

Summary
Enantiomerically enriched bispiro[γ-butyrolactone-pyrrolidin-4,4'-pyrazolone] skeletons are asymmetrically synthesized through a simple organocatalytic 1,3-dipolar cycloaddition reaction.
Abstract
Bispirocyclic scaffolds are one of the important structural subunits in many natural products that exhibit diverse and attractive biological activities. Recently, we have developed an efficient organocatalytic strategy, which provides facile access to a variety of enantiomerically enriched bispiro[γ-butyrolactone-pyrrolidin-4,4'-pyrazolone] skeletons. In this paper, we demonstrate a detailed protocol for the asymmetric synthesis of drug-like bispirocyclic compounds with two spirocyclic carbon centers via an organocatyltic 1,3-dipolar cycloaddition reaction. Spirocyclization synthons α-imino γ-lactones and alkylidene pyrazolones are prepared first, which are then subjected to a cycloaddition reaction in the presence of a bifunctional squaramide organocatalyst to afford the desired bispirocycles in high yields and excellent stereoselectivities. Chiral high-performance liquid chromatography (HPLC) is carried out to determine the enantiomeric purity of the products, and the d.r. value is examined by proton nuclear magnetic resonance (1H NMR). The absolute configuration of the product is assigned according to an X-ray crystallographic analysis. This synthetic strategy allows scientists to prepare a diversity of bispirocyclic scaffolds in high yields and excellent diastereo- and enantioselectivities.
Introduction
Chiral spirocyclic compounds found prevalent in natural products, chiral ligands and organometallic complexes have emerged as attractive synthetic targets due to their structural complexity and biological activity1,2,3. Specifically, bispirocyclic scaffolds, featured by three rings with two rigid spirocenters, are structural subunits in many natural products with important biological activities4,5. Consequently, the construction of compounds with stereocontrolled, optically pure bispirocyclic skeletons has drawn great attention over the last few decades. A large number of spirocyclic compounds and their derivatives have been synthesized successfully through organometallic approaches and organocatalytic approaches, for example, asymmetric cycloadditions such as 1,3-dipolar cycloadditions and Diels-Alder reactions6,7,8. However, these molecules are mostly monospirocyclic structures, while bispirocyclic structures are less reported on and limited to the construction of indole-based bispirocycles.
In order to obtain more structurally diverse bispirocyclic compounds, the versatility of cycloaddition synthons for the asymmetric construction of spirocyclic centers has been explored9,10,11. Especially with bifunctional squaramide organocatalysts, azomethine ylide12,13,14, such as α-imino γ-lactones, and dipolarophiles, such as alkylidene pyrazolones15,16,17, are able to undergo a simple 1,3-dipolar cycloaddition to construct bispirocyclic skeletons with multiple stereocenters, making them the perfect spirocyclization synthons (Figure 1). After the optimization of the structure of organocatalyst and reaction solvent, this cycloaddition process efficiently affords the desired product with high yields and excellent enantio- and diastereoselectivity. Moreover, this reaction exhibits a relatively high structural tolerance on a broad scope of cycloaddition synthons with diverse functional groups18. This new method provides an efficient access to a variety of highly functionalized drug-like compounds with two quaternary spirocenters via a simple organocatalytic cycloaddition, shining lights on its application in the structural diversity-oriented synthesis of this intriguing class of compounds.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
CAUTION: Please consult all relevant material safety data sheets (MSDS) before use. Chemicals and solvents used were of reagent grade and were used without further purification. All reactions involving air or moisture-sensitive reagents or intermediates were performed under an argon atmosphere.
1. Preparation of α-Arylidiene Pyrazolinone Species
- Preparation of pyrazolones
- Add 40 mL of glacial acetic acid to a 250 mL round-bottom flask from a graduated cylinder at room temperature. Stir the solution while adding hydrazine (1 equivalent, 1.58 mol/L) and methyl acetoacetate (1 equivalent, 1.58 mol/L). Equip the flask with a reflux condenser.
NOTE: This concentration is used because a lower concentration leads to a slower reaction rate. - Heat the reaction flask to 120 °C in an oil bath while stirring for 3 h. After cooling the reaction flask down to ambient temperature, remove the magnetic stir bar, using a stir bar retriever. Concentrate the reaction mixture, using a rotary evaporator at 60 °C. Avoid spilling the reaction mixture because of negative pressure.
