

Summary
Immunolabeling metoder til at analysere forskellige populationer af mikrotubuli i udviklingslandene zebrafisk hjernen er beskrevet her, som anvendes bredt til andre væv. Den første protokol skitserer en optimeret metode for immunolabeling stabile og dynamiske mikrotubuli. Den anden protokol indeholder en metode til at billede og kvantificere spirende mikrotubuli specifikt.
Abstract
Mikrotubuli (MTs) er dynamisk og skrøbelige strukturer, der er udfordrende at billedet i vivo, navnlig med hvirveldyr embryoner. Immunolabeling metoder er beskrevet her til at analysere forskellige populationer af MTs i zebrafisk embryo udvikling neuralrøret. Mens der er fokus på neurale væv, er denne metode generelt gældende for andre væv. Procedurerne, der er optimeret for tidligt til midten af somitogenesis fase embryoner (1 somite til 12 somites), men de kan tilpasses til en række andre faser med relativt mindre justeringer. Den første protokol indeholder en metode til at vurdere den geografiske fordeling af stabile og dynamiske MTs og udføre en kvantitativ analyse af disse befolkninger med billedbehandling software. Denne tilgang supplerer eksisterende værktøjer til image mikrotubulus dynamik og distribution i realtid, ved hjælp af transgene linjer eller forbigående udtryk af mærket konstruktioner. Ja, sådanne værktøjer er meget nyttige, men de ikke let skelner mellem dynamisk og stabil MTs. Evnen til at billede og analysere disse særskilte mikrotubulus befolkninger har stor betydning for forståelse underliggende celle polarisering og morfogenese mekanismer. Den anden protokol beskriver en teknik til at analysere spirende MTs specifikt. Dette opnås ved at indfange de novo vækstegenskaber af MTs over tid, efter mikrotubulus depolymerization med stoffet nocodazole og en restitutionsperiode efter narkotika udvaskning. Denne teknik endnu ikke er anvendt til undersøgelse af MTs i zebrafisk embryoner, men er en værdifuld analyse for at undersøge in vivo funktion af proteiner involveret i mikrotubulus forsamling.
Introduction
Mikrotubuli (MTs) er polymerer af α - og β-tubulin, samles til lineære protofilaments, hvoraf flere kombinere til at danne et hult rør1,2. MTs er polariseret strukturer, med hurtigtvoksende plus ender og langsomt voksende minus ender, der er forankret på centrosome eller andre organisering af mikrotubulus center (MTOC)3. De novo MT dannelse er initieret af Nukleering på γ-tubulin ring kompleks (γ-TURC), som indeholder en skabelon til MT forsamling4. I en given celle, kan to populationer af MTs skelnes der tur til forskellige priser. Dynamisk MTs udforske deres cellulære miljø ved at skifte mellem faser af vækst og svind i en proces kendt som dynamiske ustabilitet5. I modsætning til dynamisk MTs, stabil MTs er ikke-voksende og har en længere halveringstid end dynamisk MTs6.
Årtiers forskning i cellebiologi har leveret en avanceret vifte af værktøjer til at studere MT struktur og funktion og resulterede i en omfattende viden om disse cytoskeletal elementer. For eksempel, MTs spiller en central rolle i etablering og vedligeholdelse af celle polaritet, som ikke kun kan tilskrives deres iboende polaritet, men også til en differentieret subcellulært fordeling af stabil versus dynamiske MTs7, 8. derimod langt mindre forstås om MT arkitektur og funktion i mere komplekse tredimensionale (3D) miljøer, såsom hvirveldyr embryoner, delvis på grund af udfordringen af imaging MT cytoskelettet med høj opløsning. Trods denne begrænsning, den seneste generation af normal god landbrugspraksis-udtrykker transgene linjer at mærke MTs eller forbigående udtryk af fluorescently markeret MT markører har øget vor forståelse af de dynamiske ændringer, som MTs gennemgå og deres cellulære og udviklingsmæssige rolle i zebrafisk embryo. Hele MT netværket kan være afbildet i transgene linjer i hvilke tubulin er enten direkte mærket9 eller tubulin polymerer er indirekte mærket ved hjælp af MT-associerede proteiner Doublecortin-lignende-kinase (Dclk) eller Ensconsin (EMTB)10, 11. Andre linjer (og konstruktioner) er blevet genereret, aktiverer vurdering af MT iboende polaritet ved specielt mærkning MT plus ender eller centrosome-forankrede minus ender11,12,13, 14. magt af disse værktøjer ligger i evnen til at studere MT dynamics i live, udvikle organismer. Sådanne undersøgelser har for eksempel afsløret den rumlige og dynamisk fordeling af MTs i specifikke cellepopulationer, orientering af mitotiske spindler i væv gennemgår morfogenese (en indikator af flyet af celledeling), polariteten af MT polymer da det drejer sig om processer såsom celle strækforlængelse og migration, og MT vækst bestemmes af comet hastighed9,13,15. Begrænsning af disse værktøjer er, at de ikke let diskriminerer mellem stabile og dynamiske MT populationer.
Tegning fra de rige celle biologi litteratur, er immunolabeling metoder til billede stabile og dynamiske MTs i zebrafisk embryo beskrevet her, der er komplementær til brugen af transgene linjer. Den udbredte brug af sådanne immunolabeling metoder i zebrafisk er blevet lidt hæmmet af vanskeligheden i at bevare MT integritet under proceduren fiksering. Protokol 1 beskriver en optimeret metode til immunolabeling samlet, dynamisk, og stabil MTs i cross dele af den tredje zebrafisk baghjernen. Derudover er en ligetil metode ved hjælp af kommercielt tilgængelige software beskrevet for at kvantificere disse MT populationer. Stabil MTs adskiller sig fra dynamiske MTs baseret på flere posttranslationelle modifikationer af α-tubulin, som acetylation og detyrosination, der akkumulerer på stabil MTs over tid16,17. I zebrafisk embryo opstår acetylation ciliaere og cytoskeletale MTs, men ikke stabilt interphase MTs18, begrænse nytten af denne markør til en delmængde af stabiliseret MTs. Derimod synes detyrosination at forekomme på alle stabile MTs i zebrafisk embryo18. Denne posttranslationel modifikation udsætter carboxy-terminal glutaminsyre α-tubulin (detyrosinated tubulin)18 og kan påvises ved hjælp af anti-Glu-tubulin19. Selv om detyrosination sker fortrinsvis på stabil MTs, angiver eksperimentelle bevismateriale, at denne posttranslationel modifikation er et resultat af, snarere end en årsag til MT stabilitet16. Den gensidige MT befolkning, sammensat af dynamiske MTs, udmærker sig ved hjælp af en antistof, anti-Tyr-tubulin, der specifikt genkender tyrosinated form af α-tubulin19. Efter immunolabeling med disse markører og konfokal imaging, kvantitativ analyse af MTs (længde, antal og relativ overflod) kan udføres i definerede regioner i udviklingslandene neuralrøret. En strømlinet metode er fastsat her til at udføre denne analyse ved hjælp af 3D-billedbehandling software. Denne metode kan anvendes til løsning af spørgsmål vedrørende morfogenese og etablering eller modning af celle polaritet20. Faktisk, udarbejdelse af polariseret arrays af stabil MTs ledsager mange udviklingsmæssige begivenheder, herunder fotoreceptor morfogenese21, epithelialization af celler i udviklingslandene neurale rør18 og axon dannelse8.
