

Summary
मैं TASSER पाइपलाइन का उपयोग प्रोटीन के संरचनात्मक और कार्यात्मक लक्षण वर्णन आधारित कंप्यूटर के लिए दिशा - निर्देश में वर्णित है. क्वेरी प्रोटीन अनुक्रम से शुरू, 3D मॉडल का उपयोग एकाधिक सूत्रण संरेखण उत्पन्न कर रहे हैं और संरचनात्मक विधानसभा सिमुलेशन चलने का. तत्पश्चात् कार्यात्मक inferences ज्ञात संरचना और कार्यों के साथ प्रोटीन के मैचों के आधार पर तैयार कर रहे हैं.
Abstract
जीनोम अनुक्रमण परियोजनाओं प्रोटीन अनुक्रम के लाखों है, जो उनकी संरचना और समारोह के ज्ञान की आवश्यकता होती है उनके जैविक भूमिका की समझ में सुधार ciphered है. हालांकि प्रयोगात्मक विधियों इन प्रोटीनों की एक छोटा सा अंश के लिए विस्तृत जानकारी प्रदान कर सकते हैं, कम्प्यूटेशनल मॉडलिंग प्रोटीन अणु जो प्रयोगात्मक uncharacterized हैं के बहुमत के लिए की जरूरत है. मैं - TASSER सर्वर प्रोटीन संरचना और समारोह के उच्च संकल्प मॉडलिंग के लिए एक ऑन - लाइन कार्यक्षेत्र है. एक प्रोटीन अनुक्रम को देखते हुए, मैं TASSER सर्वर से एक ठेठ उत्पादन माध्यमिक संरचना भविष्यवाणी शामिल है, प्रत्येक अवशेषों की विलायक पहुंच, मुताबिक़ टेम्पलेट सूत्रण और संरचना संरेखण द्वारा पता लगाया प्रोटीन, पाँच पूर्ण लंबाई तृतीयक संरचनात्मक मॉडल, और संरचना के आधार पर भविष्यवाणी एंजाइम के वर्गीकरण के लिए कार्यात्मक एनोटेशन, जीन सत्तामीमांसा नियम और प्रोटीन ligand बाध्यकारी साइटों. सभी भविष्यवाणियों विश्वास स्कोर के साथ टैग कर रहे हैं जोबताता है कि कैसे सटीक भविष्यवाणियों प्रयोगात्मक डेटा जानने के बिना कर रहे हैं. अंत उपयोगकर्ताओं के विशेष अनुरोध को सुविधाजनक बनाने के लिए, सर्वर चैनल प्रदान करने के लिए उपयोगकर्ता द्वारा निर्दिष्ट अवशेषों अंतर दूरी को स्वीकार करने और नक्शे संपर्क करने के लिए interactively मैं - TASSER मॉडलिंग बदल, यह भी उपयोगकर्ताओं को निर्दिष्ट करने के लिए टेम्पलेट के रूप में किसी भी प्रोटीन की अनुमति देता है, या किसी टेम्पलेट बाहर संरचना विधानसभा सिमुलेशन के दौरान प्रोटीन. संरचनात्मक जानकारी या मैं TASSER भविष्यवाणियों की गुणवत्ता में सुधार लाने के उद्देश्य के साथ प्रयोगात्मक सबूत जैविक अंतर्दृष्टि के आधार पर उपयोगकर्ताओं द्वारा एकत्र किया जा सकता है है. सर्वर प्रोटीन संरचना के लिए सबसे अच्छा कार्यक्रमों और हाल ही में समुदाय चौड़ा CASP प्रयोगों में समारोह भविष्यवाणियों के रूप में मूल्यांकन किया गया था. वहाँ वर्तमान में कर रहे हैं> 100 से अधिक देशों से 20,000 पंजीकृत वैज्ञानिकों ने ऑन - लाइन मैं TASSER सर्वर का उपयोग कर रहे हैं.
Protocol
विधि सिंहावलोकन
प्रतिमान अनुक्रम संरचना करने के लिए समारोह के बाद, संरचना और समारोह मॉडलिंग के लिए मैं TASSER 1-4 प्रक्रिया के लगातार कदम चार शामिल है: (एक) 5 LOMETS द्वारा टेम्पलेट पहचान, (ख) प्रतिकृति के द्वारा reassembly टुकड़ा संरचना मुद्रा मोंटे कार्लो सिमुलेशन 6, (ग) परमाणु स्तर संरचना 7 रेमो और FG-8 एमडी का उपयोग शोधन, और (घ) संरचना के आधार पर समारोह का उपयोग कर व्याख्याओं cofactor 9 .
टेम्पलेट पहचान: एक क्वेरी उपयोगकर्ता द्वारा प्रस्तुत अनुक्रम के लिए, अनुक्रम पहले मेटा - सूत्रण सर्वर स्थानीय रूप से स्थापित LOMETS द्वारा एक प्रतिनिधि PDB संरचना और पुस्तकालय के माध्यम से लड़ी पिरोया है. सूत्रण एक अनुक्रम संरचना संरेखण प्रक्रिया है जो इसी तरह की संरचना है या क्वेरी प्रोटीन के रूप में इसी तरह के संरचनात्मक आकृति शामिल कर सकते हैं टेम्पलेट प्रोटीन की पहचान के लिए प्रयोग किया जाता है. मुताबिक़ templ के कवरेज बढ़ानेखाया detections, LOMETS कई राज्य के-the-कला अलग सूत्रण के तरीके को कवर एल्गोरिदम को जोड़ती है. चूंकि अलग सूत्रण कार्यक्रमों विभिन्न स्कोरिंग सिस्टम और संरेखण संवेदनशीलता है, प्रत्येक सूत्रण कार्यक्रम से उत्पन्न सूत्रण संरेखण की गुणवत्ता सामान्यीकृत Z स्कोर, जो के रूप में परिभाषित किया गया है द्वारा मूल्यांकन है: 
जहां Z स्कोर मानक विचलन इकाइयों में सभी कार्यक्रम द्वारा उत्पन्न alignments के सांख्यिकीय मतलब के सापेक्ष स्कोर है, और जेड 0 विशेष कार्यक्रम Z स्कोर सूत्रण बेंचमार्क 5 बड़े पैमाने पर परीक्षण के आधार पर निर्धारित किया है 'अच्छा अंतर cutoff 'और' बुरा टेम्पलेट्स '. एक उच्च Z स्कोर के साथ एक टेम्पलेट का मतलब है कि शीर्ष टेम्पलेट्स एक संरेखण काफी अन्य टेम्पलेट्स, जो आम तौर पर अर्थ है कि संरेखण एक अच्छा मॉडल से मेल खाती है है की तुलना में सबसे अधिक स्कोर है. यदि शीर्ष सूत्रण टेम्पलेट्स का सबसे हायसामान्यीकृत gh Z-स्कोर, अंतिम मैं TASSER मॉडल की सटीकता आमतौर पर उच्च है . हालांकि, अगर प्रोटीन बड़ी है और सूत्रण alignments के कवरेज क्वेरी प्रोटीन के एक छोटे से क्षेत्र तक ही सीमित है, एक उच्च सामान्यीकृत Z स्कोर जरूरी पूर्ण लंबाई मॉडल के लिए एक उच्च सटीकता मॉडलिंग नहीं मतलब है. प्रत्येक सूत्रण कार्यक्रम से शीर्ष दो सूत्रण संरेखण एकत्र कर रहे हैं और संरचना विधानसभा के अगले कदम के लिए प्रयोग किया जाता है.
