande tamanho do produto de PCR. No entanto, se uma banda correspondente ao tamanho esperado do tipo selvagem não é observada, em seguida, tipicamente, esta estirpe é um nocaute marcado.
- primers de design que abrangem a região do knockout usando software de desenho de primer (como Primer3). iniciadores de desenho a partir de ~ 200 pb de cada lado da região de nocaute.
- Se uma banda de tipo selvagem ainda está presente, em seguida, re-raia a pressão sobre aBG11 fresco + canamicina (30 ug / ml) em placa de ágar e repetir a PCR. Repetir o processo de re-estrias até que o mutante é segregada para que nenhuma banda de tipo selvagem é observado na reacção de PCR.
Nota: O aumento da quantidade de canamicina para uma concentração de 50 ug / ml, em seguida, 100 ug / ml, por vezes, é essencial, a fim de segregar um mutante marcada totalmente. - Se a tensão mostra um perfil mutante marcada via PCR, em seguida, re-raia em um BG11 fresco + canamicina (30 ug / ml) em placa de ágar. Use esta estirpe para gerar o nocaute não marcado.
Nota: O protocolo pode ser utilizado para gerar mutantes marcadas com apenas uma cassete de resistência a antibióticos ou seja, através da substituição do / cassete sacB NPT1 com apenas a cassete de NPT1 pUC18K 20 entre os fragmentos esquerdo e direito..
5. Geração de Unmarked Synechocystis Mutantes
- Configurar uma nova cultura da knockout marcado por inoculação de um circuito cheio de cells em 30-50 ml de meio BG11. Crescer a cultura durante 2-3 dias a 750nm OD = 0,2 a 0,6.
- Centrifugar 10 ml da cultura a 2.300 x g durante 5 min e desprezar o sobrenadante. Lavar uma vez com meio BG11.
Nota: Não ressuspender as células em vortex como isto pode resultar em perda de pili que são essenciais para a absorção de ADN. Ressuspender as células por pipetagem suave. - Adicionar BG11 para um volume final de 200 ul. Transferência de células para um tubo de 14 ml de fundo redondo.
- Adicionar 1 mg de DNA do plasmídeo para as células B e misturar agitando ligeiramente.
- Incubar as amostras durante 4-6 h. Coloque tubos para baixo horizontalmente.
Nota: As células podem ser misturada brevemente tocando cada 1-2 horas, mas isto não é essencial. As amostras podem ser colocadas em um incubador com agitação, embora isto não melhorar a eficiência. - Adicionar 1,8 ml de meio BG11 e incubar as amostras durante um total de 4 dias, com agitação. Este tempo é suficiente para permitir que a recombinação ocorra nas múltiplas cópias cromossómicas.
- alíquotas de chapa da mistura de transformação em BG11 / 5% placas de agar de sacarose. Placa 50 ul, 10 ul e 1 ul por placa de ágar. Se um relvado colónia aparece em todas estas placas de agar diluir ainda mais a solução e aliquota em placas frescas. Colónias deve ser visível após aproximadamente 7 dias.
- Remendo 30-50 colónias individuais sobre BG11 + canamicina (30 ug / ml) placas de agar primeiros e BG11 / 5% de placas de agar de sacarose segundo, utilizando um palito de extremidade embotada. Todas as bactérias que crescem em BG11 / 5% de sacarose placas mas as placas não BG11 + canamicina são potenciais nocautes isolado. As bactérias que crescem em ambas as placas são susceptíveis de ser resistente à sacarose devido a uma mutação no gene sacB.
- Verifique nocautes não marcadas usando os mesmos iniciadores e método que foi utilizado para verificar os furos marcados. Ex cpcC1C2for e cpcC1C2rev (Tabela 2) para verificar o nocaute não marcado cpcC1C2. Um nocaute não marcado irá mostrar uma banda em um gel de agarose correspondente tO tamanho do tipo selvagem menos a região eliminada (Figura 2).
- Se a estirpe mostra um perfil não marcado mutante por meio de PCR (passo 4.11.2) e electroforese em gel (Figura 2), em seguida, re-raia numa placa de agar BG11 fresco sem antibióticos.