Protokol 2 beskriver en i vivo tilpasning af en celle biologi analyse til at analysere MTs under deres forsamling fase (Nukleering/anchorage og vækst)22,23. Spirende MTs er nukleeret på centrosome og efterfølgende forankret til subdistal vedhæng af mor centriole23. En metode til at analysere spirende MT genvækst efter depolymerization er beskrevet. Denne protokol indeholder oplysninger om nocodazole behandlingen af depolymerize MTs, drug udvaskning procedure og efterbehandling tilbagebetalingsperioden. MT re-vækst overvåges med regelmæssige mellemrums indlæg udvaskning af immunolabeling med markører for samlede MTs (anti-β-tubulin) sammen med markører for centrosome (anti-γ-tubulin) og kerne (4', 6-diamidino-2-phenylindole (DAPI)), efter de generelle procedurer beskrevet i protokol 1. MT depolymerization trin i denne protokol er afgørende, da det giver mulighed for vurdering af de novo MT vækst og ikke forlængelse af allerede eksisterende MTs. Denne teknik er derfor adskiller sig fra andre offentliggjorte procedurer til at måle MT vækstrater (i mangel af depolymerization) ved hjælp af en plus spids markør såsom end bindende protein 3 sammenvoksede til grøn fluorescerende proteiner (EB3-NGL), som vist i Tran et al., 201211. Desuden, denne analyse er især nyttig til at analysere embryoner defekt i de novo MT forsamling, som de tidligere rapporterede NEDD1 mutanter, rekruttering af γ-tubulin til centrosome er nedsat, hvilket resulterer i ufuldstændig neuralrøret dannelse og neuronal defekter24.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
etik erklæring: procedurerne beskrevet nedenfor følger University of Maryland Baltimore County dyrs pleje retningslinjer.
1. analyse af stabile og dynamiske MTs ved hjælp af Immunolabeling (protokol 1)
- Manuel dechorionation af embryoer før fiksering
- få frisk opfostrede embryoner ved at hælde off overskydende system vand og derefter indsamle resterende embryoner i en plastik petriskål (Se Tabel of Materials).
- Fjerne snavs fra systemet vand og overførsel embryoner til en ny fad fyldt med embryo medium (Se Tabel af materialer) at sikre, at embryoner udvikler i et rent miljø.
- Tillade embryoner til at udvikle sig til den ønskede scene i en temperatur-kontrolleret inkubator på 28,5 ° C.
- Sted embryoner yngre end 24 timer efter befrugtning (hpf) i en glasskål forud for dechorionation.
Bemærk: Dechorionate embryoner før fiksering at maksimere hurtige penetration af fiksativ og Bevar MT integritet. Brug embryo medium i stedet for system vand til at give den ekstra Ca 2 + kræves under dechorionation. - Manuelt fjerne chorions fra embryoner i petriskålen, ved hjælp af fine pincet under et dissekere mikroskop.
- Klem et lille område i den runde, gennemsigtig chorion, der omkranser embryo med et par pincet og forsigtigt trække pincet fra hinanden til at skabe et brud i membranen.
- Forstørre åbningen af fint nysgerrige på de bristede chorion med pincet. Pas på ikke at røre fosteret med pincet, så det kan briste.
- Fiksering af iscenesatte embryoner
- overførsel iscenesat, dechorionated embryoner til 1,5 mL centrifugeglas. Fjern så meget embryo medium som muligt ved hjælp af et glas Pasteur pipette.
Bemærk: Udføre fiksering og narkotika behandlinger på young (mid-somitogenesis) embryoner, før dannelsen af neurale centers mægle smerte sensation, som kræver ingen yderligere procedure at lindre smerter under aktiv dødshjælp. Udviklingsmæssige stadier er som defineret i Kimmel mfl., 1995 25. De 4-5 og 11-12 somite faser blev brugt til at hente billeder til figur 2 og 3. - Forberede 4% PARAFORMALDEHYD (PFA) /MT forsamling buffer (MAB) fiksativ (Se Tabel af materialer) ved at kombinere 1 mL 8% PFA per 1 mL 2 X MAB og tilføje 2 µL 100% Triton X-100 per 1 mL af samlede.
Forsigtig: Brug handsker under håndtering opløsninger indeholdende PFA og Triton X-100, som er hudirriterende. - Fix embryoner i 1 mL 4% PFA/MAB fiksativ i 5 min på 28,5 ° C. aspirat fiksativ med en pipette, erstatte det med 1 mL frisk fiksativ, og Inkuber i 3 timer ved stuetemperatur (RT) på en rocker.
Bemærk: Prøver skal være faste hurtigt ved deres biologiske temperatur (28,5 ° C for zebrafisk) at forhindre temperatur-afhængige MT depolymerization.
- overførsel iscenesat, dechorionated embryoner til 1,5 mL centrifugeglas. Fjern så meget embryo medium som muligt ved hjælp af et glas Pasteur pipette.
- Opsug fiksativ og tilsættes 1 mL 1 x Tris-bufferet saltvand med NP40 (TBS-NP40) buffer. Forsigtigt agitere på RT på en rocker tre gange i 5 min. Opbevare embryoner ved 4 ° C i 1 mL frisk 1 X TBS-NP40 for ikke mere end 7 dage.
Forsigtig: Brug handsker når du håndterer opløsninger indeholdende NP-40, en hudirriterende. - Sectioning embryoner for immunolabeling
- varme RT 4% lavt smeltepunkt (LMP) Agarosen indlejring medium i en lukket beholder, indtil løsningen bliver klart ved hjælp af en varmeplade indstillet til 50 ° C placeret tæt på et dissekere mikroskop . Hold beholderen er lukket mellem prøver og opvarmet hele indlejring processen (trin 1.4.4-1.4.6).
- Overførsel embryoner fra 1,5 mL centrifugeres rør til en petriskål ved hjælp af et glas pipette og fyld den med 1 X TBS-NP40.
- Fjerne store æggeblomme celler fra somitogenesis fase embryoner (4-5 og 11-12 somites) i petriskålen ved hjælp af fine pincet under forstørrelse af en dissekere mikroskop 26. Hold embryonet hale bud med et par pincet og skræl væk æggeblomme celler med andre par for at bevare baghjernen væv. Overføre de-yolked embryoner til et område med gratis æggeblomme vragrester petriskål.
Bemærk: Integrere embryoner i Agarosen-fyldt støber individuelt at forhindre for tidlig hærdning af LMP agarosegelelektroforese. - Fyld en 12 mm x 5 mm x 3 mm godt skære formen med 200 µL smeltet LMP Agarosen ved hjælp af en mikropipette. Udfør trin 1.4.5.-1.4.6. hurtigt (inden for 20 s at udfylde formen) at integrere embryoet før LMP Agarosen køler til RT og størkner.