- Add 20 mL of deionized water to the reaction flask and transfer the solution into a separatory funnel. Extract the aqueous layer 3x with ethyl acetate (30 mL). Combine the organic layers in the separatory funnel and wash them 2x with brine (50 mL).
- Dry the combined organic layers over anhydrous sodium sulfate for 1 h and, then, remove the sodium sulfate by gravity filtration.
- Remove the solvent on a rotary evaporator at reduced pressure and at 35 °C.
- After removing all the solvent, apply the pyrazolone species when performing section 4.
- Add 40 mL of glacial acetic acid to a 250 mL round-bottom flask from a graduated cylinder at room temperature. Stir the solution while adding hydrazine (1 equivalent, 1.58 mol/L) and methyl acetoacetate (1 equivalent, 1.58 mol/L). Equip the flask with a reflux condenser.
- Preparation of α-Arylidiene Pyrazolinones
- Add pyrazolone (1 equivalent, 0.49 mol/L), benzaldehyde (1 equivalent, 0.49 mol/L), magnesium oxide (0.5 g, 0.6 equivalent), and a magnetic stir bar into an oven-dried 100 mL round-bottom flask under N2 atmosphere.
- Add anhydrous acetonitrile (40 mL) to the reaction flask, using an airtight syringe, and then, equip the flask with a reflux condenser. Heat the reaction flask to 120 °C in an oil bath while stirring for 12 h.
- Monitor the progress of the reaction by thin layer chromatography (TLC), using petroleum ether:ethyl acetate (2:1 [v/v], retention factor Rf = 0.86) as an eluent.
- After the complete consumption of pyrazolone, cool the reaction flask down to room temperature. Filter off the magnesium oxide through a Celite plug.
- Remove the excess acetonitrile by using a rotary evaporator under reduced pressure and at 35 °C. Purify the residue by column chromatography on silica gel eluting with petroleum ether:ethyl acetate (10:1 to 8:1 [v/v]) to provide the crude product.
- Add the crude product into a 100 mL Erlenmeyer flask equipped with a magnetic stir bar, and then, add a minimum volume of 95% ethanol. Place the flask on a hot plate and bring it to a gentle boil until the entire solid is just dissolved. Take the flask off the hot plate and cool it slowly without any agitation.
NOTE: When the mixture is cooled to room temperature, the corresponding α-arylidiene pyrazolinone is formed as pure crystals.
2. Synthesis of α-imino γ-lactones species
- Add α-amino-γ-butyrolactone hydrobromide (1 equivalent, 0.41 mol/L), magnesium sulfate (1 equivalent, 0.41 mol/L), triethylamine (1 equivalent, 0.41 mol/L), and a magnetic stir bar into an oven-dried 100 mL round-bottom flask under N2 atmosphere.
- Add 36 mL of anhydrous dichloromethane to the reaction flask, using an airtight syringe. Stir the reaction mixture at room temperature for 1 h. Add the corresponding thiophene-2-carbaldehyde (1.1 equivalent, 0.45 mol/L) to the solution and stir for another 12 h.
- Monitor the progress of the reaction by TLC, using petroleum ether:ethyl acetate (4:1 [v/v]) as an eluent until the complete consumption of the lactone species has occurred, and then, filter off the reaction mixture, using a filter paper with a pore size of 30−50 μm.
- Add 5 mL of deionized water to the resulting mixture and separate the organic layer from the aqueous phase. Extract the aqueous phase 2x with dichloromethane (30 mL). Combine the organic layers in the separatory funnel and wash them 2x with brine (50 mL).
- Dry the combined organic layers over anhydrous sodium sulfate for 1 h, and then, remove the sodium sulfate by gravity filtration. Remove the solvent on a rotary evaporator at reduced pressure and at 35 °C.
- After removing all the solvent, apply the α-Imino γ-lactones species when performing section 4.
3. Synthesis of bifunctional squaramide catalyst C519
NOTE: For the synthesis of organocatalysts 5C, see Figure 2.
