



Overview
Source: Laboratory of Dr. Henrik Sundén – Chalmers University of Technology
Nuclear magnetic resonance (NMR) spectroscopy is a vital analysis technique for organic chemists. With the help of NMR, the work in the organic lab has been facilitated tremendously. Not only can it provide information about the structure of a molecule but also determine the content and purity of a sample. Compared with other commonly encountered techniques for organic chemists — such as thermal analysis and mass spectrometry (MS) — NMR is a non-destructive method that is valuable when recovery of the sample is important.
One of the most frequently used NMR techniques for an organic chemist is proton (1H) NMR. The protons present in a molecule will behave differently depending on its surrounding chemical environment, making it possible to elucidate its structure. Moreover, it is possible to monitor the completion of a reaction by comparing NMR spectra of the starting material to that of the final product.
This video exemplifies how NMR spectroscopy can be used in the everyday work of an organic chemist. The following will be shown: i) preparation of an NMR sample. ii) Using 1H NMR to monitor a reaction. iii) Identifying the product obtained from a reaction with 1H NMR. The reaction that will be shown is the synthesis of an E-chalcone (3) from an aldehyde (1) and a ketone (2) (Scheme 1).1
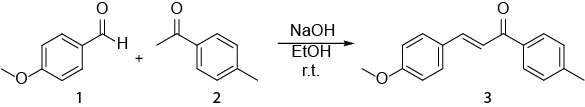
Scheme 1. Synthesis of (2E)-3-(4-methoxyphenyl)-1-(4-methylphenyl)-2-propen-1-one.
Principles
Nuclei with an odd mass or atomic number will have a property called spin, making it possible to use NMR to detect elements such as hydrogen (1H), carbon (13C), and phosphorus (31P). The spins are random and spins in random directions; however, by applying an external magnetic field these nuclei will align themselves either with or against the applied magnetic field. These two states have different energy levels: a low-energy state and a high-energy state. Irradiation with electromagnetic radiation will cause the low energy state to flip over to the high-energy state. When the radiation is ceased, the nuclei will undergo relaxation and a free inductive decay (FID) is obtained. The FID is Fourier transformed to give the peaks of the NMR spectrum. Different elements will have different frequencies leading to different chemical shifts (Table 1). The NMR spectra can provide different types of information about a compound; the integral of a peak provides the number of protons represented by it, the coupling constant (J-coupling) gives the correlation of the protons, and the splitting pattern of a peak tells how many other NMR-active nuclei are nearby (often referred to as 'neighbors').
Since 1H NMR measures 1H nuclei it is important to use deuterated solvents; otherwise the signal of interest will be lost in the noise of the solvent signal.
| Type of proton | Shift (δ, ppm) | Type of carbon | Shift (δ, ppm) |
| 1° Alkyl, RCH3 | 0.8–1.2 | 1° Alkyl, RCH3 | 0–40 |
| 2° Alkyl, R2CH2R | 1.2–1.5 | 2° Alkyl, R2CH2R | 10–50 |
| 3° Alkyl, RCHR2 | 1.4–1.8 | 3° Alkyl, RCHR2 | 15–50 |
| Allylic, R2C=CRCH3 | 1.6–1.9 | Alkene, C=C | 100–170 |
| Ketone, RC(=O)CH3 | 2.1–2.6 | Aryl, C in aromatic ring | 100–170 |
| Ether, ROCH2R | 3.3–3.9 | Alcohol or ether, R3COR | 50–90 |
| Alcohol, HOCH2R | 3.3–4.0 | Carboxylic acid or ester, RC(=O)OR |
160–185 |
| Vinylic, R2C=CH2 | 4.6–5.0 | Aldehyde or ketone, RC(=O)R |
182–215 |
| Vinylic, R2C=CRH | 5.2–5.7 | ||
| Aromatic, ArH | 6.0–8.5 | ||
| Aldehyde RC(=O)H | 9.5–10.5 | ||
| Alcohol hydroxyl, ROH | 0.5–6.0 | ||
| Carboxylic, RC(=O)OH | 10–13 |
Table 1. Common proton and carbon NMR chemical shifts.2
Subscription Required. Please recommend JoVE to your librarian.
Procedure
1. Preparation of NMR Starting Material
- Add ~10 mg starting material to a clean NMR tube.
