

Summary
Linee guida per computer basato su caratterizzazione strutturale e funzionale di proteine utilizzando l'I-Tasser pipeline è descritto. Partendo dalla sequenza della proteina query, i modelli 3D sono generate utilizzando più allineamenti threading e iterativo simulazioni di assemblaggio strutturale. Inferenze funzionali sono successivamente redatto in base alle corrispondenze di proteine con struttura nota e funzioni.
Abstract
Progetti di sequenziamento del genoma sono cifrati milioni di sequenza della proteina, che richiedono la conoscenza della loro struttura e funzione di migliorare la comprensione del loro ruolo biologico. Anche se i metodi sperimentali in grado di fornire informazioni dettagliate per una piccola parte di queste proteine, la modellazione computazionale è necessario per la maggior parte delle molecole proteiche che sono sperimentalmente caratterizzati. L'I-Tasser server è un banco di lavoro on-line ad alta risoluzione di modellazione della struttura e funzione delle proteine. Data una sequenza proteica, un output tipico del I-Tasser server include la previsione della struttura secondaria, prevede l'accessibilità di ogni residuo solvente, proteine omologhe modello di threading e rilevato da allineamenti struttura, fino a cinque modelli terziario full-length strutturale, e la struttura a base di annotazioni funzionali per la classificazione degli enzimi, Gene Ontology termini e le interazioni proteina-ligando siti di legame. Tutte le previsioni sono contrassegnati con un punteggio di confidenza cheracconta di come le previsioni sono accurate senza conoscere i dati sperimentali. Per facilitare le richieste particolari degli utenti finali, il server fornisce canali di accettare specificato dall'utente tra residui distanza e contattare le mappe di modificare interattivamente la I-Tasser di modellazione, ma permette anche agli utenti di specificare le proteine come modello, o per escludere qualsiasi modello proteine durante le simulazioni di montaggio della struttura. Le informazioni strutturali potrebbero essere raccolti dagli utenti sulla base di evidenze sperimentali o dati biologici con lo scopo di migliorare la qualità di I-Tasser previsioni. Il server è stato valutato come il migliore dei programmi per la struttura delle proteine e le previsioni funzione nel recente a livello di comunità esperimenti CASP. Ci sono attualmente> 20.000 scienziati registrati da oltre 100 paesi che utilizzano l'on line I-Tasser server.
Protocol
Metodo panoramica
Seguendo la sequenza-to-struttura a funzione paradigma, l'I-Tasser procedura di 1-4 per la struttura e la funzione di modellazione prevede quattro passaggi consecutivi di: (a) identificazione modello da LOMETS 5; (b) struttura frammento rimontaggio di replica- scambio di simulazioni Monte Carlo 6; (c) struttura atomica raffinatezza livello usando REMO 7 e FG-MD 8; e (d) la struttura a base di interpretazioni funzione utilizzando cofattore 9.
Identificazione di modelli: Per una sequenza di interrogazione da parte degli utenti, la sequenza viene prima threaded attraverso una libreria rappresentante struttura PPB per un LOMETS installato localmente meta-threading server. Threading è una sequenza-struttura procedura di allineamento usato per identificare le proteine modello che possono avere struttura simile o che contengono simili motivo strutturale, come la proteina query. Per aumentare la copertura delle omologhe Templrilevamenti mangiato, LOMETS combina più state-of-the-art algoritmi che coprono diverse metodologie di filettatura. Poiché i diversi programmi di threading hanno diversi sistemi di punteggio e sensibilità l'allineamento, la qualità degli allineamenti generati filettatura da ogni programma viene valutato da filettatura normalizzata Z-score, che è definito come: 
dove Z-score è il punteggio in unità di deviazione standard rispetto alla media statistica di tutti gli allineamenti generati dal programma, e Z 0 è un programma specifico Z-score di taglio determinata sulla base larga scala test di benchmark filettatura da 5 a differenziare 'buono 'e' cattivi 'modelli. Un modello con un alto Z-score significa che i modelli hanno un alto punteggio di allineamento significativamente più alto rispetto alla maggior parte degli altri modelli, il che implica che l'allineamento di solito corrisponde ad un buon modello. Se la maggior parte dei modelli top di filettatura hanno hinormalizzato gh Z-score, l'accuratezza della finale I-Tasser modello è generalmente elevato. Tuttavia, se la proteina è grande e la copertura degli allineamenti filettatura è limitata ad una piccola regione della proteina query, un alto normalizzato Z-score non significa necessariamente una elevata precisione di modellazione per il full-length modello. Prime due schieramenti filettatura da ogni programma di filettatura sono raccolti ed utilizzati per la fase successiva di montaggio della struttura.