6. armazenamento de longo prazo de cepas
- Defina-se uma cultura fresca da estirpe através da inoculação de um ciclo completo de células em 30-50 ml de meio BG11. Crescer a cultura durante 3-4 dias a 750nm OD = 0,4 a 0,7.
- Lave as células uma vez com BG11 e ressuspender em ~ 2 ml de BG11.
- Adicionar 0,8 ml de células concentradas para um tubo. Em seguida, adicionar 0,2 ml de 80% de glicerol esterilizado por filtração.
- Opcional: adicionar 0,93 ml de células concentradas para outro tubo. Adicionar 0,07 ml de DMSO a este tubo.
CUIDADO: DMSO é tóxico e deve ser manuseado com proteção adequada. - Armazenar ambos os tubos a -80 ° C. Para reviver tensões remover o tubo e raspar algumas células com um dente sem corteescolher em uma placa de ágar sem antibióticos. Streak como normal usando um loop estéril.

Figura 1: construção do plasmídeo para a geração de orifícios marcadas e não marcadas, por exemplo cpcC1 e cpcC2 em Synechocystis (a) uma região do genoma de Synechocystis em que (B) e cpcC1 cpcC2 e genes adjacentes estão localizados.. Realçada em preto é a região do genoma a ser eliminado no mutante. (C) sítios do genoma que são amplificados por PCR. A "região de flanqueamento (indicada a azul) e 3 '5 região flanqueadora (indicados a vermelho) são amplificados com os locais de endonucleases de restrição para clonagem em pUC19. A 5 '(ou 3') região flanqueadora é excisado de pUC19 e inserido no pUC19 + 3 '(ou 59;) flanqueando plasmídeo região para gerar o plasmídeo B. (D) A NPT1 / cassete sacB de pUM24 é extirpado através de Bam HI digestão e inserido entre 5 'e 3' flanqueando regiões para gerar o plasmídeo A. Por favor clique aqui para ver uma maior versão desta figura.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
O plasmídeo concepção é crítica para a geração bem sucedida de ambos os mutantes marcadas e não marcadas. A Figura 1 dá um exemplo do plasmídeo A e B usadas para gerar um mutante de deleção nos genes de Synechocystis cpcC1 e cpcC2 13. Em cada caso, as regiões f lanqueadoras 5 'e 3' são aproximadamente 900-1.000 pb. flanqueando reduzidas pode ser utilizado, embora o menor temos testadas com sucesso tem sido aproximadamente 500 pb. O plasmídeo B, também pode conter uma cassete de genes entre a 5 'e 3' ~ 1 kb regiões que flanqueiam ou uma versão modificada da sequência do gene nativo.