संरचना विधानसभा अनुकार चलने का सूत्रण प्रक्रिया के बाद, क्वेरी अनुक्रम गठबंधन और unaligned क्षेत्रों सूत्रण में विभाजित है. सूत्रण संरेखण में निरंतर टुकड़े टेम्पलेट्स से excised और संरचना विधानसभा के लिए सीधे इस्तेमाल किया, जबकि unaligned पाश क्षेत्रों आदितः मॉडलिंग द्वारा निर्मित कर रहे हैं. संरचना विधानसभा प्रक्रिया एक जाली प्रतिकृति मुद्रा मोंटे कार्लो 6 सिमुलेशन द्वारा निर्देशित प्रणाली पर किया जाता है. मैं TASSER बल हाइड्रोजन बो क्षेत्र शामिल हैं10 बातचीत, ज्ञान आधारित सांख्यिकीय ऊर्जा PDB 11, अनुक्रम आधारित 12 SVMSEQ से संपर्क भविष्यवाणियों, और स्थानिक LOMETS 5 सूत्रण टेम्पलेट्स से एकत्र मजबूरी में जाना जाता प्रोटीन संरचनाओं से व्युत्पन्न शर्तों nding. गठनात्मक सिमुलेशन के दौरान कम तापमान प्रतिकृतियां में उत्पन्न decoys 13 SPICKER द्वारा clustered रहे हैं कम मुक्त ऊर्जा राज्यों के ढांचे की पहचान . शीर्ष समूहों के क्लस्टर centroids सभी संकुल संरचनात्मक decoys के 3 डी निर्देशांक औसत और अंतिम मॉडल पीढ़ी के लिए इस्तेमाल किया द्वारा प्राप्त कर रहे हैं. क्लस्टरिंग और प्रक्रिया अनुकरण steric संघर्ष को हटाने और आगे वैश्विक टोपोलॉजी परिष्कृत करने के लिए दो बार दोहराया जाता है.
परमाणु स्तर के मॉडल निर्माण और शोधन: क्लस्टर SPICKER क्लस्टरिंग के बाद प्राप्त centroids प्रोटीन (प्रत्येक अपनी सी α और द्रव्यमान का केंद्र पक्ष श्रृंखला द्वारा प्रतिनिधित्व अवशेषों) मॉडल और घंटे कम कर रहे हैंएवेन्यू सीमित जैविक आवेदन. कम मॉडल से पूर्ण परमाणु मॉडल का निर्माण दो चरणों में किया जाता है. पहले चरण में 7 रेमो सी अल्फा निशान से एच बंधन नेटवर्क के अनुकूलन के द्वारा पूर्ण परमाणु मॉडल का निर्माण किया है. दूसरे चरण में, रेमो पूर्ण परमाणु मॉडल आगे FG 14-एमडी, जो रीढ़ मरोड़ कोण, बंधन लंबाई, और पक्ष श्रृंखला रोटामर झुकाव में सुधार के द्वारा परिष्कृत कर रहे हैं, आणविक गतिशील सिमुलेशन के द्वारा, के रूप में संरचनात्मक से खोजा टुकड़े द्वारा निर्देशित पंक्ति द्वारा PDB संरचनाओं टीएम. FG-एमडी परिष्कृत मॉडल तृतीयक संरचना भविष्यवाणियों के लिए अंतिम मॉडल के रूप में मैं TASSER द्वारा किया जाता है.
उत्पन्न मॉडल की गुणवत्ता विश्वास स्कोर (सी स्कोर), जो जेड LOMETS सूत्रण संरेखण के स्कोर और मैं TASSER सिमुलेशन के अभिसरण, गणितीय के रूप में तैयार के आधार पर परिभाषित किया गया है के आधार पर अनुमान है: 
जहां
सी स्कोर मैं TASSER मॉडल की गुणवत्ता के साथ एक मजबूत सहसंबंध है. सी - स्कोर और प्रोटीन की लंबाई के संयोजन से, पहले मैं TASSER मॉडल की सटीकता टीएम स्कोर और 2 RMSD 15 के लिए एक के लिए 0.08 के औसत त्रुटि के साथ अनुमान लगाया जा सकता है. सामान्य में, सी -> स्कोर के साथ मॉडल - 1.5 के लिए एक सही गुना की उम्मीद कर रहे हैं. यहाँ, RMSD और TM स्कोर दोनों मॉडल और देशी संरचना के बीच topological समानता की अच्छी तरह से जाना जाता है उपाय. टीएम के स्कोर valu[0 1] तों रेंज, जहां एक उच्च स्कोर इंगित करता है एक बेहतर संरचना मैच 16,17. लेकिन निचले क्रम मॉडल (यानी 2 एन डी -5 वें मॉडल) के लिए, टीएम स्कोर और RMSD के साथ सी - स्कोर के संबंध बहुत कमजोर है (~ 0.5), और निरपेक्ष मॉडल गुणवत्ता के विश्वसनीय आकलन के लिए नहीं किया जा सकता है.
पहला मॉडल मैं TASSER सिमुलेशन में हमेशा सबसे अच्छा मॉडल है? इस सवाल का जवाब लक्ष्य प्रकार पर निर्भर करता है. आसान लक्ष्य के लिए, पहली मॉडल आम तौर पर सबसे अच्छा मॉडल और सी स्कोर आम तौर पर मॉडलों के बाकी की तुलना में ज्यादा है. हालांकि, मुश्किल लक्ष्य है, जहां सूत्रण महत्वपूर्ण टेम्पलेट हिट नहीं है के लिए, पहली मॉडल जरूरी बेहतरीन मॉडल नहीं है और मैं TASSER वास्तव में सबसे अच्छा टेम्पलेट और मॉडल का चयन करने में कठिनाई है. इसलिए यह कठिन लक्ष्य के लिए सभी 5 मॉडल का विश्लेषण करने और उन्हें प्रायोगिक जानकारी और जैविक ज्ञान के आधार पर चयन करें की सिफारिश की है.
फंक्शन predictions: अंतिम चरण में, अंतिम FG-एमडी से उत्पन्न 3D मॉडल प्रोटीन समारोह के तीन पहलुओं की भविष्यवाणी, अर्थात् करने के लिए उपयोग किया जाता है:) एनजाइम आयोग (ईसी) 18 नंबर और (ख) जीन (जाओ) आंटलजी 19 नियम और ( ग) छोटे अणु ligands के लिए बाध्यकारी साइटों. सभी तीन पहलुओं के लिए कार्यात्मक व्याख्याओं cofactor, जो प्रोटीन PDB में जाना जाता संरचना और कार्यों के साथ वैश्विक और स्थानीय टेम्पलेट प्रोटीन समानता के आधार पर समारोह की भविष्यवाणी के लिए एक नया दृष्टिकोण है का उपयोग कर उत्पन्न कर रहे हैं. सबसे पहले, भविष्यवाणी मॉडल की वैश्विक टोपोलॉजी कार्यात्मक टेम्पलेट पुस्तकालयों के खिलाफ मिलान संरचनात्मक संरेखण प्रोग्राम का उपयोग कर 20 TM-संरेखित. अगला, अपने वैश्विक संरचना समानता के आधार पर लायब्रेरी से सबसे लक्षित मॉडल करने के लिए इसी तरह की प्रोटीन का एक सेट का चयन कर रहे हैं, और एक व्यापक स्थानीय खोज साइट सक्रिय / बाध्यकारी क्षेत्र के निकट संरचना और अनुक्रम समानता को पहचानने के लिए किया जाता है. परिणामी वैश्विक और स्थानीय समानता स्कोर रैंक करने के लिए इस्तेमाल किया जाता हैटेम्पलेट (कार्य homologues) प्रोटीन और एनोटेशन (ईसी संख्या और जीन सत्तामीमांसा 19 संदर्भ) शीर्ष स्कोरिंग हिट पर आधारित हस्तांतरण. इसी तरह, ligand बाध्यकारी साइट के अवशेष और ligand बाध्यकारी मोड शीर्ष स्कोरिंग समारोह 9 टेम्पलेट्स में जाना जाता है ligand बाध्यकारी साइट के अवशेषों के साथ क्वेरी के स्थानीय संरेखण पर आधारित अनुमानित हैं.