- Bruge fine pincet til at overføre en de-yolked embryo af tailbud fra petriskålen til Agarosen-fyldt støber mod sin tilspidset ende under et dissekere mikroskop.
- Brug fine pincet til at orientere embryo i formen, sådan at vibratome nedskæringer i den ønskede fly. Oprette tværgående sektioner af orientere fosteret, sådan at baghjernen væv løber parallelt med længden af mold med sin dorsale overflade vender kanten og dens anteriore overflade vender ud mod den koniske region ende. Gentag trin 1.4.4-1.4.6 for de resterende embryoner.
- Tillad Agarosen indlejring for at størkne i 5 min på RT.
- Generere 40 µm sektioner af den højeste akse af Agarosen indlejret embryoner (trin 1.4.1-1.4.7) ved hjælp af en vibratome med kontinuerlig skæring parabol fyldt med 1 x TBS-NP40. Overføre dele af interesse til en 24-godt plade i 500 µL 1 x TBS-NP40 ved hjælp af fine pincet. Placere dele af kun ét embryon pr. brønd.
Bemærk: Se du henviser til 18 for flere detaljer. Sikre at sektioner forbliver hydreret på alle tidspunkter i mindst 250 µL af buffer og rock ved lav hastighed (10-25 rpm) for resterende skridt til at forhindre adskillelse fra Agarosen indlejring. Vaske-og rengøringsmidler i blokering og vask løsninger bør reducere overfladespænding af det flydende miljoe og tillade nedsænkningen af sektionerne. Kontroller, at sektioner forbliver i brøndene under og efter alle manipulationer. Forsigtig for at undgå ved et uheld genudsætning sektioner under vasker.
- Fjerne bufferen og tilføje 500 µL blokerende løsning. Rock i mindst 1 time på RT.
Bemærk: Brug en blokerende løsning, der indeholder 5% sera fra værten arter af hver sekundær antistof skal anvendes (Se Tabel of Materials). - Incubate i 300 µL primære antistoffer fortyndet i blokerende buffer i 36-72 timer ved 4 ° C på en rocker. Vaskes to gange i 600 µL 1 x TBS-NP40 på en rocker for 30 min hver, på RT.
Bemærk: Dobbelt-label sektioner ved at inkubere i primære antistoffer mod samlede MTs (anti-β-tubulin, eller anti-α-tubulin) og stabil MTs (anti-Glu-tubulin) eller dynamisk MTs (anti-Tyr tubulin). Vælg primære antistoffer, der er blevet rejst i forskellige vært arter, når dobbelt mærkning for alt og post-translationally ændret α-tubulin populationer. Der henvises til Tabel af materialer for antistof fortyndinger. - Incubate i 300 µL af fluorophore-konjugeret sekundære antistoffer fortyndet i blokerende buffer på en rocker i 16-24 timer, ved 4 ° C i mørke. Vaskes to gange i 600 µL 1 x TBS-NP40 på en rocker for 30 min hver, på RT.
Bemærk: Wrap multi godt skålen indeholdende sekundær antistof i folie fra dette punkt og fremefter og efter hver manipulation at forhindre dæmper. Vælg sekundær antistoffer, der reagerer med værten immunglobulin af det primære antistof. Vælg sekundær antistof fluorophores, som har særskilt, ikke-overlappende emission spektre. Der henvises til Tabel af materialer for antistof fortyndinger. - Ruger embryoner i 500 µL af DAPI løsning på en rocker for 30 min på RT. Wash tre gange i TBS-NP40 vuggende på RT for 5 min.
Bemærk: Nukleare mærkning giver cellulære kontekst for MT kvantificering udført i trin 1.12. - Placere en dråbe af montering medium med anti-fade agent på midten af et støvfrit dias. Brug fine pincet til at overføre dele til montering medium slipværktøjet. Placer en støv-fri coverslip på toppen af prøven. Gemme diasene i et tørt, mørkt og køligt sted, pakket ind i folie, indtil imaging udføres.
Bemærk: Cirkling sektioner på bagsiden af det dias ved hjælp af en bøde-tippes permanent markør inden imaging vil hjælpe med at identificere sektioner ved brug af mikroskop. - Konfokal Imaging
- Mount sektioner på en inverteret laser scanning Konfokal mikroskop ved at anbringe diaset på scenen med coverslip mod målet. Bestemme den passende optik (mål, laser og Kanalindstillinger som få og udligne) på en kontrol dias og holde dem konsekvent mellem prøver 27. Undgå oversaturating pixel for at forhindre datatab.
- Fange Z-stakke Konfokal billeder ved hjælp af kanalindstillinger for den valgte sekundær antistof fluorophores og Gem billede filer 27. Erhverve Z-stakke for hvert afsnit.
Bemærk: Kopiere de parametre, der bruges til at erhverve billeder i figur 2 og 3 ved hjælp af indstillingerne for følgende erhvervelse: tilstand = XYZ; objektive forstørrelse = 63 X olie fordybelse linse; objektive numerisk blænde = 1,4; Z-trin = 0,1 µm; Z-dybde = 16,23 µm. Brug de følgende Kanalindstillinger: DAPI excitation med 20% UV-range laser, emission filterområdet = 430-480 nm, photomultiplier (PMT) gevinst = 525 V og ydelse offset =-1.72%; 448 nm fluorophore (Se Tabel af materialer) excitation med 20% 488 nm laser, emission filterområdet = 493-573 nm, ydelse gevinst = 689 V og ydelse offset =-0.2%; 594 nm fluorophore excitation med 32% 594 nm laser, emission filterområdet = 608-706 nm, ydelse gevinst = 768 V og ydelse offset =-6.8%. - Gemme raw datafiler med entydige, beskrivende filnavne og oprette en kopi til redigering i billed analyse software.
- Samling af Z-stakke til at vise maksimale fremskrivninger
- åbne data fil kopi ved hjælp af offentlige domæne 3-D billede analyse software (fx ImageJ). Kontroller, at hver kanal vises som en enkelt billedsekvens (Z-stacks).
- Split billede kanaler ved hjælp af følgende menu: “ billeder/farve/Split kanaler ”.
- Oprette en flettede billede af overliggende kanaler af interesse ved hjælp af følgende menu: " billeder/farve/Flet kanaler. " Vælg 594 nm, 488 nm og DAPI kanaler skal falsk farvet rød, grøn og blå,. Check " Opret sammensat " og marker " OK " 28.
Bemærk: Udelade DAPI kanal for at bedre formidle detaljer specifikke for MTs i en maksimal projektion som i figur 2 og 3, ved kun at vælge falske farver for de andre to kanaler. - Undersøge de flettede Z-stakken og noter af start- og holdninger af de indre bedste Z-fly til alle synlige kanaler. Afskedige de ydre Z-fly, der typisk har suboptimal signal på grund af de ujævne overflader af afsnittet. Henviser du henviser til 29 for detaljer.