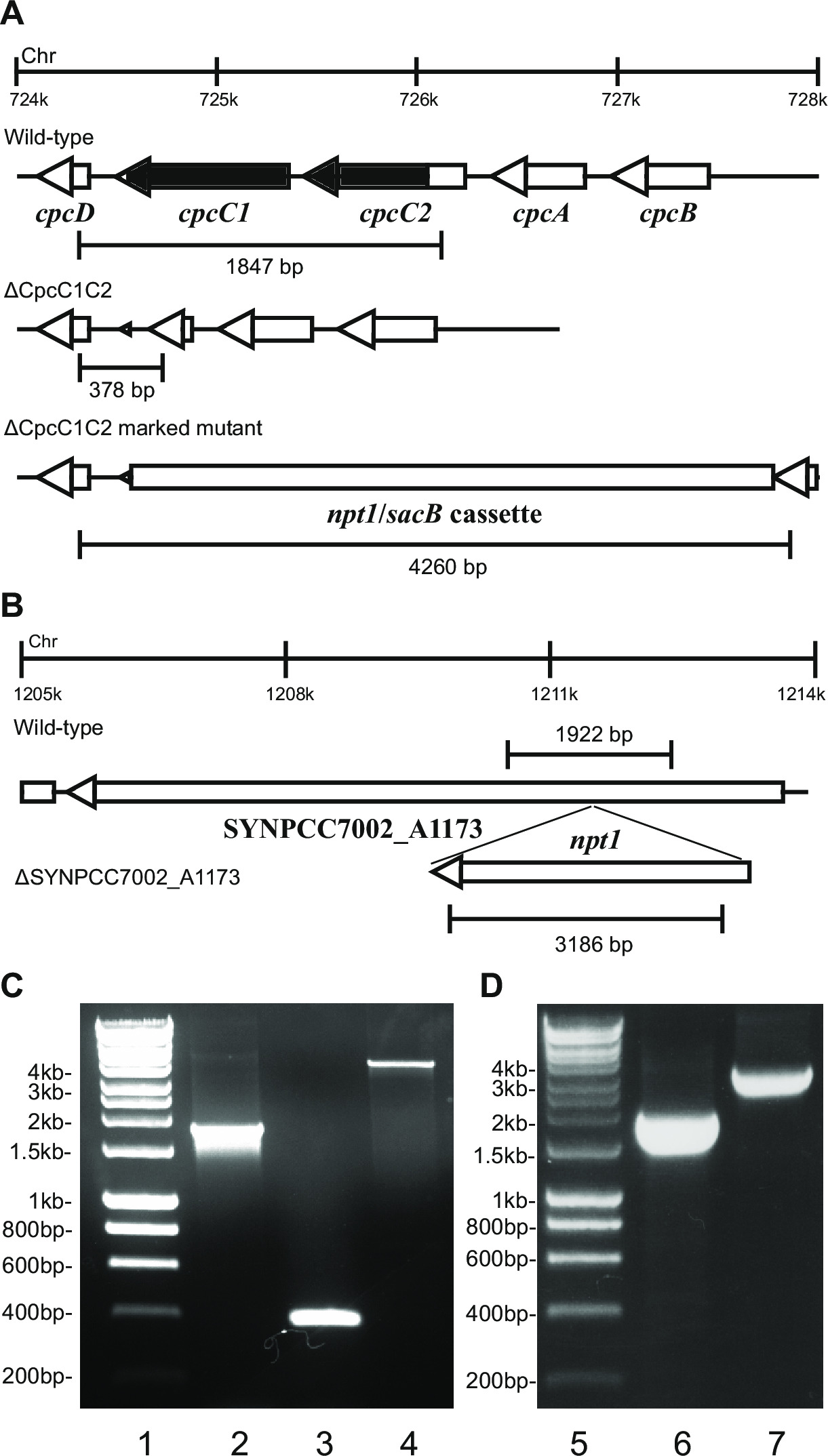
Figura 2: Verificação de mutantes marcados e não marcados, por exemplo cpcC1 / cpcC2
Após a transformação do plasmídeo A para as células, tipicamente várias centenas de colónias aparecerá em uma placa após cerca de 7-10 dias. Colónias são <1 mm de diâmetro e não vai aumentar de tamanho para as próximas semanas. Por isso, é crítica a utilização de um palito de extremidade embotada para remover a colónia e raia-o em um BG11 + canamicina placa de ágar fresco. Aproximadamente metade das colônias manchado de re vai crescer depois de 4-6 dias. Se os genes não são essenciais e mutantes demonstram crescimento similar à estirpe do tipo selvagem sob a luz contínua de 20-40 mol fótons m -2 s-1 (por exemplo terminais mutantes oxidase em Lea-Smith et al., 2013 14) (Figura 3), em seguida, todos os cromossomos devem conter uma cópia do t ele NPT1 / cassete sacB sequência inserida, tal como determinado através de PCR. Se os genes não são essenciais e mutantes demonstram um fenótipo de crescimento lento sob luz contínua de 20-40 umol fotões m-2 s-1 (por exemplo, ficobilissoma mutantes deficientes em Lea-Smith et al., 2014 13) (Figura 3), em seguida vários ciclos de re-estrias em placas de agar com BG11 progressivamente maiores quantidades de canamicina são essenciais para a obtenção de um mutante marcada segregada. Uma vez que um mutante segregada é obtido este deve ser re-semeadas em uma BG11 mais canamicina placa de agar fresco para garantir que a segregação é completa. Se rodadas repetidas de estrias não resultar em uma segregada marcada mutante, em seguida, o gene é provável essencial para a sobrevivência. A Figura 4 apresenta um esboço das etapas experimentais envolvidos na geração mutante não marcada.