Iterativo di assemblaggio simulazione struttura: in seguito alla procedura di threading, la sequenza query viene divisa in regioni threading allineati e non allineati. Frammenti di continuo allineamento filettatura sono asportati dai modelli e utilizzati direttamente per l'assemblaggio struttura, mentre le regioni ciclo non allineate sono costruiti da modelli ab initio. La procedura di assemblaggio struttura viene eseguita su un sistema reticolo guidato dallo scambio replica simulazioni Monte Carlo 6. L'I-Tasser campo di forza include idrogeno-boending interazioni 10, basata sulla conoscenza termini energetici statistici derivati da strutture proteiche note nel PPB 11, sequenza a base di previsioni contatto SVMSEQ 12, e vincoli territoriali raccolti da LOMETS 5 modelli di threading. Le esche conformazionali generato nella bassa temperatura repliche durante le simulazioni sono raggruppate da Spicker 13 a identificare le strutture di bassa energia libera stati. Centroidi dei cluster Cluster superiore sono ottenuti facendo la media delle coordinate 3D di tutte le esche cluster strutturali e utilizzato per la generazione del modello finale. La simulazione e la procedura di clustering sono ripetuti due volte per la rimozione di scontri sterico e perfezionare ulteriormente la topologia globale.
A livello atomico costruzione del modello e raffinatezza: il centroidi dei cluster ottenuti dopo il clustering Spicker sono ridotte modelli proteici (ogni residuo rappresentata dal suo α C e catena laterale centro di massa) e have limitata applicazione biologica. La costruzione di full-atomico modello dai modelli riduzione avviene in due fasi. Nella prima fase, REMO 7 viene utilizzato per la costruzione completa-atomica modelli C-alfa tracce ottimizzando la H-bond reti. Nella seconda fase, REMO full-atomico modelli vengono ulteriormente raffinate dal FG MD-14, che migliora l'angolo di torsione spina dorsale, lunghezze di legame e catena laterale orientamenti rotamero, da simulazioni di dinamica molecolare, guidato dai frammenti strutturali ricercato dalla strutture PDB da TM-align. La FG-MD modelli raffinati sono usati come modelli finali per le previsioni struttura terziaria da I-Tasser.
La qualità dei modelli generati sono stimati sulla base di un punteggio di confidenza (C-score), che è definito in base alla Z-score degli allineamenti LOMETS filettatura e la convergenza di I-Tasser simulazioni, matematicamente formulato come: 
dove
Il C-score ha una forte correlazione con la qualità dei modelli I-Tasser. Grazie alla combinazione di C-score e la lunghezza delle proteine, la precisione del primo I-Tasser modelli possono essere stimati con un errore medio di 0,08 per il TM-score e 2 Å per il RMSD 15. In generale, i modelli con C-score> - 1,5 sono tenuti ad avere una piega giusta. Qui, RMSD e TM-musica che entrambe le misure ben noti di somiglianza topologica tra il modello e la struttura nativa. TM-score Valugamma es in [0, 1], dove un punteggio più alto indica una struttura migliore corrispondenza 16,17. Tuttavia, per i modelli di rango inferiore (cioè 2 ° -5 ° modelli), la correlazione di C-score con TM-score e RMSD è molto più debole (~ 0,5), e non possono essere utilizzati per la stima affidabile del modello di qualità assoluta.