समारोह की गुणवत्ता (ईसी और जाओ शब्द) मैं TASSER में भविष्यवाणी कार्यात्मक अनुरूपता स्कोर (FH-स्कोर) जो क्वेरी और टेम्पलेट के बीच वैश्विक और स्थानीय समानता का एक उपाय है के आधार पर मूल्यांकन किया जाता है, और के रूप में परिभाषित किया है: 
जहां सी स्कोर भविष्यवाणी मॉडल के रूप में Eq में परिभाषित की गुणवत्ता के एक अनुमान है. (2), टीएम स्कोर मॉडल और टेम्पलेट प्रोटीन के बीच वैश्विक संरचनात्मक समानता के उपाय, RMSD अली से 20 TM-संरेखित structurally गठबंधन क्षेत्र में टेम्पलेट और संरचना मॉडल के बीच RMSD, Cov संरचनात्मक संरेखण है (अर्थात संरचनात्मक गठबंधन क्वेरी लंबाई से विभाजित अवशेषों का अनुपात) के कवरेज का प्रतिनिधित्व करता है, आईडी अली अनुक्रम संरेखण टीएम पंक्ति में पहचान है. चुनाव आयोग संख्या भविष्यवाणियों के लिए अनुमानित विश्वास स्कोर क्वेरी और टेम्पलेट के बीच एक परिभाषित स्थानीय क्षेत्र के भीतर सक्रिय साइट मैच (ACM), के मूल्यांकन के रूप में गणना के लिए एक शब्द भी शामिल हैं: 
घ = 0 3.0 Å, घ द्वितीय, एन अली, जहां एन टी टेम्पलेट स्थानीय क्षेत्र के भीतर मौजूद अवशेषों की संख्या का प्रतिनिधित्व करता है गठबंधन क्वेरी टेम्पलेट अवशेषों जोड़े की संख्या है मैं गठबंधन अवशेषों वें जोड़ी के बीच सी α दूरी है दूरी cutoff, एम ii गठबंधन अवशेषों का ith जोड़ी के बीच BLOSUM स्कोर है. सामान्य में, FH-स्कोर रेंज में है. [0 5] और ACM स्कोर 0 [2 के बीच,], जहां उच्च स्कोर अधिक विश्वास कार्यात्मक कार्य का संकेत है. ACM स्कोर भी स्थानीय संरचना और अनुक्रम समानता ligand बाध्यकारी साइटों, जो बी एस स्कोर के रूप में संदर्भित किया जाता है के पास के मूल्यांकन के लिए प्रयोग किया जाता है.
1. प्रोटीन अनुक्रम प्रस्तुत
- मैं TASSER वेब पेज पर जाएँ http://zhanglab.ccmb.med.umich.edu/I-TASSER के लिए संरचना और समारोह मॉडलिंग प्रयोग के साथ शुरू .
- कॉपी और प्रदान के रूप में अमीनो एसिड अनुक्रम चिपकाने या "ब्राउज़ करें" बटन पर क्लिक करके सीधे अपने कंप्यूटर से इसे अपलोड. सर्वर वर्तमान में मैं TASSER 1500 अवशेषों के साथ दृश्यों को स्वीकार है. 1500 अवशेषों से अधिक प्रोटीन आमतौर पर बहु - डोमेन प्रोटीन, कर रहे हैं और अलग - अलग डोमेन में मैं-TASSER के लिए भेजने से पहले विभाजित किया जा सिफारिश की.
- अपने ई - मेल पता (अनिवार्य) और नौकरी (वैकल्पिक) के लिए एक नाम प्रदान करें.
- उपयोगकर्ता वैकल्पिक बाह्य अंतर-Res निर्दिष्ट कर सकते हैंidue संपर्क / दूरी मजबूरी, एक अतिरिक्त टेम्पलेट में जोड़ने या संरचना मॉडलिंग की प्रक्रिया के दौरान कुछ टेम्पलेट प्रोटीन बाहर. "चर्चा" अनुभाग में इन विकल्पों का उपयोग कर के बारे में अधिक जानें.
- अनुक्रम प्रस्तुत करने के लिए, "भागो TASSER मैं" बटन पर क्लिक करें. ब्राउज़र एक पुष्टिकरण निर्दिष्ट उपयोगकर्ता जानकारी, नौकरी पहचान संख्या (नौकरी आईडी) और एक वेबपेज जहाँ परिणाम काम के पूरा होने के बाद जमा किया जाएगा करने के लिए एक लिंक प्रदर्शित पृष्ठ के लिए निर्देशित किया जाएगा. उपयोगकर्ता इस लिंक को बुकमार्क या नीचे भविष्य में संदर्भ के लिए नौकरी पहचान संख्या नोट हो सकता है.
2. परिणामों की उपलब्धता
- मैं TASSER पर कतार पृष्ठ पर जाकर अपने पेश की नौकरी की स्थिति की जाँच करें http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php . खोज टैब पर क्लिक करें और नौकरी आईडी नंबर या क्वेरी के लिए अपने पेश की जाती नौकरी खोज अनुक्रम का उपयोग करें.
- संरचना और समारोह के मो के बादdeling समाप्त हो गया है, एक अधिसूचना ई - मेल भविष्यवाणी संरचनाओं और एक वेब लिंक युक्त छवि आप को भेजा जाएगा. इस लिंक पर क्लिक करें या 1.5 चरण में बुकमार्क लिंक को खोलने के परिणाम को देखने और डाउनलोड.
3. माध्यमिक संरचना और विलायक पहुँच भविष्यवाणियों
- FASTA स्वरूपित क्वेरी अनुक्रम परिणाम पृष्ठ के शीर्ष पर प्रदर्शित की जाँच करें. यदि किसी भी अतिरिक्त संयम / टेम्पलेट अनुक्रम सबमिशन, वेबपेज उपयोगकर्ता निर्दिष्ट जानकारी प्रदर्शित करने के लिए एक कड़ी के दौरान निर्दिष्ट किया गया था भी देखा जा सकता है (चित्र 1 ए).
- अल्फा हेलिक्स (एच), बीटा किनारा (एस) या कुंडल (सी) और भविष्यवाणी की विश्वास प्रत्येक अवशेषों के लिए (0 = कम 9 उच्च =) स्कोर: माध्यमिक संरचना के रूप में प्रदर्शित भविष्यवाणी जांच करते हैं. नियमित रूप से माध्यमिक संरचना की लंबी हिस्सों भविष्यवाणियों (एच या एस) के साथ इस क्षेत्र के लिए देखो, प्रोटीन में कोर क्षेत्र का अनुमान है. प्रोटीन की स्ट्रक्चरल वर्ग माध्यमिक संरचनाओं तत्वों के वितरण पर भी विश्लेषण कर सकते हैं आधारित हो. अलहां, तो प्रोटीन में कुंडल तत्वों की लंबी क्षेत्रों आमतौर पर असंरचित / बेक़ायदा क्षेत्रों से संकेत मिलता है.
- भविष्यवाणी क्वेरी में दफन पता लगाने और विलायक उजागर क्षेत्रों के लिए विलायक पहुँच (चित्रा 1C). के मान से 0 (दफन अवशेषों) 9 (उजागर अवशेषों) विलायक पहुँच रेंज की भविष्यवाणी की. क्षेत्र ज्यादातर दफन अवशेषों से युक्त करने के लिए प्रोटीन के कोर क्षेत्र में चित्रित करने के लिए इस्तेमाल किया जा सकता है, जबकि विलायक उजागर और हाइड्रोफिलिक अवशेषों के साथ क्षेत्रों में संभावित जलयोजन / कार्यात्मक साइटों रहे हैं.
4. तृतीयक संरचना भविष्यवाणियों
- नीचे स्क्रॉल करने के लिए क्वेरी प्रोटीन की भविष्यवाणी तृतीयक संरचनाओं, एक इंटरैक्टिव Jmol एप्लेट (चित्रा 2) में प्रदर्शित देखने. एप्लेट पर क्लिक करें प्रदर्शित संरचना का प्रकटन बदलने के वाम, विशिष्ट क्षेत्र में ज़ूम, भविष्यवाणी मॉडल में विशिष्ट अवशेषों प्रकार का चयन करें या अंतर - अवशेषों दूरी की गणना.
- लंबी असंरचित क्षेत्रों की उपस्थिति के लिए मॉडल का विश्लेषण. ये regions आमतौर पर प्रोटीन में बेक़ायदा क्षेत्रों के अनुरूप या टेम्पलेट संरेखण की कमी से संकेत मिलता है. इन क्षेत्रों में आम तौर पर कम मॉडलिंग और एन एंड सी टर्मिनस क्षेत्र मॉडलिंग सटीकता में सुधार होगा से मॉडलिंग के दौरान सटीकता इन क्षेत्रों को हटाने है.