- Dissolve the starting material in ~0.7 mL deuterated solvent (example given CDCl3). A suitable height of the solvent for a good spectrum is 4.5-5 cm.
- Cap the NMR tube carefully and write the sample name on the cap.
- Shake the sample gently to ensure that all material had dissolved. Take care to avoid contact between the solvent and the cap, which could lead to possible contamination of the sample.
- Insert the NMR tube carefully into a spinner. The spinner will rotate once inserted into the magnet to ensure that the entire sample experiences a homogenous magnetic field. Clean the outside of the NMR tube and spinner with 2-propanol and lab tissues in order to remove fingerprints and dirt.
- Place the spinner in a sample depth gauge to ensure that the bottom of the NMR tube is not inserted too far into the NMR probe as that could possibly damage the spectrometer. Different probes have different sample depths and the user should be aware of the specific depth gauge.
- Place the sample in the NMR spectrometer. Here a Varian 400 MHz spectrometer, equipped with an autosampler, was used.
- After completion of the NMR measurement, process the spectrum and assign the peaks in the spectrum.
2. Preparation of 3 M NaOH and Chalcone Synthesis
- Add 60 mg NaOH to a 50-mL volumetric flask.
- Dissolve the NaOH by adding deionized water to half of the flask. Dilute the solution further by adding more water until the mark is reached.
- Add 10 mL ethanol to a 50-mL round-bottom flask equipped with a magnetic stir bar.
- Thereafter, add 680.5 mg 4-methoxybenzaldehyde and 5 mL NaOH solution prepared in step 2.1 to the same flask.
- Add 671 mg 4-methylacetophenone subsequently to the stirred solution and cap the flask and stir at room temperature.
- Monitor the progress of the reaction by 1H NMR at 30 min intervals (see step 3) until full consumption of starting materials.
- Add 5 mL water when the reaction has reached completion (~3 h). Filter the resulting precipitate and wash it with 20 mL 1:2 ethanol/water. Let the precipitate air dry.
- Calculate the yield of the obtained product. Prepare an NMR sample according to step 1.2.2.7. Check purity with 1H NMR. If not pure, purify the product via recrystallization with ethanol.
- Add approximately 3 drops of reaction mixture to an NMR tube using a Pasteur pipette and rinse the pipette with deuterated solvent.
- Repeat steps 1.2–1.8.
3. Brief Interpretation of an NMR Spectrum
- Process the spectrum with a suitable program (example given MestReNova).
- Correlate the different peaks to the NMR shifts in Table 1. The chemical shifts gives a hint of what type of environment the protons exists in.
- Integrate the peaks to give the number of hydrogens corresponding to each peak. Integration of all peaks gives a relative number of total protons.
- Evaluate the splitting of the proton-peaks, which indicate the number of neighbors.
- Measure the J-coupling to see how the protons are connected to each other.
Nuclear magnetic resonance, or NMR, spectroscopy is an important method for determining the molecular structure and purity of samples in organic chemistry.
In NMR spectroscopy samples are exposed to a strong magnetic field. Upon exposure certain nuclei transition, or resonate, between discreet energy levels. The energy gap between these levels can be measured and visualized as spectra. This data can be used to elucidate the chemical structure of the sample.
Not all nuclei have the required properties to be NMR-active. Common isotopes studied are 1H, 2H, 13C, 19F, and 31P.
This video will introduce the principles behind NMR, step through example preparations of NMR samples from different stages of a chemical reaction, and discuss several applications.
In the NMR instrument, liquid nitrogen and helium are used to cool a super-conducting magnet. The magnet applies a constant magnetic field to a sample. Within the sample, atomic nuclei with an odd number of protons and/or neutrons will align either with the field, adopting a low-energy state, or against it, adopting a high-energy state.
The energy difference between the two levels is the resonant frequency, which depends on the strength of the applied field and the type of nucleus. For the magnets used in NMR, the value is in the radio frequency, or RF, range.
An RF coil excites the sample with a radio frequency pulse, moving the low energy nuclei to the higher state, before reverting back. The coil detects these changes in magnetization, which are displayed as peaks.
The strength of NMR lies in its ability to distinguish nuclei, in this case hydrogen, by their chemical environment. Electrons on neighboring atoms will block, or "shield", the nuclei from some of the magnetic field. This effective field changes the resonant frequency for the particular nuclei, called a chemical shift. In ethanol the methylene, hydroxyl, and methyl protons all have unique resonance frequencies. Determining the area under each peak elucidates the number of each type of proton.