È il primo modello sempre il miglior modello in I-Tasser simulazioni? La risposta a questa domanda dipende dal tipo di destinazione. Per i bersagli facili, il primo modello è di solito il miglior modello e la sua C-score è di solito molto più alto rispetto al resto dei modelli. Tuttavia, per obiettivi duri, in cui filettatura non ha colpi modello significativo, il primo modello non è necessariamente il miglior modello e I-Tasser ha effettivamente difficoltà a selezionare i migliori modelli e modelli. Si raccomanda pertanto di analizzare tutti i 5 modelli per obiettivi rigidi e selezionarle sulla base delle informazioni sperimentali e conoscenze biologiche.
Funzione predictions: Nell'ultimo passaggio, definitivo modelli 3D generati da FG-MD vengono utilizzati per prevedere tre aspetti della funzione della proteina, e cioè: a) enzima della Commissione (CE) numeri 18 e (b) Gene Ontology (GO) 19 termini e ( c) siti di legame per i ligandi di piccole molecole. Per tutti e tre gli aspetti, le interpretazioni funzionali sono generati utilizzando cofattore, che è un nuovo approccio per predire la funzione delle proteine, basata sulla similarità globale e locale alle proteine modello nel PPB con struttura nota e funzioni. In primo luogo, la topologia globale dei modelli previsti viene confrontato con librerie di modelli funzionali utilizzando il programma di allineamento strutturale TM-align 20. Successivamente, una serie di proteine più simili ai modelli di destinazione vengono selezionati dalla biblioteca in base alla loro somiglianza struttura globale, e una vasta ricerca locale viene eseguita per identificare similarità struttura e sequenza vicino attiva / regione vincolante sito. Il punteggio risultante somiglianza globale e locale sono utilizzati per classificare leproteine modello (omologhi funzionali) e trasferire l'annotazione (numeri CE e Gene Ontology 19 termini) basato sulla hit punteggio più alto. Allo stesso modo, i residui del sito di legame e la modalità di legame sono dedotte in base all'allineamento locale di query con nota residui del sito di legame nei modelli top funzione segnando 9.
La qualità della funzione (CE e GO termine) la previsione in I-Tasser viene valutata sulla base del punteggio omologia funzionale (Fh-score) che è una misura di similarità globale e locale tra la query e modello, ed è definito come: 
dove C-score è una stima della qualità del modello previsto ai sensi Eq. (2), TM-score misura la somiglianza strutturale globale tra il modello e le proteine modello; RMSD ali è il RMSD tra il modello e la struttura modello nella regione strutturalmente allineate da TM-align 20; Cov rappresenta la copertura del tracciato strutturale (ovvero il rapporto tra i residui strutturalmente allineati diviso per la lunghezza query); ID ali è l'identità sequenza nella TM-align allineamento. Il punteggio di confidenza stimato per le previsioni numero CE comprende anche un termine per valutare la corrispondenza del sito attivo (ACM) tra domanda e il modello all'interno di una regione definita locale, calcolato come: 
t dove N rappresenta il numero di residui di template presenti all'interno del locale, Ali N è il numero della linea query modello coppie di residui, D II è la distanza C α tra i due esimo di residui allineati, d 0 = 3.0 Å è il taglio a distanza, M ii è il punteggio BLOSUM tra due-esimo di residui allineati. In generale, la Fh-score è nel range [0, 5] e il punteggio ACM è compresa tra [0, 2], Dove punteggi più alti indicano incarichi funzionali più sicuri. Punteggio ACM è utilizzato anche per valutare la struttura locale e somiglianza sequenza vicino al ligando siti di legame, che viene indicato come BS-score.
1. Presentazione della sequenza della proteina
- Visita il I-Tasser pagina web all'indirizzo http://zhanglab.ccmb.med.umich.edu/I-TASSER per iniziare con la struttura e la modellazione esperimento funzioni.
- Copia e incolla la sequenza di aminoacidi nella forma fornita o direttamente caricare dal tuo computer cliccando sul pulsante "Sfoglia". I-Tasser server accetta attualmente le sequenze con fino a 1500 residui. Le proteine più di 1500 i residui sono di solito multi-dominio proteine, e sono raccomandati per essere suddiviso in singoli domini prima di presentare al I-Tasser.
- Fornire il proprio indirizzo e-mail (obbligatorio) e un nome per il lavoro (opzionale).