- मॉडल के PDB स्वरूपित संरचना फ़ाइलें "डाउनलोड माडल" लिंक पर क्लिक करके डाउनलोड करें. आप संरचनात्मक सुविधाओं के आगे के विश्लेषण के लिए किसी भी आणविक दृश्य सॉफ्टवेयर (Pymol जैसे, Rasmol आदि) में इन फ़ाइलों को खोल सकते हैं.
- विश्लेषण संरचना मॉडलिंग का विश्वास (सी स्कोर) स्कोर के लिए भविष्यवाणी की संरचनाओं की गुणवत्ता का अनुमान है. सी स्कोर मान (2 Eq.) श्रेणी में आम तौर पर कर रहे हैं [-5, 2], जिसमें एक उच्च स्कोर बेहतर गुणवत्ता के एक मॉडल को दर्शाता है. अनुमानित टीएम स्कोर और पहली मॉडल के RMSD "मॉडल 1 की अनुमानित सटीकता" के रूप में दिखाया गया है. लंबे प्रोटीन के लिए, यह मॉडल TM-स्कोर पर आधारित गुणवत्ता का मूल्यांकन करने की सिफारिश की है, के रूप में टीएम स्कोर RMSD की तुलना में topological परिवर्तन के प्रति संवेदनशील है. < li> सी स्कोर का विश्लेषण करने के लिए लिंक, क्लस्टर आकार और सभी मॉडलों के क्लस्टर घनत्व "सी स्कोर के बारे में अधिक" पर क्लिक करें. अनुमानित टीएम स्कोर और RMSD केवल पहली मॉडल मैं TASSER के लिए प्रस्तुत कर रहे हैं, क्योंकि कम रैंक मॉडलों के सी - स्कोर जोरदार टीएम स्कोर या RMSD के साथ सहसंबद्ध नहीं है. निचले क्रम मॉडल की गुणवत्ता आंशिक रूप से उनके क्लस्टर घनत्व और क्लस्टर पहली मॉडल के सापेक्ष आकार के आधार पर मूल्यांकन किया जा सकता है, बड़ा क्लस्टर और उच्च घनत्व से मॉडल जिसमें औसतन देशी संरचना करने के लिए करीब रहे हैं.
- निम्न सी स्कोर भविष्यवाणियों आमतौर पर एक कम सटीकता भविष्यवाणी संकेत मिलता है. अधिकांश ऐसे मामलों में, क्वेरी प्रोटीन पुस्तकालय में एक अच्छा टेम्पलेट का अभाव और आदितः मॉडलिंग (यानी अवशेषों> 120) सीमा से परे एक आकार है . इन मामलों में, उपयोगकर्ताओं को अतिरिक्त स्थानिक मजबूरी के लिए तलाश करने और उन्हें उपयोग करने के लिए मैं TASSER मॉडलिंग (चर्चा अनुभाग देखें) में सुधार कर सकते हैं. यह भी हमारे क्वार्क सर्वर से दृश्यों को प्रस्तुत करने के लिए प्रोत्साहित किया जाता है (क्वार्क / ">) http://zhanglab.ccmb.med.umich.edu/QUARK/ एक शुद्ध आदितः मॉडलिंग के लिए यदि प्रोटीन आकार के नीचे 200 के अवशेष.
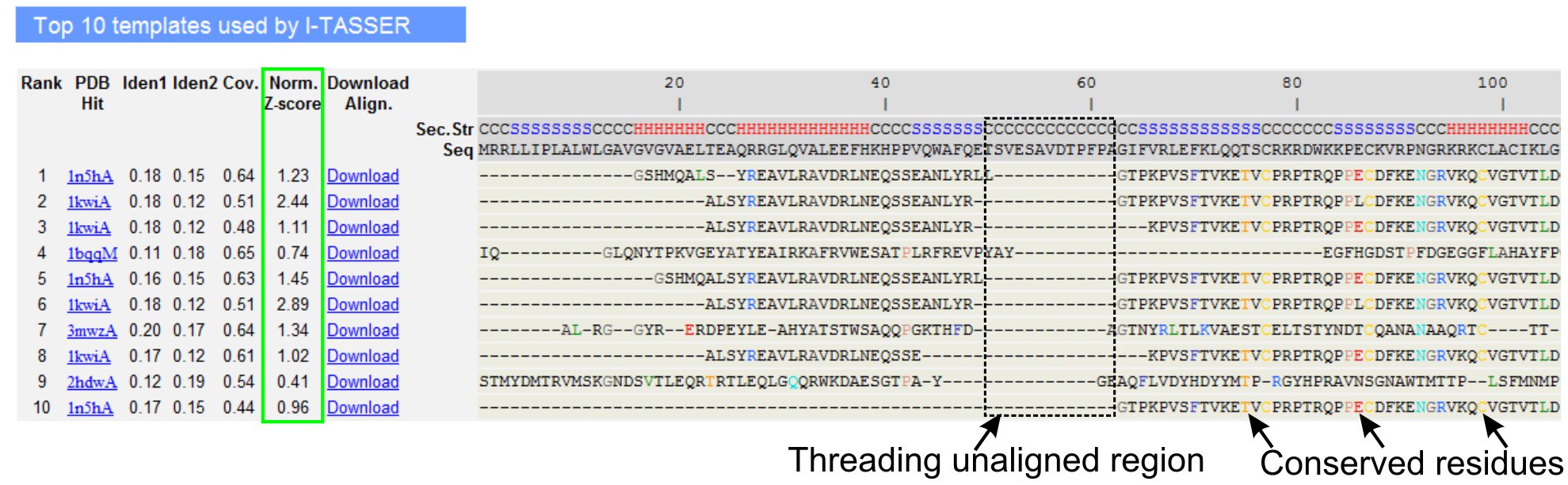
5. LOMETS लक्ष्य टेम्पलेट संरेखण
- नीचे स्क्रॉल करने के लिए क्वेरी प्रोटीन की शीर्ष दस सूत्रण टेम्पलेट्स, LOMETS सूत्रण (चित्रा 3) कार्यक्रम के द्वारा की पहचान का विश्लेषण. सामान्यीकृत (1 Eq.) Z-स्कोर, 'नॉर्म में दिखाया गया है. 'जेड स्कोर स्तंभ, सूत्रण संरेखण की गुणवत्ता का विश्लेषण. एक normalized> 1 Z स्कोर के साथ alignments एक विश्वास संरेखण को दर्शाता है और सबसे अधिक संभावना क्वेरी प्रोटीन के रूप में एक ही गुना है.
- सूत्रण गठबंधन क्षेत्र (स्तंभ 'IDEN 1.') और पूरी श्रृंखला (स्तंभ 'IDEN 2') करने के लिए क्वेरी और टेम्पलेट प्रोटीन के बीच समरूपता आकलन के लिए अनुक्रम पहचान का विश्लेषण करें. उच्च अनुक्रम पहचान क्वेरी टेम्पलेट और प्रोटीन के बीच विकासवादी संबद्धता का एक संकेतक है.
- सूत्रण गठबंधन रंग में दिखाया गया है नेत्रहीन विपक्ष की पहचान के अवशेष देखेंerved अवशेषों / क्वेरी और टेम्पलेट प्रोटीन में रूपांकनों. सूत्रण गठबंधन क्षेत्र में एक उच्च अनुक्रम पहचान पूरी श्रृंखला संरेखण की तुलना में भी संरक्षित संरचनात्मक / क्वेरी में आकृति डोमेन की उपस्थिति इंगित करता है.
- देखने के द्वारा सूत्रण संरेखण के कवरेज का आकलन 'Cov. स्तंभ और संरेखण निरीक्षण. यदि शीर्ष alignments के कवरेज कम है और केवल क्वेरी प्रोटीन या क्वेरी अनुक्रम के एक लंबे खंड के लिए अनुपस्थित के एक छोटे से क्षेत्र तक ही सीमित है, तो क्वेरी प्रोटीन आमतौर पर एक से अधिक डोमेन होता है और यह करने के लिए अनुक्रम और मॉडल को विभाजित करने के लिए सिफारिश की है व्यक्तिगत डोमेन (चित्रा 3).