- Preparation of 3-((3,5-bis(trifluoromethyl)phenyl)amino)-4-methoxycyclobut-3-ene-1,2-dione (compound 1)
- Add 3,4-dimethoxycyclobut-3-ene-1,2-dione (1 equivalent, 0.63 mol/L), 3,5-bis(trifluoromethyl)aniline (1.1 equivalent, 0.69 mol/L), 20 mL of methanol, and a magnetic stir bar into an oven-dried 100 mL round-bottom flask under N2 atmosphere.
- Stir the mixture at room temperature for 48 h. The formation of yellow precipitate is an indication that the reaction is taking place.
- Filter the reaction solution through a funnel fitted with filter paper and wash the solid product 3x with methanol (15 mL). Dry the yellow solid in vacuo overnight to afford the final products as yellow solid.
- Synthesis of catalyst C5
- Add 3-((3,5-bis(trifluoromethyl)phenyl)amino)-4-methoxycyclobut-3-ene-1,2-dione (compound 1; 1 equivalent, 0.2 mol/L) and (S)-(6-methoxyquinolin-4-yl)((1S,2R,4S,5R)-5-vinylquinuclidin-2-yl)methanamine (compound 2; 1 equivalent, 0.2 mol/L) and a magnetic stir bar into a 25 mL round-bottom flask under N2 atmosphere.
- Add anhydrous dichloromethane (5 mL), using an airtight syringe. Stir the mixture at room temperature for 48 h.
- Monitor the progress of the reaction by TLC, using dichloromethane:methanol (10:1 [v/v], Rf = 0.49) as an eluent. After the reaction is completed, concentrate the reaction mixture, using a rotary evaporator at 40 °C.
- Purify the residue by column chromatography on silica gel eluting with dichloromethane:methanol (20:1 [v/v]) to provide the desired product.
4. Asymmetric synthesis of bispirocyclic compounds
- Dry a 50 mL round-bottom reaction flask containing a magnetic stir bar. Remove the flask from the oven and cool it to room temperature by blowing on it with inert gas before use.
- Add α-arylidiene pyrazolinone (1 mmol, 1 equivalent, 0.1 mol/L) and α-imino γ-lactones (1.2 mmol, 1.2 equivalent, 0.12 mol/L) to the 50 mL round-bottom flask under N2 atmosphere.
- Add anhydrous ethyl ether (10 mL) to the reaction flask, using an airtight syringe. Then, add the corresponding organocatalyst (0.1 equivalent, 0.01 mol/L) to the solution and stir the reaction mixture at 40 °C.
- Monitor the progress of the reaction by TLC, using petroleum ether:ethyl acetate (4:1 [v/v], Rf = 0.51) as an eluent.
NOTE: The spots of the starting materials and products were visualized using a hand-held 254 nm UV lamp. - After the reaction was completed, concentrate the reaction mixture, using a rotary evaporator at 40 °C.
- Purify the residue by column chromatography on silica gel eluting with petroleum ether:ethyl acetate (4:1 [v/v]) to provide the final product.
- Characterize the final product by 1H and 13C NMR spectra, using a 400 MHz NMR spectrometer. Determine the ee values of the product, using a chiral column.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Various hydrogen-bond donor bifunctional organocatalysts were examined in the presence of organocatalysts in dichloromethane (DCM) at 25 °C (Table 1). The representative synthetic process of organocatalysts is shown in Figure 1. The screening of different organocatalysts (Table 1, entries 1-6) resulted in C5 with excellent stereoselectivity (94% ee, >20:1 d.r., entry 5) and the best yield (85% yield). A further optimization of the solvents (Table 1, entries 7-11) suggested that Et2O was preferable in this synthetic process.
Significantly, to examine the generality of the reaction, a variety of substituents of two spirocyclization synthons with different functional groups were tested successfully using the optimized model reaction conditions, resulting in the desired bispirocycles with good to excellent yields and stereoselectivity. The scope of the pyrazolinone 1a includes a replacement of the phenyl group on α-arylidiene with a wide range of aryl, naphthyl, and thienyl groups, substrates with different substituents such as ethyl, decyl, tert-butyl, and benzyl group at 3-position, and substituents with different electronic properties on the aryl ring at 1-position. Besides, to explore the substrate scope of imino lactone 2a, the cyclic imino ester moiety was substituted with a 5-methylthiophenyl, a phenyl or 2-naphthyl group, providing bispirocycles with similar yields and considerable stereoselectivities.