/54001/54001fig3highres.jpg "Width =" 700 "/>
Figura 3:. O crescimento de mutantes de Synechocystis Os exemplos de mutantes que demonstram (a) O crescimento semelhante à do tipo selvagem e (B) um crescimento mais lento do que o de tipo selvagem. O mutante ΔCOX carece de citocromo-oxidase devido ao apagamento dos genes CtaC1D1E1. O mutante ΔCyd carece de quinol oxidase devido ao apagamento dos genes CydAB. O mutante de oliva carece de uma porção do ficobilissoma devido a deleção dos genes CpcABC1C2D. As amostras em (B) foram borbulhar com ar para facilitar o crescimento. Reproduzido a partir de dados publicados em Lea-Smith et al, 2013 14 e 2014 13 (www.plantphysiol.org; de Copyright Sociedade Americana de biólogos da planta).. Por favor clique aqui para ver uma versão maior desta figura.
por exemplo, Figura 2. Se uma cassete do gene está a ser inserido no cromossoma, em seguida, tipicamente uma proporção mais elevada de canamicina são observadas colónias resistentes resistentes e sacarose. Estes mutantes podem crescer em sacarose, devido a uma mutação no gene sacB. Se não há colónias resistentes à sacarose e sensíveis à canamicina são geradas, em seguida, a cassete de gene é prejudicial para a célula.

Figura 4: Geração de mutantes marcados e não marcados em Synechocystis detalhando (A) Representação esquemática de recombinação e (B) passos experimentais envolvidos na geração de mutantes.. Um plasmídeo é primeiro misturado com as células. Após a incubação em placas de agar contendo canamicina, as colónias em que um evento de recombinação ocorre entre as 5 'umD 3 'que flanqueiam regiões (indicados a azul e vermelho, respectivamente) e a sequência homóloga no cromossoma, são isolados. Além disso, o / cassete sacB NPT1 entre a 5 'e 3' que flanqueiam regiões está inserido no cromossoma. A seguir a segregação um mutante marcada é gerado. Células mutantes marcadas são então misturados com o plasmídeo B, que pode conter ou (C) 1: as regiões f lanqueadoras 5 'e 3'; 2: 5 'e 3' que flanqueiam regiões com uma cassete de expressão contendo genes de interesse inserida entre estas sequências; 3: 5 'e 3' que flanqueiam regiões com a sequência de tipo selvagem com as alterações de nucleótidos desejadas inseridos entre estas sequências. Um segundo evento de recombinação homóloga ocorre entre o 5 'e 3' que flanqueiam regiões e as regiões homólogas no cromossoma, resultando na remoção do NPT1 / sacB cassete e, ou o knockout não marcado ou de um mutante com uma inserção ou alterado selvagem Tiporegião e introduzida no cromossoma. Por favor clique aqui para ver uma versão maior desta figura.