- पर क्लिक करके PDB स्वरूपित अनुक्रम संरचना संरेखण फाइलें डाउनलोड लिंक "डाउनलोड संरेखित करें. ये संरेखण फ़ाइल को किसी भी आणविक दृश्य सामग्री अनुभाग में सूचीबद्ध कार्यक्रम में खोला जा सकता है, और संरचना मॉडलिंग (1.4 चरण) के दौरान अतिरिक्त मजबूरी को जोड़ने के लिए भी इस्तेमाल किया जा सकता है.
6.PDB में स्ट्रक्चरल analogs
- परिणाम पृष्ठ के अगले तालिका (चित्रा 4) देखें संरचनात्मक संरेखण प्रोग्राम द्वारा पहले भविष्यवाणी मॉडल के शीर्ष दस संरचनात्मक analogs, के रूप में पहचान निर्धारित 20 TM-संरेखित. एक> 0.5 TM-स्कोर इंगित करता है कि पता लगाया एनालॉग और मॉडल एक समान टोपोलॉजी और क्वेरी 16 प्रोटीन की संरचनात्मक परिवार वर्ग / प्रोटीन का निर्धारण करने के लिए इस्तेमाल किया जा सकता है, जबकि TM-स्कोर के साथ उन 0.3 <एक यादृच्छिक संरचना समानता का मतलब है.
- अनुक्रम पहचान का विश्लेषण और संरचनात्मक गठबंधन क्षेत्र में RMSD में दिखाया गया है और 'IDEN एक' मॉडल में स्थानिक रूपांकनों के संरक्षण और संरचनात्मक अनुरूप का आकलन करने के लिए स्तंभों 'एक RMSD'. दिखने में संरेखण में रंग और गठबंधन अवशेषों जोड़े का निरीक्षण करने के लिए इन संरचनात्मक संरक्षित अवशेष और रूपांकनों की पहचान.
- के बारे में उनकी संरचनात्मक वर्गीकरण (SCOP PDB 'PDB मारो' के लिए RCSB वेबसाइट पर जाएँ और अधिक जानने के कॉलम में दिखाया कोड पर क्लिक करें, कैथ और PFAM) और कार्यात्मक जानकारी (ईसी संख्या, संबद्ध शर्तों और बाध्य ligand जाओ).
7. फंक्शन cofactor का उपयोग भविष्यवाणी
- परिणाम पृष्ठ में नीचे स्क्रॉल करने के लिए क्वेरी प्रोटीन के लिए कार्यात्मक व्याख्याओं का विश्लेषण. प्रोटीन कार्य तीन संदर्भ तालिकाओं में enumerated हैं, प्रदर्शित: एनजाइम आयोग (ईसी) संख्या, जीन सत्तामीमांसा (जाओ) संदर्भ में, और ligand बाध्यकारी साइटों.
- , 'एक IDEN' और 'एक RMSD' टीएम स्कोर '' Cov. प्रत्येक तालिका में स्तंभों वैश्विक संरचना समानता और मॉडल और पहचान कार्यात्मक homologues (टेम्पलेट्स) के बीच स्थानिक पैटर्न के संरक्षण के मानकों का विश्लेषण करने के लिए.
8. एनजाइम आयोग संख्या भविष्यवाणी
- शीर्ष पांच क्वेरी प्रोटीन की संभावित एंजाइम homologues में दिखाया देखें तालिका (चित्रा 5) "चुनाव आयोग अनुमानित संख्या". चुनाव आयोग संख्या में इन टेम्पलेट्स का उपयोग कर भविष्यवाणी के आत्मविश्वास का स्तर 'चुनाव आयोग स्कोर' कॉलम में दिखाया गया है. Benchma पर आधारित23 विश्लेषण, क्वेरी और टेम्पलेट प्रोटीन के बीच कार्यात्मक समानता (ईसी संख्या के पहले 3 अंक) rking मज़बूती से चुनाव आयोग के स्कोर> 1.1 का उपयोग कर व्याख्या कर सकते हैं.
- टेम्पलेट्स, जो क्वेरी प्रोटीन के रूप में इसी तरह गुना (यानी टीएम स्कोर 0.5>) के बीच समारोह का सर्वसम्मति (ईसी संख्या) के लिए देखो. यदि एकाधिक टेम्पलेट्स एक ही चुनाव आयोग संख्या और 1.1> चुनाव आयोग स्कोर है, भविष्यवाणी के आत्मविश्वास का स्तर बहुत अधिक है. हालांकि, अगर चुनाव आयोग स्कोर उच्च है, लेकिन वहाँ पहचान हिट के बीच आम सहमति की कमी है, तो भविष्यवाणी कम विश्वसनीय हो जाता है और उपयोगकर्ताओं के लिए जाओ अवधि भविष्यवाणियों से परामर्श करने के लिए सिफारिश कर रहे हैं.
- चुनाव आयोग संख्या पर उपलब्ध कराई लिंक ExPASy एनजाइम डेटाबेस पर जाएँ और समारोह का विश्लेषण करने के लिए, विस्तार में टेम्पलेट प्रोटीन की प्रतिक्रिया उत्प्रेरित, सह कारक आवश्यकताओं और चयापचय मार्ग सहित, पर क्लिक करें.
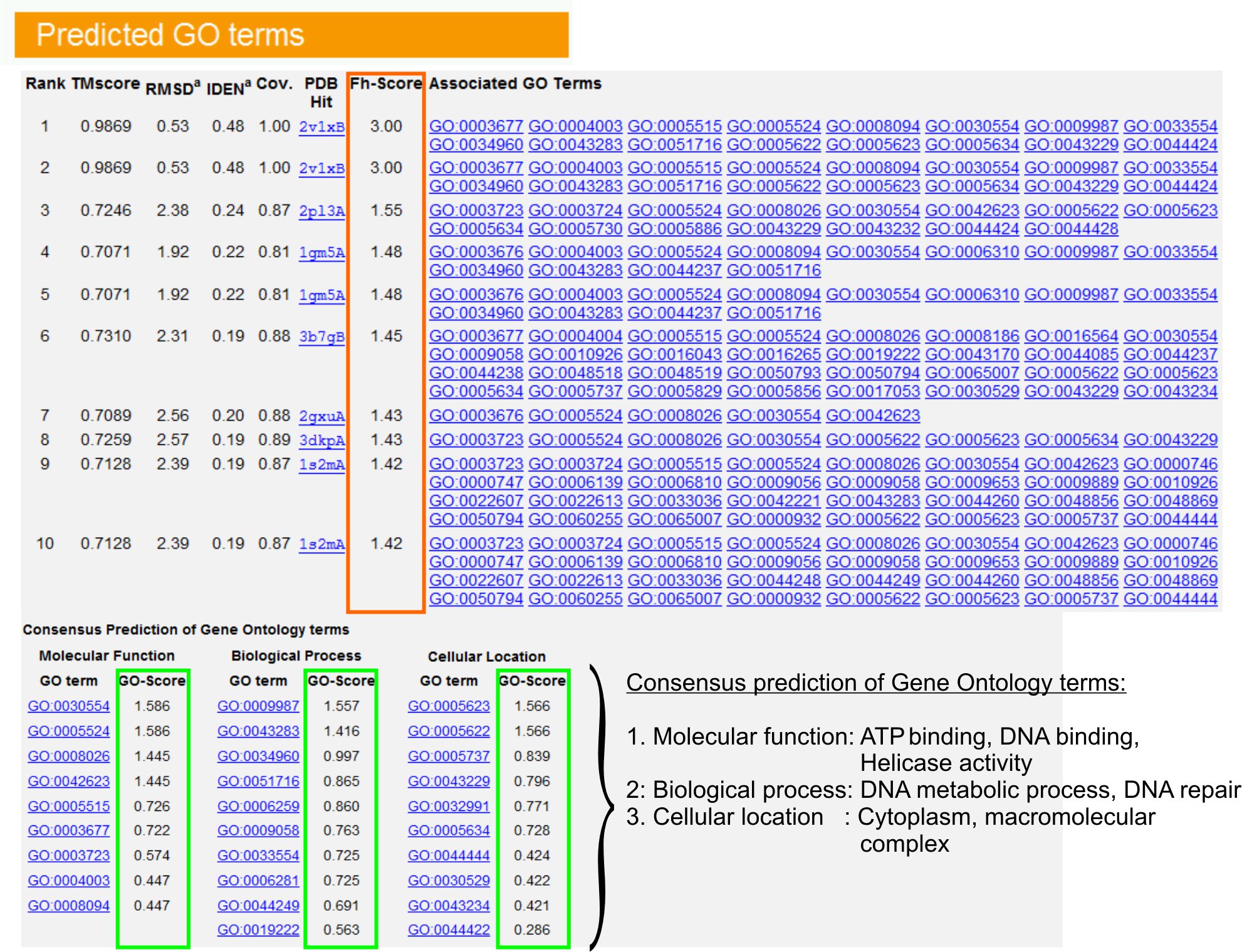
9. जीन सत्तामीमांसा (जाओ) अवधि भविष्यवाणियों
- देखें "अनुमानित शर्तों जाओ (चित्र तालिका6 ure) PDB लायब्रेरी में शीर्ष दस क्वेरी प्रोटीन की homologues, जीन (जाओ) आंटलजी शर्तों के साथ एनोटेट की पहचान करने के लिए. प्रत्येक प्रोटीन आमतौर पर एकाधिक जाओ शर्तों के साथ जुड़ा हुआ है, अपनी आणविक (एमएफ) कार्यों, जैविक प्रक्रियाओं (बीपी) और सेलुलर घटक (सीसी) का वर्णन है. प्रत्येक शब्द पर क्लिक करें Amigo वेबसाइट पर जाएँ और इसकी परिभाषा और वंश का विश्लेषण.