The structure of bispirocyclic products was confirmed by 1H and 13C NMR spectroscopy. To characterize the optical purity and stereoselectivity of the stereoisomer products, the ee values were determined using chiral HPLC and the d.r. values were determined by 400 MHz 1H NMR. Reprehensive HPLC characterization results of compound 3e are given in Figure 3. To explore the structural relativity, X-ray crystallography was used to analyze 3e, revealing the absolute configuration of the product 3e as (5S,6R,7R,13R). The single-crystal structure of 3e is shown in Figure 4. CCDC 1590396 contains the crystallographic data of 3e, which can be obtained free of charge from the Cambridge Crystallographic Data Centre (www.ccdc.cam.ac.uk/data_request/cif).
For example, the characterization data for bispirocyclic product (3e) were as follows: Rf = 0.51 (4:1 [v/v], petroleum ether/EtOAc); 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 7.2 Hz, 2H), 7.35-7.31 (m, 2H), 7.26-7.22 (m, 2H), 7.19-7.15 (m, 2H), 6.98-6.93 (m, 3H), 6.90-6.88 (m, 1H), 5.03 (d, J = 11.6 Hz, 1H), 4.57 (s, 1H), 4.46-4.40 (m, 1H), 4.09 (td, J = 8.8, 2.0 Hz, 1H), 3.80 (d, J = 11.6 Hz, 1H), 2.67-2.61 (m, 1H), 2.35-2.27 (m, 4H); 13C NMR (100 MHz, CDCl3) δ 178.8, 171.9, 163.9, 161.4, 158.5, 137.1, 136.0, 131.3, 131.2, 128.8, 127.0, 125.8, 125.4, 125.0, 119.7, 116.3, 116.1, 70.8, 67.4, 66.0, 64.1, 57.5, 34.6, 13.6; 19F NMR (376 MHz, CDCl3) δ 112.8. The ee value was determined by HPLC analysis, hexane/2-propanol 80/20, flow rate = 1.0 mL/min, 254 nm, tr = 8.63 min (major); HRMS (ESI) Calcd for C26H23N3O3SF+ [M+H]+ 476.1439, found 476.1446.
| Entry | Catalyst | Solvent | Yield[a] (%) | d.r.[c] | ee[d] (%) |
| 1 | C1 | DCM | 81 | >20:1 | 94 |
| 2 | C2 | DCM | 82 | 12:1 | 90 |
| 3 | C3 | DCM | 30 | 9:1 | 0 |
| 4 | C4 | DCM | 78 | >20:1 | 71 |
| 5 | C5 | DCM | 85 | >20:1 | 94 |
| 6 | C6 | DCM | 70 | 12:1 | 93 |
| 7 | C5 | Toluene | 79 | >20:1 | 95 |
| 8 | C5 | THF | 73 | 15:1 | 89 |
| 9 | C5 | CHCl3 | 71 | >20:1 | 93 |
| 10 | C5 | DCE | 81 | 18:1 | 91 |
| 11 | C5 | Et2O | 88 (83[b]) | >20:1 | 98 |
| [a] The yields were determined by 1H NMR analysis of crude product using 4-iodoanisole as the internal standard. [b] Isolated yield. [c] Ratio is determined by 1H NMR. [d] Determined by chiral HPLC. |
|||||
Table 1: Optimization of the reaction condition. The table has been modified from Chen et al.18.

Figure 1: Model reaction between 1a and 2a. Structures of bifunctional organocatalysts (C1-C6) are listed. This figure shows reactions performed with 1a (0.10 mmol), 2a (0.12 mmol), and catalyst (10 mol%) in solvent (1 mL) at room temperature for 8-72 h. For detailed experimental procedures, see the protocol. This figure has been modified from Chen et al.18. Please click here to view a larger version of this figure.