| Receitas da solução | |
| Químico | Quantidade (g) |
| 100x BG11 (por L) | |
| NaNO3 | 149,6 |
| MgSO 4 .7H 2 O | 7,49 |
| CaCl 2 .2H 2 O | 3.6 |
| Ácido cítrico | 0,6 |
| Adicionar 1,12 ml de 0,25 M de Na 2 EDTA, pH 8,0 | |
| 0,25 M de Na2EDTA, pH 8,0 (100 ml) | |
| na 2 | 9.3 |
| Oligoelementos (100 ml) | |
| H 3 BO 3 | 0,286 |
| MnCl 2 .4H 2 O | 0,181 |
| ZnSO 4 .7H 2 O | 0,022 |
| Na 2 MoO 4 .2H 2 O | 0,039 |
| CuSO 4 .5H 2 O | 0,008 |
| Co (NO 3) 2 .6H 2 O | 0,005 |
| Estoque de ferro (100 ml) | |
| citrato de amónio férrico | 1.11 |
| De partida de fosfato (por 100 ml) | |
| K 2 HPO 4 | 3.05 |
| Na 2 CO 3 estoque (por 100 ml) | |
| Na 2 CO 3 | 2 |
| Tampão de TES, pH 8,2 (100 ml) | |
| TES | 22,9 |
| NaHCO3 estoque (100 ml) | |
| NaHCO3 | 8.4 |
| HEPES, pH 8,2 (por 500 ml) | |
| HEPES | 119.15 |
| A vitamina B 12 (por 50 ml) | |
| cyanocobalamin | 0,02 |
| Media Luria Bertani (por 500 ml) | |
| caldo de Luria Bertani | 12,5 |
| 1 M de MgCl2 (100 ml) | |
| MgCl 2 .6H 2 O | 20,33 |
| MnCl 2 .4H 2 O | 0,395 |
| CaCl 2 .2H 2 O | 1,47 |
| 2- (N-morfolino) de hidrato de ácido etanossulfónico, ácido 4-Morpholineethanesulfonic (MES) | 0,4265 |
| Solução A + glicerol | |
| 10 ml de solução A | |
| 1,5 ml de glicerol |
Tabela 1: As soluções utilizadas neste estudo.
| cartilha | Seqüência |
| cpcC1C2leftfor | GTAC TCTAGA GCGGCTAAATGCTACGAC |
| CPCC1C2leftrev | GATC GGATCC GCGGTAATTGTTCCCTTTGA |
| cpcC1C2rightfor | GATC GAGCTC TGCACTGGTCAGTCGTTC |
| cpcC1C2rightrev | GACT GAATTC ATCGTTGCTTGAACGGTCTC |
| frente M13 | TGTAAAACGACGGCCAGT |
| reverso M13 | CAGGAAACAGCTATGAC |
| cpcC1C2for | GTTTTCATTGGCATCGGTCT |
| cpcC1C2rev | ATGTCCCAGGAACGACTGAC |
| A1173for | AGCAAACCGTTTTTGTGACC |
| A1173rev | TGCAAGGTGGCGAACTGTAT |
Tabela 2:. Os iniciadores utilizados em locais de endonucleases de restrição deste estudo encontram-se sublinhados.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Os passos mais críticos na geração de mutantes não marcados são: 1) um projeto cuidadoso plasmídeo para garantir que apenas a região de destino é alterada; 2) garantir que as amostras permanecem axênico, especialmente quando cultivadas em sacarose; 3) para a geração de células chapeamento mutante marcada inicialmente transformado em placas de agar BG11 falta antibióticos, seguido por adição de ágar antibióticos mais 24 horas mais tarde; 4) cultura marcada mutantes para 4 dias completos antes de plaqueamento em BG11 além de agar de sacarose placas: 5) assegurando que os mutantes marcados são totalmente segregados e 6) confirmando completamente o genótipo de cepas mutantes. Para esta última etapa, os iniciadores adicionais concebidos para amplificar parte da região eliminada, pode ser utilizado para garantir que ele tiver sido removido. Southern blotting, enquanto laborioso, pode também ser usado. No entanto, nossa experiência é que o procedimento descrito neste artigo é suficiente para a verificação adequada de mutantes. Este procedimento tem também sido usado para gerar mutantes marcadas em Synechococcus elongatus PCC7942. No entanto, a transformação repetida deste cianobactéria demonstrou ser um desafio.