- Visualisere de flettede Z-stak som en enkelt 2D-billede ved at udføre en maksimal intensitet projektion af Z-stakken ved hjælp af følgende 3D-billede analyse menu sekvens: " billeder/stak/Z-projektet. " Angiv start- og sluttider holdninger af den indre bedste Z-fly fra trin 1.11.3 som den " Start skive " og " Stop skive, " henholdsvis. Vælg " Max intensitet " som projektionen type og klik " OK ". Henviser du henviser til 28 for flere detaljer.
- Analysere MT mærkning
- åbne kommercielle 3-D billede analyse software. Vælg " oprette biblioteket " og angive et beskrivende navn for billedbiblioteket. Klik på " oprette. " træk HUDLØS billedfiler genereret fra Konfokal mikroskop ind i biblioteket. Større filer kræver mere tid til at overføre.
- Vælg en fil til at analysere. Vælg " udvidet fokus " fra den " Se " menu for at vise de kanal-fusionerede billede i hovedvinduet.
- Justere tærskel ved at trække værktøjet skyderen for hver kanal til venstre eller højre, indtil baggrunden signal er reduceret og den sande signal er robust. Observere, hver kanal viser et rigtigt signal til molekylet mærket (fx, DAPI kanal viser aflange eller mitotiske kerner, men ikke auto-fluorescens fra cytoplasmaet eller Agarosen).
- Vælg den " Freehand Region af interesse (ROI) " værktøj og redegøre for det pågældende område, der skal analyseres. Vælg de " handlinger " fane efterfulgt af " afgrøde til valg af " til at beskære billedet. Gem den beskårne billedfil under et nyt navn. Klik på den " målinger " fane til at oprette protokol for filtrering specifikke objekter relevante for 3-D analyse.
- Træk " finde objekter " til protokol-vinduet. Omdøbe den første protokol " DAPI. " Vælg DAPI kanal i dropdown-menuen. Træk følgende indstillinger til DAPI protokol og placere dem nedenfor " finde objekter " i følgende rækkefølge (tabel 1): " udfylde huller i ObjectsŔ " Separat rørende ObjectsŔ " Udelukke objekter af SizeŔ " Udelukker ikke rører ROIs ".
Bemærk: Målet med indstillingerne i tabel 1 er du først angive en tærskel, at udsmid signaler hvis fordeling og størrelse er uforenelige med størrelsen af objekter ved at blive analyseret. For eksempel, fjerne et signal ikke stor nok til at være en kerne, når tælle kerner. - Udføre sekvens (trin 1.12.9) at samle den resterende filtre for β-tubulin og andre markører ved hjælp af indstillingerne i tabel 1.
- Vælg " foranstaltning " i bunden af hver protokol. Vælg " intensitet og volumen måling " og " skelet længde " for alle tubulin mærkning, men kun tidligere for DAPI signal.
- Tegner en ROI rundt i regionen skal måles. Overholde målene under den " Resumé " fanen efter softwaren behandler regionen. Kopiere data og gemme dem i en brugbar regneark, som i tabel 2. Oprette en back-up kopi af regnearket til senere analyse.
- Vælg målinger af interesse (eksempelvis længde af MT bundt, antallet af MT bundter/kerne, som afslørede med forskellige markører) i regnearket og analysere for at bestemme gennemsnittene for hver gruppe.
Bemærk: Den gennemsnitlige MT bundt længde = summen af de ' betyde skelet længde for β-tubulin ' for hvert embryon, divideret med det samlede antal af embryoner. Henvise til række 20 i tabel 2. Formatere regnearket, så variabler og eksperimentelle grupper gengivelsesegenskaberne let.
2. De Novo MT forsamling Assay (protokol 2)
- konstruktion og test multi godt gennemstrømnings-apparatet ( figur 1) 2 dage før eksperimentet.
Bemærk: Apparatet muliggør samtidige udvaskning af flere eksperimentelle grupper efter nocodazole behandling ved hjælp af leverancer fra Tabel af materialer. Silikone sealer kræver mindst 24 h af tørretid før det ikke indebærer nogen toksicitet risiko at embryonerne.- Split et 50 mL centrifugeglas i halve på langs, ved hjælp af en jig eller båndsav.
- Skære 7 halvcirkler med en radius på 3 cm ud af 70 µm nylon mesh og trimme dem til at passe stramt ind i en halvdelen af Splits centrifugeres tube. Lim halvcirkler i centrifugeglasset parallelt med 10 mL trindeling markeringer ved hjælp af akvariet-safe silicium sealer. Lade enheden tørre i 2 dage og skyl af iblødsætning i et bægerglas med vand til 2-3 h.
- Line top (gevind) årets cut centrifugeglasset med modeling ler således, at højden af væsken opbevares i gennemstrømnings-enheden har en dybde på ¼ tommer ( figur 1, 2 og 3).
- Forbered udvaskning apparatet ved at fjerne stemplet fra en 50 mL sprøjte og indsætte 12 inches af fine slangen i spidsen. Skub slangen, som det vil gå og forsegle omkring leddet ved hjælp af modellering ler.
- Før våd den trådnet ved hjælp af embryo medium at give væske til at køre gennem hele gennemstrømnings-enheden. Vinkel enhed på isen så at væske pools i alle rum, men stadig tømmes ud foran hvor ler rand er placeret. Ved hjælp af en ring fod, suspendere apparatet udvaskning over gennemstrømnings-enhed på is ( figur 1).
- Chill 200 mL af embryo medium på is og hæld nok udvaskning apparatet til at sikre, at alle luftbobler er ryddet og at flowet er ca 7 mL/min. Juster flowet ved at ændre højden af sprøjten.
- Enzymatisk dechorionate embryoner
- gøre en brugsopløsning af ikke-specifik protease ved fortynding af 1 mL af 10 mg/mL non-specifik protease lager i 20 mL embryo medium.
- Udføre kemiske dechorionation på embryoner 1 time før tidspunkt, hvor de forventes at nå den ønskede udviklingsstadiet. Fordøje chorions ved at fjerne embryo medium fra 100 mm petriskåle, der indeholder iscenesat embryoner og tilføje 20 mL af ikke-specifik protease brugsopløsning.
- Incubate embryoner ved 37 ° C i 5 min.
Bemærk: Ikke overstige 5 min eller bruge en højere koncentration af ikke-specifik protease løsning, da dette vil resultere i embryoner falder fra hinanden en gang behandlet med nocodazole. - Hurtigt afpipetteres ud non-specifik protease og genopfylde retter med ca 25 mL embryo medium. Gentag engang.
- Ved hjælp af en 1-mL glas pipette, overførsel embryoner yngre end 24 hpf til glas retter for at beskytte dem mod skade.
- Komplet dechorionation ved manuelt at fjerne chorions ved hjælp af et par fine pincet, som beskrevet i trin 1.1.5.
- Placer glas petriskåle, der indeholder dechorionated embryoner i en 28,5 ° C inkubator i mindst 30 min., indtil de når den ønskede udviklingsstadiet.
- Depolymerize de eksisterende MTs
- forberede en brugsopløsning af 5 µg/mL nocodazole ved at kombinere 50 µL 1 mg/mL stock nocodazole med 10 mL is kold embryo medium.