- स्तंभ FH स्कोर (कार्यात्मक अनुरूपता स्कोर) का विश्लेषण करने के लिए क्वेरी और टेम्पलेट प्रोटीन के बीच कार्यात्मक समानता का उपयोग और इन प्रोटीनों से कार्यात्मक एनोटेशन के हस्तांतरण की आत्मविश्वास के स्तर का अनुमान है. हमारी बेंच मार्किंग अध्ययन में 23, देशी जाओ शर्तों के 50% सही ढंग से पहले की पहचान की 0.8 के एक cutoff FH-स्कोर का उपयोग कर 56% की एक समग्र सटीकता के साथ करने से टेम्प्लेट से पहचाना जा सकता है.
- टेम्पलेट्स के बीच समारोह की सहमति का विश्लेषण "जाओ पदों की सहमति भविष्यवाणी" तालिका देखें. ये आम कार्यों क्वेरी के जाओ शर्तों (एमएफ, बीपी और सीसी) की भविष्यवाणी करने के लिए उपयोग किया जाता हैप्रोटीन और आत्मविश्वास के स्तर की अवधि भविष्यवाणियों जाओ (जाओ स्कोर) का आकलन. बेंच मार्किंग 23 परीक्षण के आधार पर, सबसे अच्छा झूठी सकारात्मक और नकारात्मक झूठी दरों GO के स्कोर cutoff के साथ = 0.5 भविष्यवाणियों के लिए प्राप्त कर रहे हैं भविष्यवाणी की गहरी आंटलजी स्तर पर कवरेज घटते साथ.
10. प्रोटीन ligand बाध्यकारी साइट भविष्यवाणियों
- शीर्ष क्वेरी प्रोटीन के लिए दस ligand बाध्यकारी साइट भविष्यवाणियों को देखने के लिए पृष्ठ के नीचे करने के लिए नीचे स्क्रॉल. अनुमानित बाध्यकारी साइटों भविष्यवाणी ligand रचना है कि शेयर आम बाध्य जेब की संख्या के आधार पर क्रमबद्ध हैं. सबसे अच्छा बाध्यकारी साइट की पहचान पहले से ही Jmol एप्लेट में प्रदर्शित है. अन्य भविष्यवाणियों का विश्लेषण और कल्पना ligand बातचीत अवशेषों रेडियो बटन पर क्लिक करें.
- स्तंभ बी एस के स्कोर का विश्लेषण करने के लिए मॉडल और टेम्पलेट बाध्यकारी साइट के बीच स्थानीय समानता का मूल्यांकन करने के लिए. बेंचमार्क 9 के आधार पर, बी एस स्कोर 1.1> उच्च अनुक्रम और संरचना सिम इंगित करता हैभविष्यवाणी मॉडल और टेम्पलेट में जाना जाता बंधनकारी साइट में बाध्यकारी साइट के पास ilarity.
- "डाउनलोड" लिंक पर क्लिक करके परिसर के PDB स्वरूपित संरचना फ़ाइल डाउनलोड करें. उपयोगकर्ता किसी भी आणविक दृश्य कार्यक्रम में इन फ़ाइलों को खोलने और interactively भविष्यवाणी बाध्यकारी साइट और अपने स्थानीय कंप्यूटर पर ligand प्रोटीन बातचीत देख सकते हैं.
11. प्रतिनिधि परिणाम

चित्रा 1 मैं TASSER परिणाम पृष्ठ का एक अंश दिखा (ए) FASTA क्वेरी अनुक्रम स्वरूपित, (बी) माध्यमिक संरचना और संबद्ध विश्वास स्कोर की भविष्यवाणी की, और (सी) अवशेषों की विलायक पहुँच की भविष्यवाणी की. विश्लेषण कोर क्षेत्र और क्वेरी में संभावित जलयोजन साइट सियान और लाल आयत में डाला जाता है, क्रमशः.

चित्रा 2.

चित्रा 3. मैं TASSER परिणाम पृष्ठ LOMETS 5 सूत्रण कार्यक्रमों द्वारा शीर्ष दस पहचान सूत्रण टेम्पलेट्स और संरेखण दिखाने का एक उदाहरण है. सूत्रण संरेखण की गुणवत्ता सामान्यीकृत Z-स्कोर (हरे रंग में प्रकाश डाला) है, जहां एक मूल्य 1> एक विश्वास संरेखण को दर्शाता है के आधार पर मूल्यांकन किया जाता है. टेम्पलेट में निरपेक्ष अवशेषों है कि इसी क्वेरी अवशेषों के लिए समान हैं रंग में हाइलाइट किया जाता है संरक्षित अवशेषों / आकृति की उपस्थिति का संकेत है, जबकि संरेखण की कमी के अधिकांश में शीर्ष टेम्पलेट्स इंगित करता है क्वेरी प्रोटीन और unaligned अवशेषों में एकाधिक डोमेन की उपस्थिति डोमेन linker क्षेत्रों के अनुरूप 3 आंकड़ा के संस्करण पूर्ण आकार देखने के लिए यहाँ क्लिक करें.

चित्रा 4. परिणाम पृष्ठ के शीर्ष दस पहचान संरचनात्मक analogs और संरचनात्मक संरेखण, द्वारा की पहचान दिखा एक उदाहरण 20 संरचनात्मक संरेखण कार्यक्रम टीएम - संरेखित. में दिखाया analogs की रैंकिंग संरचनात्मक संरेखण TM-स्कोर (नीले रंग में हाइलाइट) पर आधारित है. एक> 0.5 TM-स्कोर इंगित करता है कि दोनों की तुलना संरचनाओं एक समान टोपोलॉजी है, जबकि एक 0.3 <टीएम के स्कोर दो यादृच्छिक संरचनाओं के बीच समानता का मतलब है. संरचनात्मक गठबंधन अवशेषों जोड़े उनकी संपत्ति एमिनो एसिड पर आधारित रंग में हाइलाइट किया जाता है, जबकि unaligned क्षेत्रों से संकेत कर रहे हैं "-" है.ove.com/files/ftp_upload/3259/3259fig4large.jpg "> यहाँ क्लिक करें आंकड़ा 4 पूर्ण आकार संस्करण को देखने.