Se mutantes marcados não podem ser segregadas, em seguida, diferentes condições ambientais altas concentrações de CO 2, com pouca luz (<20 umol fotões m-2 s-1) ou nutrientes adicionais (isto é, glucose) pode ser testado. Por exemplo, a adição de glucose é essencial, a fim de gerar mutantes fotossistema II 21. Se mutantes marcados não segregar completamente, em seguida, o gene é provavelmente essencial para a viabilidade. No entanto, existem exemplos na literatura em que alguns grupos de pesquisa têm sido incapazes de knockout de um gene (por exemplo, Vipp em Synechocystis) 22, apenas para outros grupos de mostrar mais tarde que o gene não é essencial 23. Isto pode ser devido a diferenças nas estirpes de tipo selvagem ou projeto plasmídeo incorreta, resultando em efeitos polares diante, genes essenciais adjacentes. Se um mutante não totalmentesegregar nós recomendamos que o plasmídeo contendo a cassete NPT1 de pUC18K 20 entre os fragmentos esquerdo e direito ser utilizado para a transformação. É mais fácil de verificar a presença de bandas que correspondem ao tipo selvagem e mutante por PCR, uma vez que este fragmento tem cerca de 1,2 kb, em comparação com os 3,8 kb NPT1 / cassete de sacB. Este resultado é uma peça importante de evidência demonstrando que o gene é essencial.
Geração de mutantes não marcados com cassetes de expressão inserida é geralmente mais desafiador do desenvolvimento de estirpes knockout. Em geral, expressar genes sob o controlo do promotor forte 13 cpcBAC1C2D. Em alguns casos, isto pode diminuir as hipóteses de sucesso de inserção da cassete do gene, se a sobre-expressão de uma proteína é prejudicial para a célula. Os promotores mais fracos deve então ser testado. De um modo geral observou-se que quanto maior for a cassete do gene é, mais difícil é para emSERT-lo no genoma. Nós não temos sido capazes de inserir cassetes de genes maiores que 5 kb. Além disso, deverá ser tomado em escolhendo locais para inserir cassetes de expressão no genoma. Locais neutras que não afectam a viabilidade ou crescimento celular deve ser usado. Exemplos incluem em Synechocystis e phaAB phace, que codificam as proteínas que codificam a via biossintética polihidroxibutirato 24,25. Mais recentemente uma extensa lista de sites neutros em Synechocystis foi identificado 26.
Geração de mutantes não marcados em cianobactérias é um processo lento, levando cerca de 5-7 semanas, se todas as medidas estão a realizar corretamente. Isto é mais lento do que o método padrão de geração de orifícios marcadas utilizados pela maioria dos grupos de pesquisa investigaram cianobactérias. No entanto, a flexibilidade de ser capaz de introduzir outras mutações em mutantes não marcados parcialmente compensa este, uma vez que os plasmídeos adicionais containing uma variedade de cassetes que conferem resistência a antibióticos diferentes, não tem que ser construído. Para efeitos de investigação a capacidade de mutação múltiplos genes é por vezes necessário, a fim de caracterizar completamente o papel funcional de proteínas. Por exemplo, foi identificado um fenótipo deletério apenas mediante eliminação dos dois electrões pias oxidase terminais localizadas na membrana tilacoide, uma vez que a perda de apenas um destes complexos pode ser compensada pela actividade da outra 14. Desenvolvimento de uma estirpe para aplicações industriais também vai exigir várias modificações a uma estirpe, não apenas para a introdução de genes estranhos, mas também para aumentar a eficiência fotossintética, a otimização da colheita de luz e supressão de vias concorrentes para o substrato desejado.
O principal fator limitante da velocidade da geração mutante não marcada é o momento de divisão lenta de espécies de cianobactérias modelo, entre 8-20 horas, dependendo das condições de luz. Under intensidades de luz mais altas e concentrações de CO 2, o crescimento é mais rápido. No entanto, existe o risco de que as estirpes mutantes que não podem tolerar qualquer luz elevada ou CO 2 vai ser seleccionado contra, ou que as estirpes mutantes irá sofrer alterações indesejáveis antes da caracterização fenotípica. Por conseguinte, isso não é recomendado. No entanto, seria altamente vantajoso se um protocolo mais rápido para gerar mutantes não marcado foi desenvolvido. No geral, o que facilitaria o desenvolvimento de cepas tanto para a pesquisa básica e aplicações aplicadas. Tais estirpes poderia ser usado para o biocombustível, biomassa ou de produção química ou na compreensão de muitos aspectos da bioquímica de cianobactérias, genética e fisiologia.