Since instruments with different magnetic strengths will shift the resonant frequencies, they are referenced to a standard molecule added to the sample, often tetramethylsilane, or TMS. The chemical shift of the frequencies is very small, often reported in parts per million, or ppm.
When using a strong magnet with high resolution, peaks will sometimes split into subpeaks. This is caused by the neighboring nuclei, some of which are aligned with the magnetic field, some against; further changing the effective field applied to the nuclei. In ethanol, the 2 methylene protons split the methyl peak twice into a triplet, and the 3 methyl protons split the methylene peak thrice into a quartet. The distance of the splitting, or J-coupling, is related to the distance of the nuclei, aiding in qualitative discovery.
Now that you understand the basic principles behind NMR, let's review an example procedure that uses proton NMR to monitor the synthesis of chalcone from an aldehyde and ketone.
Begin by using a Pasteur pipette to add a tiny amount of the starting material to a beaker. Move into the fume hood, and dilute the starting material with 0.7 mL of deuterated solvent. Deuterated solvents are used, as deuterium's resonant frequency is outside the range for protons.
Use a Pasteur pipette to add 0.7 mL of the diluted starting material into a clean 5 mm NMR tube, filling the bottom 4.5–5 cm. Cap the NMR tube and label it. Gently shake the tube, taking care to avoid contact between the sample and the cap. Then, insert the tube into the spinner.
Clean the outside of tube and spinner using 2-propanol and lab tissues. Then place the sample assembly into the depth gauge and calibrate the insertion depth.
After calibration, load the sample assembly into the NMR spectrometer either manually or using an auto-sampler. Finally, use a computer workstation to acquire the NMR spectrum.
Generate NMR spectra using this procedure for each of the reaction's starting materials. For the synthesis of chalcone, spectra should be generated for both methoxybenzaldehyde and methylacetophenone.
Next, perform the sample synthesis by combining the starting materials and reagents in a flask to begin the reaction.
At 30 min intervals, remove a small aliquot of the reaction mixture using a Pasteur pipette and add 3 drops to a clean NMR tube.
Dilute this crude reaction product with deuterated solvent, and prepare for NMR using the previously described procedure.
As the reaction progresses, a yellow precipitate will form. When the reaction is complete, wash and filter the precipitate and generate NMR spectra for the purified reaction product.
Now that we've generated NMR spectra at each stage of the chemical reaction, let's analyze them.
The peaks of the NMR spectra for each of the starting materials are assigned to different proton groups within the molecule according to their chemical shifts and the number of protons contributing to each peak. Here, we assign the 4 major proton groups for methylacetophenone and methoxybenzaldehyde, noting the aldehyde peak between 9.5 and 10.5 ppm. By comparing the NMR spectra of the crude reaction products at different time-points, the evolution of the chemical reaction that synthesizes chalcone is elucidated. For example, the aldehyde peak from the starting material methoxybenzaldehyde is still present after 30 min of reaction, but is completely gone after 3 h, signifying the completion of the reaction.
By examining the spectrum of the purified product we can assign each peak to a proton group in chalcone's structure. For example, examining peaks 3 and 4 we see their integrals are both one, corresponding to groups containing just one proton.
Peaks 3 and 4 are so called doublets indicating one neighboring proton. Both have J-coupling constants of 16 Hz, suggesting that the protons are situated across an E-double bond. By assigning all of the NMR peaks of the purified reaction product, we confirm the synthesis of a pure chalcone.
NMR spectroscopy has a wide range of applications and is used in many scientific and medical fields.
In this application, proton NMR is used to verify the synthesis and structure of both diamidocarbene and mononoamidocarbine, whose NMR spectra have different peak-splitting patterns. These carbenes also generated seemingly different reaction products when combined with white phosphorus; DAC1 generated a bright red reaction product, whereas MAAC2 produced a bright orange product. These differences in reaction products were confirmed using a second application of NMR, 31P NMR, which generates spectra based on differences in the resonant frequency of phosphorus nuclei.
Here, nuclear magnetic resonance imaging, or MRI, was used to generate an anatomical map of the brain and to select the brain regions of interest. Then NMR spectroscopy was used to generate spectra of key metabolites. Finally, using MRI changes in brain metabolism in different experimental conditions were evaluated.