Forsigtig: Brug handsker når du håndterer nocodazole, en hudirriterende. - Udveksle embryo medium af gruppen nocodazole behandling med 10 mL koldt nocodazole brugsopløsning. Placer petriskåle på is i en passende tid til udviklingsstadiet (f.eks 1 h for 4-5 somite embryoner). Afsat ubehandlet kontrol embryoner i en petriskål på isen skal fastsættes sammen med udvaskning prøver i trin 2.3.4.1.
- Overførsel embryoner ved hjælp af en brand poleret 1 mL glas pipette til gennemstrømnings-apparater, med separate rum til hver eksperimentelle gruppe. Start nocodazole udvaskning af hælde is kold embryo medium ind i top 50 mL sprøjte.
Bemærk: Brug mindst 30 embryoner pr. eksperimentelle gruppe. Eksperimentelle grupper kunne bestå af kontrol embryoner eller en række morpholino eller RNA-indsprøjtning embryoner. Udskylning vil kræve en alt omkring 150 ml af embryo medium tilføjes hver 8-10 min. udvaskning nocodazole samtidig med at hæmme MT vækst med is. Holde embryoner på isen er afgørende for succes i denne analyse, fordi MTs er ustabile ved kolde temperaturer og kolde forsinker udviklingen i tidlige embryoner. - Tillader MTs til regrow efter 20 min for udvaskning på RT ved at overføre embryoner til glas petriskåle, der indeholder varm (28,5 ° C) embryo medium ved hjælp af en brand poleret 1 mL glas pipette. Så snart at embryonerne overføres, start en timer.
- Fastsætte kontrol og udvaskning embryonerne på 1 min., 5 min. og 10 min. når der afpipetteres ca 10 embryoner i en 1,5 mL centrifugeres rør fyldt med 1 mL 4% PFA/MAB fix (28,5 ° C) og følge vejledningen i trin 1.2.3.
- forberede en brugsopløsning af 5 µg/mL nocodazole ved at kombinere 50 µL 1 mg/mL stock nocodazole med 10 mL is kold embryo medium.
- Forberede prøverne for immunolabeling som beskrevet i afsnit 1.3-1.5.
- Immunolabel flydende sektioner og billede embryoner som beskrevet i afsnit 1.6-1.10 med følgende ændringer til primære antistof specifikationer: Brug 1: 500 kanin anti-γ-tubulin og 1:200 musen anti-β-tubulin.
- Proces og analysere billeder ved hjælp af 3D-billede analyse software, som beskrevet i trin 1.12.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Analyse af stabile og dynamiske MTs ved hjælp af immunolabeling
I protokol 1, er fordelingen af MT delpopulationer i begyndelsen (neurale køl) og slutningen (neurale stang) stadier af neuralrøret udvikling afsløret, bruger Glu-tubulin og Tyr-tubulin som markører for stabile og dynamiske MTs, henholdsvis. Dynamisk MTs dominerer i baghjernen på stadiet neurale køl (4-5 somites) (figur 2A-D). Som køl udvikler sig til neurale stangen (11-12 somites), en fase af forbedrede epithelialization, er kvalitativt færre MTs immunoreaktive med Tyr-tubulin antistof (figur 2E-H), især i den ventrale rod. Derimod Glu-tubulin er spredte og punktformet hele den neurale køl (fig. 3A-D), men er beriget med de ventrale neurale stang langs MT skrifter (figur 3E-H). Pilespidser peger på specifikke MT bundter eller strukturer, hvor mærkning er øget.
Selv om både anti-Glu-tubulin og Tyr-tubulin antistoffer blev produceret i den samme vært arter (forhindrer en dobbelt mærkning eksperiment), viser disse resultater, at stabile og dynamiske MT markører sjældent overlapper hinanden i zebrafisk baghjernen. For det første, de ventrale neurale rod har mere stabil (figur 3F) end dynamisk (figur 2F) MTs. Tendensen er vendt i den dorsale neurale stang, i overensstemmelse med en model af zebrafisk neurulation hvor den dorsale væv forbliver dynamisk indtil neuralrøret er dannet20. For det andet, mens mitotiske spindler er fuldt mærket med Tyr-tubulin antistof i den neurale køl (figur 2D, pilespidser) kun bunden af spindel, sammenfaldende med centrosome, er mærket med stabilitet markør () Glu-tubulin Figur 3 D, pilespidser). Β-tubulin immunofluorescens, fælles for begge assays, oplyser eksperimentatoren distribution af alle MTs og giver et grundlag for at afvise ikke-specifik mærkning.
Måling af objekter ved hjælp af 3D-billede analyse software resulterer i en stor mængde data, der kan organiseres i en praktisk tabel (tabel 2). For at gøre længde, tælle og området målinger, bruger vi kun et undersæt af de data, der er tilgængelig til at analysere. En af komponenterne i de data, vi ikke yderligere analysere er antallet af objekter identificeret. Dette tal bruges som en intern kvalitetskontrol, som antallet ikke bør varierer meget mellem som sektioner og forholdet mellem kerner til MTs bør holde lignende i en enkelt behandling tilstand. En outlier er en indikator, der enten analysen skal køres igen med tilpassede filtre eller at billedet er for dårligt mærket for at analysere. Således bør alle outlier billeder reanalyzed med tilpassede indstillinger. Afsnittet outlier bør undersøges for tegn på dårlig mærkning eller fysiske skader, der måtte resultere i usædvanlige objekt tæller. Når analysen er færdig og kvalitet kontrolleres, nyttige oplysninger kan inddrives fra de rå data såsom den gennemsnitlige længde af samlede MTs og stabil MTs eller forholdet mellem stabil MTs til samlede MTs (tabel 3). Ud over disse målinger, kan mange andre målinger opnås ved hjælp af 3D-billede analyse software, der kan bruges til at drage slutninger om MTs eller deres relation til andre cellulære strukturer (nucleus, centrosome, osv.).
De novo MT forsamling assay
Nocodazole behandling depolymerizes MTs resulterer i diffus mærkning (figur 4A, 4 D, og 4 G). Som MTs regrow, de strækker sig fra centrosome (fig. 4B, 4E, og 4 H), dette kan dog ikke være indlysende i et enkelt fly på grund af deres ikke-planar baner (figur 4C, 4F, og 4I). Ikke desto mindre nogle billede analyse software er i stand til at måle længder i 3-D, muliggør en vurdering af MT vækst efter nocodazole nedtonet (tabel 4). En vigtig observation, der kan opnås fra datasæt i tabel 4 er, at den gennemsnitlige længde af MTs synes at stige i tidens løb efter nocodazole udvaskning i alle regioner af neuralrøret analyseret. Som nævnt ovenfor, kan andre typer af målinger fra 3-D billede analyse software giver cellulære kontekst for at fortolke MT data (f.eks. forholdet mellem MTs per kerne).