Subscription Required. Please recommend JoVE to your librarian.
Acknowledgments
Somos gratos aos Serviços Ambientais Association Education Trust, a Biologia Sintética em Cambridge fundo Synbio e do Ministério da Justiça Social e Empoderamento, Governo da Índia, para apoio financeiro.
Materials
| Name | Company | Catalog Number | Comments |
| NaNO3 | Sigma | S5506 | |
| MgSO4.7H2O | Sigma | 230391 | |
| CaCl2 | Sigma | C1016 | |
| citric acid | Sigma | C0759 | |
| Na2EDTA | Fisher | EDT002 | |
| H3BO3 | Sigma | 339067 | |
| MnCl2.4H2O | Sigma | M3634 | |
| ZnSO4.7H2O | Sigma | Z4750 | |
| Na2MoO4.2H2O | Sigma | 331058 | |
| CuSO4.5H2O | Sigma | 209198 | |
| Co(NO3)2.6H2O | Sigma | 239267 | |
| Ferric ammonium citrate | Sigma | F5879 | |
| K2HPO4 | Sigma | P3786 | |
| Na2CO3 | Fisher | SODC001 | |
| TES | Sigma | T1375 | |
| NaHCO3 | Fisher | SODH001 | |
| HEPES | Sigma | H3375 | |
| cyanocobalamin | Sigma | 47869 | |
| Na2S2O3 | Sigma | 72049 | |
| Bacto agar | BD | 214010 | |
| Sucrose | Fisher | SUC001 | |
| Petri dish 90 mm triple vented | Greiner | 633185 | |
| 0.2 µm filters | Sartorius | 16534 | |
| 100 ml conical flasks | Pyrex | CON004 | |
| Parafilm M 100 mm x 38 m | Bemis | FIL003 | |
| Phusion high fidelity DNA polymerase | Phusion | F-530 | |
| Agarose | Melford | MB1200 | |
| DNA purification kit | MoBio | 12100-300 | |
| Restriction endonucleases | NEB | ||
| T4 ligase | Thermo Scientific | EL0011 | |
| Luria Bertani broth | Invitrogen | 12795-027 | |
| MES | Sigma | M8250 | |
| Kanamycin sulfate | Sigma | 60615 | |
| Ampicillin | Sigma | A9518 | |
| GeneJET plasmid miniprep kit | Thermo Scientific | K0503 | |
| 14 ml round-bottom tube | BD falcon | 352059 | |
| GoTaq G2 Flexi DNA polymerase | Promega | M7805 | |
| 425-600 µm glass beads | Sigma | G8772 | |
| Glycerol | Sigma | G5516 | |
| DMSO | Sigma | D8418 | |
| Fluorescent bulbs | Gro-Lux | 69 | |
| HT multitron photobioreactor | Infors |
References
- Zwirglmaier, K., et al. Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ Microbiol. 10, 147-161 (2008).
- Galloway, J. N., et al. Nitrogen cycles: past, present, and future. Biogeochemistry. 70, 153-226 (2004).
- Lea-Smith, D. J., et al. Contribution of cyanobacterial alkane production to the ocean hydrocarbon cycle. Proc Natl Acad Sci U S A. , (2015).
- Howe, C. J., Barbrook, A. C., Nisbet, R. E. R., Lockhart, P. J., Larkum, A. W. D. The origin of plastids. Philos Trans R Soc Lond B Biol Sci. 363, 2675-2685 (2008).
- Lea-Smith, D. J., Bombelli, P., Vasudevan, R., Howe, C. J. Photosynthetic, respiratory and extracellular electron transport pathways in cyanobacteria. Biochim Biophys Acta. , (2015).
- McCormick, A. J., et al. Hydrogen production through oxygenic photosynthesis using the cyanobacterium Synechocystis sp PCC 6803 in a bio-photoelectrolysis cell (BPE) system. Energy Environ. Sci. 6, 2682-2690 (2013).
- Bradley, R. W., Bombelli, P., Lea-Smith, D. J., Howe, C. J. Terminal oxidase mutants of the cyanobacterium Synechocystis sp. PCC 6803 show increased electrogenic activity in biological photo-voltaic systems. Phys Chem Chem Phys. 15, 13611-13618 (2013).