In this application, NMR was used to analyze the binding properties and propose a 3D structure of a copper-binding peptide. First, NMR spectra were compared for the peptide's unbound and copper-bound states. Then, using more advanced 2-dimensional NMR techniques, different potential conformations of the peptide's structure were evaluated. Finally, these NMR-derived structural constraints were used to develop a proposed 3-Dimensional structure for the unbound peptide.
You've just watched JoVE's introduction to NMR Analysis. You should now understand the underlying principles behind NMR spectra generation and analysis, as well as a procedure for NMR sample preparation.
Thanks for watching!
Subscription Required. Please recommend JoVE to your librarian.
Results
By comparing the spectra of the starting materials (Figures 1 and 2) to that of the final product (Figure 5) a clear difference between the spectra can be observed, indicating formation of the chalcone. The endpoint of the reaction can be determines by taking NMR samples at different time-intervals; for example, the aldehyde proton peak (C(=O)H) (1) can be seen in Figure 3 but not in Figure 4, signifying completion of the reaction after 3 h. By looking at the integrals, splitting patterns, and J-couplings of the peaks, it is possible to validate the structure of the chalcone. The integrals of the peaks (numbers under the peak, Figure 5) show the relative amount of hydrogens present which should correlate to the amount of hydrogens found in the product. Furthermore, the splitting pattern gives an indication of the number of neighbors; for example, peak (5) and (1) — both singlets — indicate no neighbors nearby with integrals of 3 correlates with the MeO- and the Me-group respectively. By comparing the chemical shifts in the spectra with Table 1 it is possible to elucidate that the MeO-group corresponds to the singlet at 3.80 ppm and the Me-group to 2.45 ppm. Moreover, formation of the double bond can be seen as two doublets (Figure 5), 7.80 and 7.44 ppm. Looking at the J-coupling of 16 Hz indicates formation of an E-alkene; the Z-alkene typically has a smaller value of 10–12 Hz.3 Assigning the aromatic peaks verifies the structure (Figure 5).4

Figure 1. Assigned 1H NMR spectra of 4-methylacetophenone Please click here to view a larger version of this figure.

Figure 2. Assigned 1H NMR spectra of 4-methoxybenzaldehyde. Please click here to view a larger version of this figure.

Figure 3. Crude 1H NMR spectra after 30 min showing the residual aldehyde peak. Please click here to view a larger version of this figure.

Figure 4. Crude 1H NMR spectra after 3 h showing no aldehyde residual peak. Please click here to view a larger version of this figure.

Figure 5. 1H NMR spectra of the obtained product after work-up. The inserted picture shows the J-couplings of the alkene. Please click here to view a larger version of this figure.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
NMR can, for example, be used to detect reaction intermediates, facilitating the work in elucidation of a reaction mechanism. With the help of NMR it is also possible to observe molecular motions and interactions important for drug development. Furthermore, NMR can give structural information about solid materials; for instance to provide a rationale for observed material properties. Other applications of NMR can be found in the field of medicine, where magnetic resonance imaging (MRI) is frequently used for medical diagnosis. NMR has also been used in metabolomics to detect different metabolites excreted by an organism thus providing a metabolic fingerprint. The uses of NMR are wide; from determining the structure of a single molecule to imaging of the human brain.
Subscription Required. Please recommend JoVE to your librarian.
References
- Ta, L., Axelsson, A., Bijl, J., Haukka, M., Sundén, H., Ionic Liquids as Precatalysts in the Highly Stereoselective Conjugate Addition of α,β-Unsaturated Aldehydes to Chalcones. Chem. Eur. J. 20 (43), 13889-13893 (2014).
- Table adapted from Graham Solomons, T. W. Fryhle, C. B., Organic Chemistry, 10th edition, Wiley, p. 387, 418 (2011).
- Clayden, J., Greeves, N., Warren, S., Wothers, P. Proton nuclear magnetic resonance. Organic Chemistry, Chapter 11, Oxford University Press, 269 (2001).
- Wu, X.-F., Neumann, H., Spannenberg, A., Schulz, T., Jiao, H., Beller, M.,Development of a General Palladium-Catalyzed Carbonylative Heck Reaction of Aryl Halides. J. Am. Chem. Soc. 132 (41), 14596-14602 (2010).