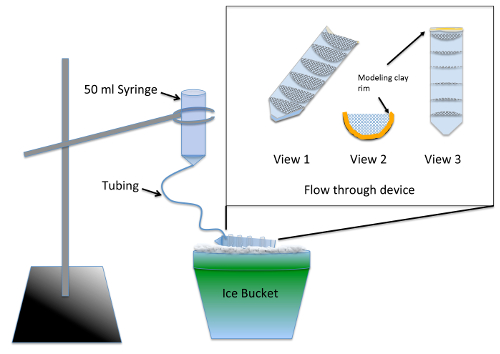
Figur 1 : Illustration af udvaskning apparater til de novo MT forsamling assay. Indsatsen er et nærbillede af gennemstrømnings-enhed fra mesh limet i et 50 mL-centrifugerør længdesnit. Trådnet compartmentalizes gennemstrømnings-enhed, således at flere eksperimentelle grupper kan behandles samtidigt. Under brug, embryo medium er føjet til sprøjten og strømmer langsomt gennem slangen til at fylde gennemstrømnings-enhed, giver en konstant skyl alle eksperimentelle grupper. Venligst klik her for at se en større version af dette tal.

Figur 2: brug af immunolabeling til billede dynamisk MTs. Dechorionated embryoner blev fastsat på relevante stadier (4-5 i A-D og 12-13 somites i E-H), paa tvaers i snit gennem baghjernen og immunolabeled med antistoffer mod β-tubulin (grøn i A og E) til at markere alle MTs og tyrosinated α-tubulin (rød i B og F) for at afsløre dynamisk MT populationer. Højdynamiske MTs kan ses i de flettede billeder (C, G) og deres højere forstørrelser (D, H) som områder hvor gul etiket er synlige (pilespidser i D, H). Skalere barer = 25 µm (A-C og E-G) og 10 µm (D og H). Venligst klik her for at se en større version af dette tal.

Figure 3: brug af immunolabeling til billede stabil MTs. Dechorionated embryoner blev fastsat, sectioned gennem baghjernen og immunolabeled på relevante stadier (4-5 somites i A -D og 12-13 somites i E-H). Stabil MTs er mærket med antistoffer mod detyrosinated formen af α-tubulin (Glu-tubulin) (rød i B og F), mens samlede MTs blev visualiseret med en generel β-tubulin antistof (grøn i A og E). Røde og gule signaler i flettede billeder (C, G) og deres højere forstørrelser (D, H) repræsenterer områder af høj MT stabilitet (pilespidser i D, H). Skalere barer = 25 µm (A-C og E-G) og 10 µm (D og H). Venligst klik her for at se en større version af dette tal.

Figur 4: brug af immunolabeling til billede spirende MTs. Dechorionated embryoner blev fastsat til 4-5 somites og paa tvaers i snit gennem baghjernen. Sektionerne var immunolabeled med β-tubulin (D, E og F) markere voksende MTs og γ-tubulin (A, Bog C) til at markere Nukleering punkt/centrosome. En dorsale region af neuralrøret er boxed i (A, D; B, E og C-F) og vist på højere forstørrelse (G, H, jeg, henholdsvis) at afsløre kerner (DAPI, blå), centrioler (γ-tubulin, red) og total MTs (β-tubulin, grønne). Hvid pilespidser: colocalization af MTs og centrioler; gul pilespidser: den anden centriole i en celle er synlige. Skalere barer = 25 µm (A-F) og 10 µm (G-jeg). Venligst klik her for at se en større version af dette tal.

Tabel 1: Standard indstillinger for filtrering objekter i 3D-billede analyse software.

Tabel 2: repræsentative rå data, der er fremstillet ved hjælp af 3D-billedanalyse software til at analysere stabil MTs. Hver kolonne repræsenterer målinger fra en enkelt sektion. Min.: mindste måling; Max: største måling; SD: standard afvigelse; SE: standard fejl.

Tabel 3: eksempler på datasæt, der kan opnås fra 3-D billede analyse software for at kvantificere stabil MTs. Vælg målinger af den gennemsnitlige længde af alt (β-tubulin) og stabil (Glu-tubulin) MTs beregnes ved at tage gennemsnittet af de gennemsnitlige skelet længde for den pågældende etiket fra alle prøver (Se tabel 2) og forholdet mellem stabile for at MTs (i alt Glu-Tubulin striber per β-tubulin striber) ved beregningen af den gennemsnitlige β-tubulin tælle divideret med den gennemsnitlige Glu-tubulin count.

Tabel 4: Eksempler på datasæt, der kan fås fra 3-D billede analyse software til at analysere de novo MT forsamling. Repræsentative resultater fra de novo MT forsamling eksperimentere, sammenligne datasæt opnået for tre opsving tid point (1, 5 og 10 min) efter nocodazole nedtonet. For hvert tidspunkt er målinger opnået for nukleare count, centrioler (γ-tubulin puncta), antallet af samlede MTs (β-tubulin striber), vist for udvalgte regioner i de afbildede analyseret (tværsnit af udvikle neuralrøret).
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Der er i øjeblikket mange metoder til imaging MT dynamics i tidlige zebrafisk udvikling, lige fra live imaging af mærkede molekyler til immunolabeling af faste væv11,12,13,14. Selvom MTs i en enkelt celle kan findes i enten dynamiske eller stabile stater, er epithelialization en proces, hvor stabiliseret MTs gradvist over tid. Ved hjælp af markører for stabile og dynamiske MTs tilbyder en måde at visualisere dette fænomen. Metoden præsenteres her udnytter styrken af 3D-billedbehandling software til at kvantificere overgangen fra dynamiske til stabil MT populationer i et tværsnit af embryonale zebrafisk væv. I protokol 2, der metoden bruges til at mærke en særskilt population af spirende MTs og følge deres Nukleering og vækst overarbejde.
MTs er notorisk vanskeligt at billede i deres oprindelige tilstand på grund af deres tilbøjelighed til at depolymerize. Således er en væsentlig del af denne metode hurtig fiksering af MTs hele hele fosteret. Dette opnås ved at starte fiksering ved fysiologisk temperatur og ved hjælp af en buffer, der både stabiliserer MTs og øger permeabilitet af embryoet. Fiksering er også vigtige som afkortede fiksering undlader at arrestere MTs mens overdrevne fiksering kan maskere epitoper, forstyrrer antistof bindende. Den foreslåede fiksering tid på 3-4 h arbejder med embryoner, der er i midten gastrulation op til 24 timer efter befrugtningen. Embryoner i yngre slutningen af tidsskalaen bør fastsættes for tættere til 3 h mens ældre embryoner skal muligvis hele 4 h. Selv med korrekt fiksering, vil MTs depolymerize med tiden så skære- og immunolabeling skal ske senest en uge efter fastsættelse.
Når vævet er korrekt fastsat, kan der opstå problemer med immunolabeling. Den mest almindelige problem støder har været ringe penetration gennem midten af væv, især, hvis alt for mange sektioner er udruget i den samme godt. Øger koncentrationen af det primære antistof og inkubering tid til primære og sekundære antistoffer, kombineret med stigende rengøringsmidler for at forbedre permeabilization for embryoner vil rette op på de fleste af immunolabeling problemerne. Hvis antistof mærkning mislykkes på grund af problemer med fiksation eller immunolabeling problemer, er det muligt at bestemme årsagen ved at undersøge det antistof mærkning mønster. Dårlig fiksering vil resultere i intens mærkning i membranen og diffuse mærkning i cytoplasmaet, mens overdrevne fiksering vil resultere i svage mærkning der bevarer MT arkitektur. Dårlig penetration af antistof, men vises som områder i centrum af væv uden mærkning.