चित्रा 5 मैं TASSER परिणाम पृष्ठ PDB लायब्रेरी में क्वेरी प्रोटीन की पहचान एंजाइम homologues दिखाने का एक उदाहरण है . चुनाव आयोग संख्या भविष्यवाणी के आत्मविश्वास के स्तर (हरे रंग में प्रकाश डाला) चुनाव आयोग स्कोर, जहां चुनाव आयोग स्कोर 1.1> कार्यात्मक क्वेरी और टेम्पलेट प्रोटीन के बीच समानता (एक ही चुनाव आयोग संख्या के पहले 3 अंक) इंगित करता है के आधार पर विश्लेषण किया है.

चित्रा 6 मैं TASSER परिणाम दिखा पृष्ठ का एक उदाहरण क्वेरी प्रोटीन के लिए अवधि भविष्यवाणियों जाओ. जीन सत्तामीमांसा टेम्पलेट लायब्रेरी में क्वेरी प्रोटीन के लिए कार्यात्मक homologues उनके FH-स्कोर (नारंगी आयत में) के आधार पर क्रमबद्ध हैं. इन शीर्ष स्कोरिंग हिट से आम कार्यात्मक सुविधाओं gener प्राप्त कर रहे हैं अंतिम क्वेरी प्रोटीन के लिए अवधि भविष्यवाणियों जाओ खा लिया. भविष्यवाणी जाओ शर्तों की गुणवत्ता का अनुमान आंकड़ा 6 संस्करण पूर्ण आकार देखने के लिए यहाँ क्लिक करें जाओ - (हरे रंग में दिखाया गया है) स्कोर है, जहां एक GO के स्कोर> 0.5 एक विश्वसनीय भविष्यवाणी इंगित करता है. पर आधारित है.

7 चित्रा मैं TASSER परिणाम पृष्ठ शीर्ष दस प्रोटीन ligand बाध्यकारी साइट cofactor 9 एल्गोरिथ्म का उपयोग कर भविष्यवाणियों दिखाने का एक उदाहरण है. भविष्यवाणी बाध्यकारी साइटों की रैंकिंग भविष्यवाणी ligand रचना है कि क्वेरी में आम बाध्यकारी जेब साझा की संख्या पर आधारित है. बी एस-स्कोर (लाल रंग में हाइलाइट) स्थानीय अनुक्रम और संरचना भविष्यवाणी की है और टेम्पलेट बाध्यकारी साइट के बीच समानता के एक उपाय है, और बाध्यकारी साइट जेब के संरक्षण का विश्लेषण करने के लिए उपयोगी है.
les/ftp_upload/3259/3259fig8.jpg "/>
8 चित्रा बाहरी संयम अवशेषों अवशेषों संपर्क / दूरी मजबूरी निर्दिष्ट करने के लिए करने के लिए इस्तेमाल किया फाइलों का एक उदाहरण है .

9 चित्रा संयम मैं TASSER सर्वर के लिए एक टेम्पलेट प्रोटीन निर्दिष्ट करने के लिए उपयोग की गई फ़ाइलों का उदाहरण है . या (बी) 3 डी स्वरूप, उपयोगकर्ता क्वेरी टेम्पलेट (ए) FASTA प्रारूप में संरेखण या तो निर्दिष्ट कर सकते हैं.

10 चित्रा एक उदाहरण मैं TASSER संरचना मॉडलिंग की प्रक्रिया के दौरान टेम्पलेट को छोड़कर के लिए इस्तेमाल किया फ़ाइल . प्रथम स्तंभ टेम्पलेट के लिए बाहर रखा जाना प्रोटीन की PDB आईडी शामिल हैं. दूसरे स्तंभ के लिए अनुक्रम पहचान cutoff जो टेम्पलेट लायब्रेरी में अन्य समान टेम्पलेट्स के लिए इस्तेमाल किया जाएगा निर्दिष्ट करने के लिए प्रयोग किया जाता है.
Discussion
ऊपर प्रस्तुत प्रोटोकॉल संरचना और समारोह मैं TASSER सर्वर का उपयोग कर मॉडलिंग के लिए एक सामान्य दिशानिर्देश है. हालांकि, यह स्वचालित प्रक्रिया प्रोटीन के अधिकांश के लिए बहुत अच्छी तरह से काम करता है, मानव हस्तक्षेप अक्सर काफी मॉडलिंग सटीकता में सुधार प्रोटीन जो PDB पुस्तकालय में करीब टेम्पलेट्स कमी के लिए विशेष रूप से, मदद. उपयोगकर्ता मैं TASSER निम्न तरीकों में मॉडलिंग के दौरान हस्तक्षेप कर सकते हैं: (क) बहु - डोमेन प्रोटीन के बंटवारे, (ख) बाहरी मजबूरी प्रदान करने के लिए संरचना विधानसभा में सुधार, और (ग) मॉडलिंग के दौरान टेम्पलेट्स हटाने.
बंटवारे प्रोटीन बहु - डोमेन:
कई लंबे प्रोटीन दृश्यों अक्सर एकाधिक डोमेन लचीला linker क्षेत्रों, जो उनकी संरचना की व्याख्या दोनों प्रयोगात्मक और कम्प्यूटेशनल तकनीकों का उपयोग कर मुश्किल बनाता द्वारा सीमित होते हैं. फिर भी, के रूप में डोमेन स्वतंत्र संस्थाओं तह कर रहे हैं और अलग आणविक समारोह में प्रदर्शन कर सकते हैं, यह हैप्रत्येक डोमेन लंबे बहु - डोमेन प्रोटीन और मॉडल अलग - अलग विभाजित वांछनीय. मॉडलिंग डोमेन केवल व्यक्तिगत भविष्यवाणी की प्रक्रिया को गति नहीं होगा, लेकिन यह भी क्वेरी-टेम्पलेट संरेखण की गुणवत्ता बढ़ जाती है, और अधिक विश्वसनीय संरचना और समारोह भविष्यवाणियों में जिसके परिणामस्वरूप.
प्रोटीन दृश्यों में डोमेन सीमाओं आज़ादी से उपलब्ध NCBI CDD 24, PFAM 25 या 26 InterProScan जैसे बाह्य ऑनलाइन कार्यक्रम का उपयोग कर भविष्यवाणी की जा सकती है. इसके अलावा, अगर LOMETS सूत्रण संरेखण क्वेरी प्रोटीन के लिए उपलब्ध हैं, डोमेन सीमाओं नेत्रहीन शीर्ष सूत्रण टेम्पलेट्स में unaligned अवशेषों की लंबी हिस्सों की पहचान (5.4 चरण देखें) द्वारा स्थित किया जा सकता है. इन unaligned क्षेत्रों ज्यादातर डोमेन linker क्षेत्रों के अनुरूप. यदि बहु - डोमेन टेम्पलेट्स सभी क्वेरी गठबंधन डोमेन के साथ टेम्पलेट PDB लायब्रेरी में पहले से ही उपलब्ध हैं, तो क्वेरी प्रोटीन पूरी लंबाई के रूप में modeled किया जा सकता है.
बाहरी मजबूरी प्रदान
ए / संपर्क दूरी मजबूरी निर्दिष्ट
प्रयोगात्मक विशेषता अंतर - अवशेषों संपर्कों / दूरी, या उदाहरण के लिए एनएमआर सेपार से जोड़ने प्रयोगों, एक संयम फ़ाइल को अपलोड करने के द्वारा निर्दिष्ट किया जा सकता है है. एक उदाहरण फ़ाइल 8 चित्रा, जहां संयम के एक स्तंभ प्रकार निर्दिष्ट में दिखाया गया है, "जिला" या "संपर्क" अर्थात्. दूरी संयम के लिए (जिला), 2 कॉलम और 4 अवशेषों पदों (i, j) होते हैं, 3 कॉलम और 5 अवशेषों और 6 स्तंभ में परमाणु प्रकार के होते हैं दो निर्दिष्ट परमाणुओं के बीच दूरी निर्दिष्ट. संपर्क मजबूरी (संपर्क), 2 स्तंभ और 3 अवशेष है जो संपर्क में होना चाहिए की स्थिति (i, j) शामिल हैं. इन अवशेषों जोड़े संपर्क के पक्ष श्रृंखला केंद्र के बीच की दूरी PDB में जाना जाता संरचनाओं में मनाया दूरी के आधार पर फैसला किया है. मैं - TASSER इन परमाणु संरचना शोधन सिमुलेशन के दौरान निर्दिष्ट दूरी के करीब जोड़े को आकर्षित करने की कोशिश करेंगे.