- Gli utenti possono specificare facoltativamente esterno inter-rescontatto idue / distanza restrizioni, add-in un modello aggiuntivo o escludere alcune proteine modello durante il processo di modellazione della struttura. Ulteriori informazioni sull'utilizzo di queste opzioni nella sezione "Discussione".
- Per inviare la sequenza, fare clic sul pulsante "Run I-Tasser". Il browser sarà indirizzato a una pagina di conferma visualizzazione utente specificato informazioni, identificazione del processo (ID lavoro) e un collegamento a una pagina web in cui i risultati saranno depositati dopo il completamento del lavoro. Gli utenti possono bookmark questo link o annotare il numero di identificazione lavoro per riferimento futuro.
2. Disponibilità dei risultati
- Controllare lo stato del tuo lavoro presentato visitando la pagina I-Tasser coda http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php . Fare clic sulla scheda ricerca e utilizzare il numero ID di lavoro o la sequenza di query per il vostro lavoro di ricerca presentato.
- Dopo che la struttura e la funzione moDeling è finito, una e-mail di notifica contenente l'immagine delle strutture previste e una web-link verrà inviato a voi. Clicca su questo link o aprire il link nei segnalibri a punto da 1,5 a visualizzare e scaricare i risultati.
3. Struttura secondaria e le previsioni accessibilità al solvente
- Controllare la sequenza in formato FASTA ricerca visualizzato sulla parte superiore della pagina dei risultati. Se qualche restrizione aggiuntiva / template è stato specificato durante l'invio sequenza, un link per visualizzare la pagina web specificata dall'utente informazione può anche essere visto (Figura 1A).
- Esaminare la predizione della struttura secondaria visualizzato come: alfa elica (H), beta filo (S) o bobina (C) e punteggio di confidenza della previsione (0 = basso, 9 = alta) per ogni residuo. Cercare regione con lunghi tratti di regolare struttura secondaria (H o S) previsioni, per stimare il core-regione della proteina. Classe strutturale della proteina può anche essere analizzato in base alla distribuzione degli elementi secondari strutture. Alcosì, lungo le regioni di elementi bobina nella proteina di solito indicano le regioni non strutturate / disordinato.
- Visualizza l'accessibilità previsto solvente (Figura 1C) per accertare le regioni sepolto e solvente esposti nella query. I valori del predetto gamma solvente accessibilità da 0 (residuo sepolto) a 9 (residui esposti). Regione per lo più interrati contenenti residui possono essere utilizzati per delineare la regione centrale della proteina, mentre le regioni con residui di solvente a vista e l'idratazione sono idrofile potenziale / siti funzionali.
4. Previsioni struttura terziaria
- Scorrere verso il basso per visualizzare le strutture terziarie di proteine previsto query, visualizzati in un applet Jmol interattiva (Figura 2). Clicca col tasto sinistro sull'applet per cambiare l'aspetto della struttura visualizzata, zoom in regione specifica, selezionare i tipi di residui specifici nel modello previsto o calcolare distanze tra residui.
- Analizzare i modelli per la presenza di lunghe regioni non strutturate. Questi regioni di solito corrispondono a regioni disordinate di proteine o di indicare la mancanza di allineamento modello. Queste regioni hanno generalmente bassa precisione la modellazione e la rimozione di queste regioni durante la modellazione da N & C-terminale regione migliorare la precisione di modellazione.
- Scarica il file in formato PDB struttura del modello, cliccando su "Scarica Modello" link. È possibile aprire questi file in qualsiasi software di visualizzazione molecolare (ad esempio PyMOL, Rasmol ecc) per ulteriori analisi delle caratteristiche strutturali.
- Analizzare il punteggio di confidenza (C-score) di modellazione struttura per valutare la qualità delle strutture previste. C-score (Eq. 2) i valori sono tipicamente nell'intervallo [-5, 2], in cui un punteggio più alto riflette un modello di qualità migliore. La stima TM-score e RMSD del primo modello è indicato come "precisione stimata del Modello 1". Per le proteine lungo, si raccomanda di valutare la qualità del modello basato su TM-score, come TM-score è più sensibile ai cambiamenti topologici di RMSD. < li> Clicca su "più su C-score" link per analizzare C-score, dimensione dei cluster e la densità gruppo di tutti i modelli. Stima TM-score e RMSD sono presentati solo per i primi I-Tasser modello, perché C-score inferiore di modelli classificato, non è fortemente correlato con TM-score o RMSD. Qualità della bassa classifica i modelli possono essere in parte valutata in base alla loro densità cluster e dimensione dei cluster rispetto al primo modello, in cui i modelli dai più grandi cluster e una maggiore densità sono in media più vicini alla struttura nativa.