Evnen til at analysere MT billeder på en meningsfuld måde er afhængig af høj kvalitet billeddannelse. For at fange MT længde i 3D, bør det mindstemål muligt for mål og numerisk blænde Z-trin anvendes. Billeder, der vises her blev fanget med en 63 X olie emersion målet med 1,4 numerisk blænde producerer følgende: pixel = 240 nm, Z-trin = 0,1 µm, Z-stack størrelse = 16.252 µm. Fordi bredden af en enkelt MT er 25 nm, groft 10 gange nedenfor beslutning for et lysmikroskop, denne metriske ikke kan være præcist målt ved hjælp af denne teknik. I stedet, kun MT længder lig med eller større end den minimale pixelstørrelse opnåelige i alle tre dimensioner kan måles. Line og/eller ramme gennemsnit kan forbedre MT signal definition. MT analyse bør reserveres til høj kvalitet sektioner. Mens væv med dårlig fiksering ikke kan afbildet og analyseret, mild overfixation kan opvejes ved omhyggeligt at øge laser intensitet og få til at opdage det svage signal samtidig opretholde en god dynamikområde. Dårlig antistof penetration, kan mens ikke optimal, rettes ved at begrænse billede erhvervelse til godt mærket regioner, hvilket resulterer i imaging en tyndere sektion (5-10 µm). Høj baggrund fra mærkning kan kompenseres for ved at justere filterindstillingerne for. Men hvis nogen af disse justeringer er færdig, det er nødvendigt at kontrollere, at tærsklen filtre acceptabelt på hvert fly af Z-stakken.
3D-billede analyse software giver mulighed for eksperimentatoren at kvantificere MT længde, areal, vinkel, overflod og andre målinger i 3D-rummet af faste væv sektioner. Metoden beskrevet her giver retninger for at opnå sådanne data ved hjælp af kommercielt tilgængelige software. De filtrering moduler kan dog tilpasset til public domain-software forbedret med relevante plugins og/eller makroer, tilrådighedsstillelse analyse til alle. Inden analyse, skal raw billeder være thresholded at undgå herunder baggrund og ikke-specifikke signaler i kvantificering. Når analysen er afsluttet og data overføres til en brugbar regneark, kan mange slutninger gøres fra datasættet. En af de beregninger, der foretages her var Glu-tubulin striber per β-tubulin striber eller forholdet mellem stabil MTs til samlede MTs, hvor 1 repræsenterer, at hele MT cytoskeleton var stabiliseret i en ROI. Hvis eksperimentatoren ønsker at supplere deres kvantitative data, generere en poleret tagged image file format (TIFF) er billede med skala barer ubesværet med 3-D billede analyse software.
Denne analyse giver mulighed for den funktionelle analyse af proteiner involveret i MT forsamling, in vivo. Hvis immunolabeling er udført på skiftevis seriel afsnit, kunne denne protokol bruges til at studere dynamisk og stabil MTs i samme embryo. I fremtiden vil ændringer som øget rengøringsmidler eller ændrede indlejring vinkler tillade brugen af disse metoder til ældre embryoner og en bredere vifte af anatomiske spørgsmål.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
Forfatterne har ikke noget at oplyse.
Acknowledgments
Konfokal mikroskop blev købt med midler fra den amerikanske National Science Foundation (NSF), grant #DBI-0722569. Forskningen blev støttet af det amerikanske nationale institutter af sundhed/National Institute of General Medical Sciences (NIH/NIGMS) grant #GM085290 og amerikanske Institut for Defense (DOD) grant #W81XWH-16-1-0466 tildeles R.M. Brewster. E. Vital blev støttet af en bevilling til UMBC fra Howard Hughes Medical Institute gennem pre college og Undergraduate Science Education Program, yde #52008090. S.P. Brown blev støttet af en US Department af uddannelse GAANN Fellowship, en Meyerhoff Graduate stipendium finansieret af NIH/NIGMS tilskud, #GM055036, og en forskning undervisningsopholdet finansieret af den amerikanske DOD grant #W81XWH-16-1-0466.
Materials
| Name | Company | Catalog Number | Comments |
| Agarose | Used to treat petridishes. Prepare 1% agarose by heating a solution of 1 gram agarose per 100 ml 1X embryo medium in a microwave until polymerized. |
||
| Kpipes | Sigma | P7643 | |
| NaCl | Sigma | S7653 | |
| Tris-HCl | Sigma | T3253-500G | |
| KCl | Sigma | P9333-500G | |
| CaCl2·2H2O | Sigma | C5080 | |
| NP-40 | American Bioanalyticals | AB01424 | |
| EGTA | Sigma | E3889-25G | |
| MgCl2 | Sigma | M2670-500G | |
| Bovine serum albumin (BSA) | Fisher | BP1605 | |
| Triton-x | American Bioanalyticals | AB02025 | |
| Anti-Fade mounting medium | Invitrogen | P10144 | |
| Mouse anti-β-tubulin | Developmental studies Hybridoma Bank | E7 | 1/200 |
| Rabbit anti-γ-tubulin | Genetex | GTX113286 | 1/500 |
| Rabbit anti-α-tubulin | Genetex | GTX108784 | 1/1000* |
| Rabbit anti-detyrosinated-tubulin | Millipore | AB3201 | 1/200-1/1000* Titrate antibody with first use of new lot. |
| Rabbit anti-tyrosinated-tubulin | Millipore | ABT171 | 1/500 |
| Mouse anti-centrin | Millipore | 04-1624 | 1/1000 |
| Goat 488 anti-rabbit | Thermofisher | A11008 | 1/500 |
| Goat 594 anti-rabbit | Thermofisher | A11012 | 1/500 |
| Goat 594 anti-mouse | Thermofisher | A11005 | 1/500 |
| Goat 488 anti-mouse | Thermofisher | A11001 | 1/500 |
| Vibratome | Vibratome | 1500 | |
| Forceps | World Precision Instruments | 555227F | |
| 100 mm petri dish | Cell treat | 229693 | |
| 35 mm petri dish | Cell treat | 229638 | |
| 50 ml falcon tube | Fisher | 14-432-22 | |
| Woven nylon mesh 70 um | Amazon.com | B0043D1SZG | |
| Micropipette | Gilson | F123602 | |
| Glass pipette | Fisher | NC-999363-9 | |
| Aquarium sealant | Amazon.com, by MarineLand | Silicone Sealer 1 oz (Tube) | |
| Ring stand | Fisher | 14-675BO | |
| Microbore PTFE Tubing, 0.022"ID | Cole-Parmer | WU-06417-21 | |
| Modeling clay | Amazon.com | Sargent Art 22-4000 | Any wax or oil based non-toxic modeling clay will suffice |
| Clamp | Fisher | 02-215-466 | |
| 60ml syringe | Fisher | 14-820-11 | |
| Embryo medium (E3) | 34.8 g NaCl 1.6 g KCl 5.8 g CaCl2·2H2O 9.78 g MgCl2·6H2O To prepare a 60X stock, dissolve the ingredients in H2O, to a final volume of 2 L. Adjust the pH to 7.2 with NaOH. Autoclave. To prepare 1X medium, dilute 16.5 mL of the 60X stock to 1 L. |
||
| Blocking Solution | 50 ml TBS-NP-40 2.5 ml normal goat serum 1 g BSA 625 µl Triton-X |
||
| TBS-NP-40 (pH 7.6) | 155 mM NaCl 10 mM Tris HCl 0.1% NP-40 |
||
| 2x MAB (pH6.4) | 160 mM KPIPES 10 mM EGTA 2 mM MgCl2 |
||
| Commercial 3-D Image processing Software | PerkinElmer | Volocity (V 6.2) | |
| Dry block heater | VWR | 12621-108 | Used as a hot plate to melt agarose in Protocol 1. |
| Dissecting Microscope | Leica | MZ12 | |
| Confocal Microscope | Leica | SP5 | |
| Flat embedding mold | emsdiasum.com | BEEM 70904-01 | |
| Public domain image processing software | NIH | ImageJ (V 1.5) | |
| * Success varies by lot number | |||
References
- Akhmanova, A., Steinmetz, M. O. Tracking the ends: a dynamic protein network controls the fate of microtubule tips. Nat Rev Mol Cell Biol. 9 (4), 309-322 (2008).