बी एक प्रोटीन संरचना टेम्पलेट निर्दिष्ट
LOMETS सूत्रण प्रोग्राम एक प्रतिनिधि PDB पुस्तकालय का उपयोग करने के लिए क्वेरी prot के लिए प्रशंसनीय सिलवटों मिलईआईऍन. हालांकि एक प्रतिनिधि संरचना पुस्तकालय का उपयोग कर के अनुक्रम संरचना संरेखण गणना करने के लिए आवश्यक समय को कम करने में मदद करता है, यह संभव है कि एक अच्छा टेम्पलेट प्रोटीन पुस्तकालय में याद किया जाता है या टेम्पलेट LOMETS सूत्रण कार्यक्रमों द्वारा पहचान नहीं हो सकता है, भले ही यह है पुस्तकालय में उपस्थित थे. इन मामलों में, उपयोगकर्ता को टेम्पलेट के रूप में वांछित प्रोटीन संरचना को निर्दिष्ट करना चाहिए.
एक अतिरिक्त टेम्पलेट के रूप में प्रोटीन की संरचना को निर्दिष्ट करने के लिए, उपयोगकर्ताओं को या तो एक PDB स्वरूपित संरचना फ़ाइल अपलोड कर सकते हैं या एक PDB पुस्तकालय में जमा प्रोटीन संरचना के PDB ID निर्दिष्ट. मैं TASSER क्वेरी टेम्पलेट का उपयोग मस्टर कार्यक्रम 23 संरेखण पैदा करते हैं और दोनों निर्दिष्ट उपयोगकर्ता टेम्पलेट और LOMETS संरचना विधानसभा अनुकार गाइड टेम्पलेट्स से स्थानिक मजबूरी इकट्ठा करेंगे. क्योंकि LOMETS मजबूरी की सटीकता लक्ष्यों के लिए अलग अलग है, LOMETS मजबूरी के वजन आसान टा (मुताबिक़) में मजबूत हैहार्ड (गैर - मुताबिक़) लक्ष्य है, जो व्यवस्थित किया गया है हमारे बेंचमार्क प्रशिक्षण में देखते है कि तुलना में rgets.
उपयोगकर्ताओं को भी अपने स्वयं क्वेरी टेम्पलेट संरेखण निर्दिष्ट कर सकते हैं. FASTA स्वरूप (चित्रा 9A) और 3 डी स्वरूप (9b चित्रा): सर्वर के दो स्वरूपों में संरेखण स्वीकार करता है. FASTA प्रारूप मानक और में वर्णित है http://zhanglab. ccmb.med.umich.edu / / FASTA . 3 डी स्वरूप मानक PDB स्वरूप (समान है http://www.wwpdb.org/documentation/format32/sect9.html ), लेकिन दो अतिरिक्त टेम्पलेट्स से व्युत्पन्न स्तंभों परमाणु रिकॉर्ड (चित्रा 9b देखें) करने के लिए जोड़ रहे हैं:
1-30 कॉलम: एटम (केवल सी अल्फा) और क्वेरी अनुक्रम के लिए नाम अवशेषों.
31-54 कॉलम: टेम्पलेट में संगत परमाणुओं से नकल क्वेरी के सी अल्फा परमाणुओं के निर्देशांक.
55-59 स्तंभ: टेम्पलेट में संगत अवशेषों संख्या संरेखण पर आधारित
टेम्पलेट में संगत अवशेषों नाम: 60-64 स्तंभ
टेम्पलेट्स प्रोटीन अपवर्जित
प्रोटीन लचीला अणु होते हैं और कई गठनात्मक राज्यों को अपनाने के लिए उनके जैविक गतिविधि को बदल सकते हैं. उदाहरण के लिए, कई प्रोटीन kinases और झिल्ली प्रोटीन की संरचना दोनों सक्रिय और निष्क्रिय रचना में हल किया गया है. इसके अलावा बाध्य ligand की उपस्थिति या अनुपस्थिति बड़े संरचनात्मक आंदोलनों का कारण बन सकती है. जबकि टेम्पलेट के सभी गठनात्मक राज्यों सूत्रण कार्यक्रमों के लिए एक जैसे हैं, यह केवल एक विशेष राज्य में टेम्पलेट्स का उपयोग कर क्वेरी मॉडल वांछनीय है. सर्वर पर एक नया विकल्प संरचना मॉडलिंग के दौरान टेम्पलेट प्रोटीन को बाहर करने के लिए उपयोगकर्ता की अनुमति देता है. यह सुविधा भी उपयोगकर्ता मॉडलिंग के लिए इस्तेमाल किया जा टेम्पलेट्स समरूपता के स्तर का चयन करने के लिए अनुमति होगी. उपयोगकर्ता टेम्पलेट प्रोटीन fr बाहर कर सकते हैंपुस्तकालय मैं - TASSER ओम द्वारा:
ए निर्दिष्ट एक अनुक्रम पहचान cutoff
मैं TASSER टेम्पलेट लायब्रेरी से मुताबिक़ प्रोटीन को अलग उपयोगकर्ता इस विकल्प का उपयोग कर सकते हैं. अनुरूपता स्तर अनुक्रम पहचान cutoff पर आधारित है, क्वेरी और टेम्पलेट क्वेरी अनुक्रम के अनुक्रम की लंबाई से विभाजित प्रोटीन के बीच समान अवशेषों की संख्या अर्थात्. उदाहरण के लिए, यदि प्रदान किए गए प्रपत्र में उपयोगकर्ता "70%" में प्रकार, सभी टेम्पलेट्स प्रोटीन है जो> 70% क्वेरी प्रोटीन मैं मैं TASSER टेम्पलेट लायब्रेरी से बाहर रखा जाना करने के लिए एक दृश्य पहचान है.
बी विशिष्ट टेम्पलेट प्रोटीन अपवर्जित करें
विशिष्ट टेम्पलेट प्रोटीन बाहर रखा जा संरचनाओं के PDB आईडी युक्त सूची अपलोड करके मैं TASSER टेम्पलेट लायब्रेरी से बाहर रखा जा सकता है. एक उदाहरण फ़ाइल चित्रा 10 में दिखाया गया है. के रूप में एक ही प्रोटीन PDB लायब्रेरी में एकाधिक प्रविष्टियाँ, मैं - TASSER से के रूप में मौजूद कर सकते हैंrver डिफ़ॉल्ट रूप से निर्दिष्ट टेम्पलेट्स (स्तंभ 1 में) के रूप में के रूप में अच्छी तरह से है कि एक पहचान है पुस्तकालय से सभी अन्य टेम्पलेट्स> 90% निर्दिष्ट टेम्पलेट्स बाहर होगा. उपयोगकर्ताओं को भी एक अलग पहचान cutoff, 70 उदाहरण%, जहां पहचान के साथ सभी टेम्पलेट्स> 70% निर्दिष्ट टेम्पलेट प्रोटीन के लिए बाहर हो जाएगा निर्दिष्ट कर सकते हैं.
Disclosures
ब्याज की कोई संघर्ष की घोषणा की.
Acknowledgments
परियोजना में भाग अल्फ्रेड पी. स्लोअन फाउंडेशन, NSF कैरियर अवार्ड (1,027,394 DBI), और जनरल मेडिकल साइंसेज के राष्ट्रीय संस्थान (GM084222 GM083107) द्वारा समर्थित है.
Materials
| Name | Company | Catalog Number | Comments |
| Material Name | Type | Company | Catalogue Number |
| FASTA formatted amino acid sequence of the protein to be modeled (see, http://www.ncbi.nlm.nih.gov/BLAST/fasta.shtml). | |||
| A personal computer with access to the internet and a web browser. | |||
| Molecular visualizing software, e.g. RASMOL or PYMOL, for analyzing the predicted tertiary structure and functional sites. |
References
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H.
- Finn, R. D.
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).

{kind=link}
{kind=link}