- Low C-score di solito le previsioni indicano una previsione a bassa precisione. Nella maggior parte dei casi, la proteina richiesta non ha un buon modello in biblioteca e ha una dimensione oltre il campo della modellazione ab initio (cioè> 120 residui). In questi casi, gli utenti possono cercare ulteriori restrizioni spaziali e utilizzarli per migliorare l'I-Tasser di modellazione (vedi sezione Discussione). E 'inoltre invitati a presentare le sequenze al nostro server QUARK (QUARK / "> http://zhanglab.ccmb.med.umich.edu/QUARK/) per una pura modellazione ab initio se le dimensioni delle proteine è inferiore a 200 residui.
5. LOMETS obiettivo l'allineamento modello
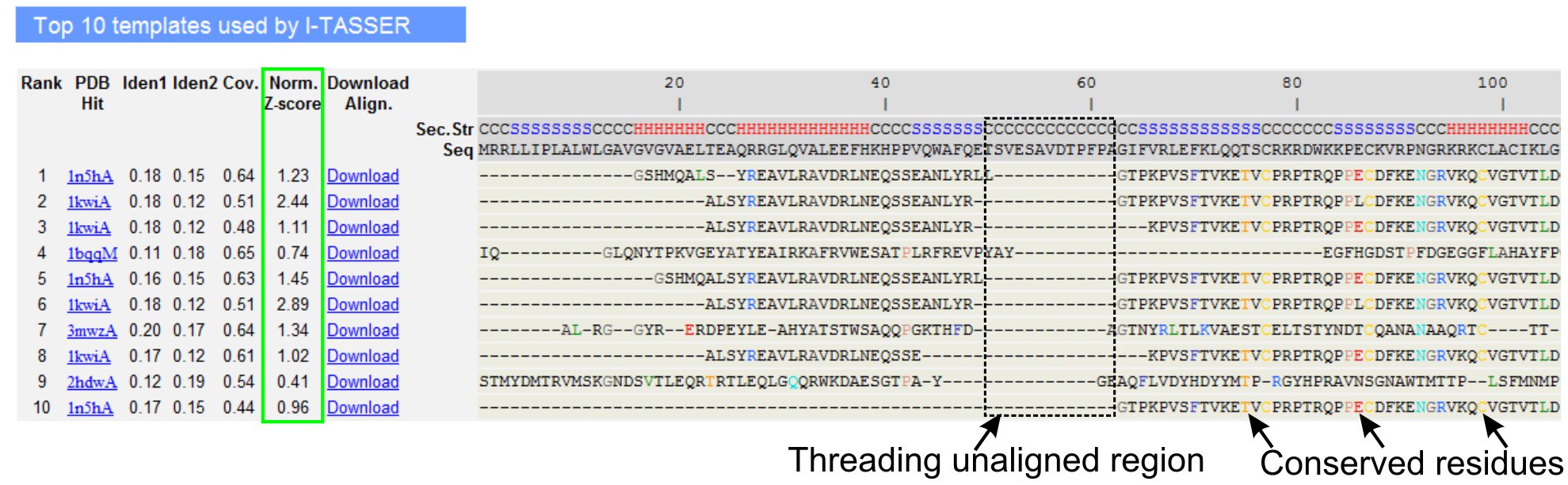
- Scorrere verso il basso per analizzare i primi dieci modelli filettatura della proteina query, come identificato da LOMETS programmi filettatura (Figura 3). Mostra il normalizzata Z-score (Eq. 1), mostrato in 'Norm. Colonna Z-score ', per analizzare la qualità degli allineamenti filettatura. Allineamenti con un normalizzata Z-score> 1 riflette un allineamento fiducioso e molto probabilmente la piega come la proteina query.