- Conde, C., Cáceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci. 10 (5), 319-332 (2009).
- Kaverina, I., Straube, A. Regulation of cell migration by dynamic microtubules. Semin Cell Dev Biol. 22 (9), 968-974 (2011).
- Kollman, J. M., Merdes, A., Mourey, L., Agard, D. A. Microtubule nucleation by γ-tubulin complexes. Nat Rev Mol Cell Biol. 12 (11), 709-721 (2011).
- Howard, J., Hyman, A. A. Growth, fluctuation and switching at microtubule plus ends. Nat Rev Mol Cell Biol. 10 (8), 569-574 (2009).
- Schulze, E., Kirschner, M. Dynamic and stable populations of microtubules in cells. J Cell Biol. 104 (2), 277-288 (1987).
- Gundersen, G. G., Kalnoski, M. H., Bulinski, J. C. Distinct populations of microtubules: Tyrosinated and nontyrosinated alpha tubulin are distributed differently in vivo. Cell. 38 (3), 779-789 (1984).
- Li, R., Gundersen, G. G. Beyond polymer polarity: how the cytoskeleton builds a polarized cell. Nat Rev Mol Cell Biol. 9 (11), 860-873 (2008).
- Asakawa, K., Kawakami, K. A transgenic zebrafish for monitoring in vivo microtubule structures. Dev Dyn Off Publ Am Assoc Anat. 239 (10), 2695-2699 (2010).
- Wühr, M., Tan, E. S., Parker, S. K., Detrich, H. W., Mitchison, T. J. A model for cleavage plane determination in early amphibian and fish embryos. Curr Biol CB. 20 (22), 2040-2045 (2010).
- Tran, L. D., Hino, H., et al. Dynamic microtubules at the vegetal cortex predict the embryonic axis in zebrafish. Development. 139 (19), 3644-3652 (2012).
- Butler, R., Wood, J. D., Landers, J. A., Cunliffe, V. T. Genetic and chemical modulation of spastin-dependent axon outgrowth in zebrafish embryos indicates a role for impaired microtubule dynamics in hereditary spastic paraplegia. Dis Model Mech. 3 (11-12), 743-751 (2010).
- Yoo, S. K., Lam, P. -Y., Eichelberg, M. R., Zasadil, L., Bement, W. M., Huttenlocher, A. The role of microtubules in neutrophil polarity and migration in live zebrafish. J Cell Sci. 125 (23), 5702-5710 (2012).
- Andersen, E. F., Halloran, M. C. Centrosome movements in vivo correlate with specific neurite formation downstream of LIM homeodomain transcription factor activity. Development. 139 (19), 3590-3599 (2012).
- Lee, S. -J. Dynamic regulation of the microtubule and actin cytoskeleton in zebrafish epiboly. Biochem Biophys Res Commun. 452 (1), 1-7 (2014).
- Bulinski, J. C., Gundersen, G. G. Stabilization and post-translational modification of microtubules during cellular morphogenesis. BioEssays. 13 (6), 285-293 (1991).
- Magiera, M. M., Janke, C. Chapter 16 - Investigating Tubulin Posttranslational Modifications with Specific Antibodies. Methods Cell Biol. 115, 247-267 (2013).
- Hong, E., Jayachandran, P., Brewster, R. The polarity protein Pard3 is required for centrosome positioning during neurulation. Dev Biol. 341 (2), 335-345 (2010).
- Westermann, S., Weber, K. Post-translational modifications regulate microtubule function. Nat Rev Mol Cell Biol. 4 (12), 938-948 (2003).
- Jayachandran, P., Olmo, V. N., et al. Microtubule-associated protein 1b is required for shaping the neural tube. Neural Develop. 11, 1 (2016).
- Nam, S. -C. Role of Tau, a microtubule associated protein, in Drosophila photoreceptor morphogenesis. Genes N Y N 2000. 54 (11), 553-561 (2016).
- Abal, M., Piel, M., Bouckson-Castaing, V., Mogensen, M., Sibarita, J. -B., Bornens, M. Microtubule release from the centrosome in migrating cells. J Cell Biol. 159 (5), 731-737 (2002).
- Delgehyr, N., Sillibourne, J., Bornens, M. Microtubule nucleation and anchoring at the centrosome are independent processes linked by ninein function. J Cell Sci. 118 (8), 1565-1575 (2005).
- Manning, J. A., Lewis, M., Koblar, S. A., Kumar, S. An essential function for the centrosomal protein NEDD1 in zebrafish development. Cell Death Differ. 17 (8), 1302-1314 (2010).
- Kimmel, C. B., Ballard, W. W., Kimmel, S. R., Ullmann, B., Schilling, T. F. Stages of embryonic development of the zebrafish. Dev Dyn Off Publ Am Assoc Anat. 203 (3), 253-310 (1995).
- Beck, A. P., Watt, R. M., Bonner, J. Dissection and Lateral Mounting of Zebrafish Embryos: Analysis of Spinal Cord Development. JoVE J Vis Exp. (84), e50703 (2014).
- FÖldes-Papp, Z., Demel, U., Tilz, G. P. Laser scanning confocal fluorescence microscopy: an overview. Int Immunopharmacol. 3 (13-14), 1715-1729 (2003).
- Ferreira, T., Rasband, W. S. ImageJ User Guide - IJ 1.46. , Available from: https://imagej.nih.gov/ij/docs/guide/ (2010).
- Z-functions - ImageJ. , Available from: https://imagej.net/Z-functions (2017).