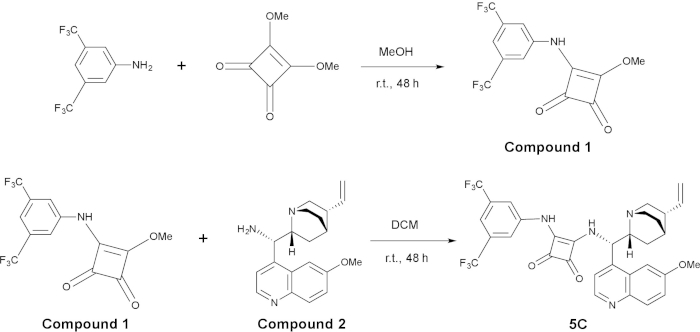
Figure 2: Synthesis of organocatalyst 5C. The top panel is the synthesis of compound 1, and the bottom panel is the synthesis of 5C from compound 1 and compound 2. Please click here to view a larger version of this figure.
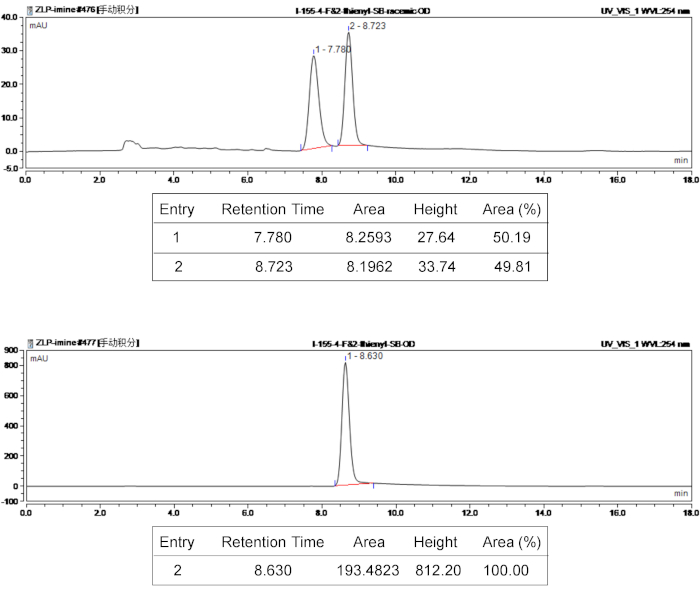
Figure 3: HPLC spectra of racemic and chiral product 3e. The top panel is the HPLC spectrum of racemic product 3e, and the bottom panel is the HPLC spectrum of chiral 3e. Please click here to view a larger version of this figure.

Figure 4: Single-crystal structure of 3e. The left structure is the single-crystal structure of 3e, and the right structure is 3e with the stereochemistry of each atom properly designated. Please click here to view a larger version of this figure.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
The successful preparation of bispiro[γ-butyrolactone-pyrrolidin-4,4'-pyrazolone] skeletons is dependent on a number of factors.
The key step of this one-step asymmetric cycloaddition process is the synergistical activation of the α-arylidiene pyrazolinone 1a and cyclic imino ester 2a by the bifunctional squaramide catalyst. It is achieved by the formation of multiple intermolecular hydrogen bonds between catalyst as a hydrogen-bond donor and two reaction substrates. Accordingly, with large steric hindrance, C5, out of all the hydrogen-bond donor bifunctional organocatalysts screened, exhibited the best stereoselectivity. The noted protocol uses 10 mol% catalyst in the model reaction. Besides, the requirement of high solubility of both substrates and catalyst is essential. As a result, the use of Et2O as the optimal solvent not only ensures that both substrates and catalyst are fully dissolved at room temperature but that they undergo a smooth cycloaddition with high yields and stereoselectivities as well. Notably, water in the reaction system would lead to poor stereoselectivity. In order to ensure a successful synthesis, it is critical to check the dryness of all of the reagents and solvent before starting the reaction.
The cycloaddition is compatible with a wide variety of substituted α-arylidiene pyrazolinone. Specifically, substituents with a different aryl group on α-arylidiene are well tolerated. Electron withdrawing aryl groups, due to their increased electrophilicity during 1,3-dipolar cycloaddition, are preferred in terms of yields and stereoselectivity. Also, substrates such as 3-position, replaced by ethyl, decyl, tert-butyl, and benzyl groups, and 1-position, functionalized by different electronic aryl rings, are highly tolerated. Moreover, substituents of the cyclic imino ester moiety on imino lactone with phenyl, thiophenyl, or naphthyl groups are also compatible with the reaction. It is notable that, in order to ensure a successful reaction, a small excess of imino lactone (1.2 equivalent) is required. In most cases, the concentrations of substrates are kept at a 0.1-0.12 mol/L scale in 1 mL of solvent. Depending on the types of substrates and catalysts, the one-step cycloaddition reaction may take 8-72 h at room temperature.