- Ducat, D. C., Way, J. C., Silver, P. A. Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 29, 95-103 (2011).
- Dismukes, G. C., Carrieri, D., Bennette, N., Ananyev, G. M., Posewitz, M. C. Aquatic phototrophs: efficient alternatives to land-based crops for biofuels. Curr Opin Biotechnol. 19, 235-240 (2008).
- Tan, L. T. Bioactive natural products from marine cyanobacteria for drug discovery. Phytochemistry. 68, 954-979 (2007).
- Volk, R. B., Furkert, F. H. Antialgal, antibacterial and antifungal activity of two metabolites produced and excreted by cyanobacteria during growth. Microbiol Res. 161, 180-186 (2006).
- Scott, S. A., et al. Biodiesel from algae: challenges and prospects. Curr Opin Biotechnol. 21, 277-286 (2010).
- Lea-Smith, D. J., et al. Phycobilisome-deficient strains of Synechocystis sp. PCC 6803 have reduced size and require carbon-limiting conditions to exhibit enhanced productivity. Plant Physiol. 165, 705-714 (2014).
- Lea-Smith, D. J., et al. Thylakoid terminal oxidases are essential for the cyanobacterium Synechocystis sp. PCC 6803 to survive rapidly changing light intensities. Plant Physiol. 162, 484-495 (2013).
- Liu, X., Sheng, J., Curtiss, R. 3rd Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci U S A. 108, 6899-6904 (2011).
- Xu, H., Vavilin, D., Funk, C., Vermaas, W. Multiple deletions of small cab-like proteins in the cyanobacterium Synechocystis sp PCC 6803 - Consequences for pigment biosynthesis and accumulation. J Biol Chem. 279, 27971-27979 (2004).
- Castenholz, R. W. Culturing methods for Cyanobacteria. Method Enzymol. 167, 68-93 (1988).
- Mitschke, J., et al. An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp PCC6803. Proc Natl Acad Sci U S A. 108, 2124-2129 (2011).
- Ried, J. L., Collmer, A. An nptI-sacB-sacR cartridge for constructing directed, unmarked mutations in gram-negative bacteria by marker exchange-eviction mutagenesis. Gene. 57, 239-246 (1987).
- Vieira, J., Messing, J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene. 19, 259-268 (1982).
- Vermaas, W. F. J., Williams, J. G. K., Rutherford, A. W., Mathis, P., Arntzen, C. J. Genetically Engineered Mutant of the Cyanobacterium Synechocystis 6803 Lacks the Photosystem-Ii Chlorophyll-Binding Protein Cp-47. Proc Natl Acad Sci U S A. 83, 9474-9477 (1986).
- Westphal, S., Heins, L., Soll, J., Vothknecht, U. C. Vipp1 deletion mutant of Synechocystis: A connection between bacterial phage shock and thylakoid biogenesis? Proc Natl Acad Sci U S A. 98, 4243-4248 (2001).
- Zhang, S. Y., Shen, G. Z., Li, Z. K., Golbeck, J. H., Bryant, D. A. Vipp1 Is Essential for the Biogenesis of Photosystem I but Not Thylakoid Membranes in Synechococcus sp PCC 7002. J Biol Chem. 289, 15904-15914 (2014).
- Taroncher-Oldenberg, G., Nishina, K., Stephanopoulos, G. Identification and analysis of the polyhydroxyalkanoate-specific beta-ketothiolase and acetoacetyl coenzyme A reductase genes in the cyanobacterium Synechocystis sp strain PCC6803. Appl Environ Microbiol. 66, 4440-4448 (2000).
- Hein, S., Tran, H., Steinbuchel, A. Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch Microbiol. 170, 162-170 (1998).
- Ng, A. H., Berla, B. M., Pakrasi, H. B. Fine tuning of photoautotrophic protein production by combining promoters and neutral sites in Synechocystis 6803, a cyanobacterium. Appl Environ Microbiol. , (2015).