It is worth noting that this cycloaddition provided a high level of stereoselectivity when the R4 substituent of imino lactone 2 was a thiolphenyl, 5-methylthiophenyl, or 2-naphthyl group. However, when the R4 substituent was replaced with other alkyl substituents or heterocyclic substituents, either a low stereoselectivity or a low reaction yield was achieved.
In summary, the presented protocol allows the direct asymmetric construction of bispiro[γ-butyrolactone-pyrrolidin-4,4'-pyrazolone] using an efficient one-step organocatalytic 1,3-dipolar cycloaddition reaction in excellent yields and a high level of stereoselectivity. Moreover, this new methodology is compatible with two synthons bearing versatile functional groups and should be useful for the synthesis of diverse therapeutic agents with bispirocyclic scaffolds.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors greatly appreciate the financial support from the National Natural Science Foundation of China (No. 21708051 to X.C.).
Materials
| Name | Company | Catalog Number | Comments |
| Acetonitrile, anhydrous, 99.9% | Innochem (China) | A0080 | |
| α-amino-γ-butyrolactone hydrobromide, 98% | Alfa Aesar | B23148 | |
| 3,5-bis(trifluoromethyl)aniline, 98+% | Adamas | 48611B | |
| Dichloromethane, 99.5% | Greagent | G81014H | |
| 3,4-dimethoxycyclobut-3-ene-1,2-dione, 98+% | Leyan (China) | 1062550 | |
| Ethanol, 99.5% | Greagent | G73537B | |
| Ethyl acetate, 99.5% | Greagent | G23272L | |
| Ethyl ether,anhydrous,99.5% | Greagent | G69159B | |
| Ethyl 3-oxobutanoate, 98% | TCI | A0649 | |
| 4-fluorobenzaldehyde, 98% | Innochem (China) | A24295 | |
| Glacial acetic acid, 99.5% | Greagent | G73562B | |
| Magnesium oxide, 99+% | Alfa Aesar | 44733 | |
| Magnesium sulfate, 98% | Greagent | G80872C | |
| Methanol, 99.5% | Greagent | G75851A | |
| Petroleum ether | Greagent | G84208D | |
| Phenylhydrazine, 98% | Innochem (China) | A57671 | |
| (S)-(6-methoxyquinolin-4-yl)((1S,2R,4S,5R)-5-vinylquinuclidin-2-yl)methanamine | DAICEL Group | 111240 | |
| Sodium sulfate,anhydrous,99% | Greagent | G82667A | |
| Thiophene-2-carbaldehyde, 98% | J & K scientific (China) | 124605 | |
| Triethylamine, 99% | J & k scientific (China) | 432915 |
References
- Rios, R. Enantioselective methodologies for the synthesis of spiro compounds. Chemical Society Reviews. 41 (3), 1060-1074 (2012).
- Khan, R. K., et al. Synthesis, isolation, characterization, and reactivity of high-energy stereogenic-at-Ru carbenes: stereochemical inversion through olefin metathesis and other pathways. Journal of the American Chemical Society. 134 (30), 12438-12441 (2012).
- Wang, X., Han, Z., Wang, Z., Ding, K. Catalytic asymmetric synthesis of aromatic spiroketals by spinphox/iridium(I)-catalyzed hydrogenation and spiroketalization of alpha,alpha'-bis(2-hydroxyarylidene) ketones. Angewandte Chemie International Edition. 51 (4), 936-940 (2012).
- Kim, N., Sohn, M. J., Koshino, H., Kim, E. H., Kim, W. G. Verrulactone C with an unprecedented dispiro skeleton, a new inhibitor of Staphylococcus aureus enoyl-ACP reductase, from Penicillium verruculosum F375. Bioorganic & Medicinal Chemistry Letters. 24 (1), 83-86 (2014).
- Mulholland, D. A., Schwikkard, S. L., Crouch, N. R. The chemistry and biological activity of the Hyacinthaceae. Natural Product Reports. 30 (9), 1165-1210 (2013).
- Tan, B., Hernandez-Torres, G., Barbas, C. F. Highly efficient hydrogen-bonding catalysis of the Diels-Alder reaction of 3-vinylindoles and methyleneindolinones provides carbazolespirooxindole skeletons. Journal of the American Chemical Society. 133 (32), 12354-12357 (2011).
- Cayuelas, A., et al. Enantioselective Synthesis of Polysubstituted Spiro-nitroprolinates Mediated by a (R,R)-Me-DuPhos.AgF-Catalyzed 1,3-Dipolar Cycloaddition. Organic Letters. 18 (12), 2926-2929 (2016).
- Lacharity, J. J., et al. Total Synthesis of Unsymmetrically Oxidized Nuphar Thioalkaloids via Copper-Catalyzed Thiolane Assembly. Journal of the American Chemical Society. 139 (38), 13272-13275 (2017).
- Liu, K., Teng, H. L., Yao, L., Tao, H. Y., Wang, C. J. Silver-catalyzed enantioselective desymmetrization: facile access to spirolactone-pyrrolidines containing a spiro quaternary stereogenic center. Organic Letters. 15 (9), 2250-2253 (2013).
- Zhu, G., et al. Asymmetric [3 + 2] Cycloaddition of 3-Amino Oxindole-Based Azomethine Ylides and alpha,beta-Enones with Divergent Diastereocontrol on the Spiro[pyrrolidine-oxindoles]. Organic Letters. 19 (7), 1862-1865 (2017).
- Sun, W., et al. Organocatalytic diastereo- and enantioselective 1,3-dipolar cycloaddition of azlactones and methyleneindolinones. Angewandte Chemie International Edition. 52 (33), 8633-8637 (2013).
- Grigg, R., Kilner, C., Sarker, M. A. B., Orgaz de la Cierva, C., Dondas, H. A. X=Y–ZH compounds as potential 1,3-dipoles. Part 64: Synthesis of highly substituted conformationally restricted and spiro nitropyrrolidines via Ag(I) catalysed azomethine ylide cycloadditions. Tetrahedron. 64 (37), 8974-8991 (2008).
- Liu, T. L., He, Z. L., Tao, H. Y., Wang, C. J. Stereoselective construction of spiro(butyrolactonepyrrolidines) by highly efficient copper(I)/TF-BiphamPhos-catalyzed asymmetric 1,3-dipolar cycloaddition. Chemistry. 18 (26), 8042-8046 (2012).
- Wang, L., Shi, X. M., Dong, W. P., Zhu, L. P., Wang, R. Efficient construction of highly functionalized spiro[gamma-butyrolactone-pyrrolidin-3,3'-oxindole] tricyclic skeletons via an organocatalytic 1,3-dipolar cycloaddition. Chemical Communications. 49 (33), 3458-3460 (2013).
- Yetra, S. R., Mondal, S., Mukherjee, S., Gonnade, R. G., Biju, A. T. Enantioselective Synthesis of Spirocyclohexadienones by NHC-Catalyzed Formal [3+3] Annulation Reaction of Enals. Angewandte Chemie International Edition. 55 (1), 268-272 (2016).
- Liu, J. Y., Zhao, J., Zhang, J. L., Xu, P. F. Quaternary Carbon Center Forming Formal [3 + 3] Cycloaddition Reaction via Bifunctional Catalysis: Asymmetric Synthesis of Spirocyclohexene Pyrazolones. Organic Letters. 19 (7), 1846-1849 (2017).
- Mondal, S., Mukherjee, S., Yetra, S. R., Gonnade, R. G., Biju, A. T. Organocatalytic Enantioselective Vinylogous Michael-Aldol Cascade for the Synthesis of Spirocyclic Compounds. Organic Letters. 19 (16), 4367-4370 (2017).
- Chen, N., et al. Asymmetric Synthesis of Bispiro[γ-butyrolactone-pyrrolidin-4,4'-pyrazolone] Scaffolds Containing Two Quaternary Spirocenters via an Organocatalytic 1,3-Dipolar Cycloaddition. European Journal of Organic Chemistry. 2018 (23), 2939-2943 (2018).
- Yang, W., Du, D. M. Highly enantioselective Michael addition of nitroalkanes to chalcones using chiral squaramides as hydrogen bonding organocatalysts. Organic Letters. 12 (23), 5450-5453 (2010).