- Analizzare l'identità sequenza nella regione filettatura-allineati ('Iden. 1' colonna) e per l'intera catena (colonna 'Iden. 2') per valutare l'omologia tra la query e le proteine modello. Identità di sequenza ad alta è un indicatore di relazione evolutiva tra la query e proteine modello.
- Vedi i residui di filettatura allineati mostrato in colorati per identificare visivamente controresidui erved / motivi nella query e le proteine modello. Una identità di sequenza più alto in filettatura allineato a regione, rispetto a tutta la catena di allineamento indica anche la presenza di conservata motivo strutturale / domini nella query.
- Valutare la copertura del tracciato threading visualizzando il 'Cov.' colonna e ispezionare l'allineamento. Se la copertura degli allineamenti top è bassa e limitata solo ad una piccola regione della proteina query o assenti per un lungo tratto della sequenza query, quindi la proteina query contiene solitamente più di un dominio e si consiglia di dividere la sequenza e il modello i domini individualmente (Figura 3).
- Scarica il PPB formattato sequenza-struttura dei file di allineamento cliccando su "Scarica Allinea" link. Questi file di allineamento può essere aperto in qualsiasi programma di visualizzazione molecolare elencati nella sezione Materiali, e può anche essere usato per aggiungere ulteriori restrizioni durante la modellazione delle strutture (Step 1.4).
6.Analoghi strutturali nel PPB
- Visualizzare la tabella seguente (Figura 4) della pagina dei risultati per determinare i primi dieci analoghi strutturali del primo modello previsto, come indicato dal programma di allineamento strutturale TM-align 20. Un TM-score> 0,5 indica che l'analogico e il modello hanno rilevato una topologia simile e può essere utilizzato per determinare la classe strutturale / proteine della famiglia delle proteine domanda 16, mentre quelli con TM-score <0,3 indica una somiglianza casuale struttura.
- Analizzare l'identità di sequenza e RMSD nella regione strutturalmente allineati mostrato in 'IDEN a' e 'RMSD un' colonne per valutare la conservazione dei motivi spaziali nel modello e l'analogo strutturale. Controllare visivamente le coppie di residui di colore e allineato in allineamento per identificare questi residui strutturalmente conservati e motivi.
- Clicca sul codice PDB indicato nella colonna 'Hit PPB' per visitare il sito RCSB e conoscere meglio la loro classificazione strutturale (SCOP, CATH e PFAM) e le informazioni funzionali (numero CE, associato GO termini e ligando legato).
7. Funzione di predizione utilizzando cofattore
- Scorrere verso il basso nella pagina dei risultati per analizzare le interpretazioni funzionali per la proteina richiesta. Le funzioni delle proteine sono enumerati in tre tabelle contesto, la visualizzazione: enzimi della Commissione (CE) numeri, Gene Ontology (GO) i termini, e ligando siti di legame.
- Vista la 'TM-score', 'RMSD a', 'IDEN a' e 'Cov.' colonne di ogni tabella per analizzare i parametri di somiglianza struttura globale e conservazione dei modelli spaziali tra modello e identificati omologhi funzionali (template).
8. Enzima Commissione il numero di previsione
- Guarda le prime cinque omologhi enzima potenziale della proteina query illustrata nella "Predicted CE numeri" tabella (Figura 5). Il livello di confidenza della previsione di numero CE utilizzando questi modelli è mostrato nella colonna 'CE-Score'. Sulla base di benchmarking analisi 23, somiglianza funzionale (prime 3 cifre del numero CE) tra la query e proteine modello può essere attendibilmente interpretato utilizzando CE-score> 1.1.
- Cercare il consenso della funzione (numeri CE) tra i modelli, che hanno la piega simile (cioè TM-score> 0,5), come la proteina query. Se tra molti modelli hanno lo stesso numero CE e CE-score> 1,1, il livello di confidenza della previsione è molto alto. Tuttavia, se il-CE punteggio è alto, ma c'è una mancanza di consenso tra i colpi identificati, poi la previsione diventa meno affidabile e gli utenti sono invitati a consultare le previsioni GO termine.
- Clicca sul link fornito sui numeri CE per visitare il database ExPASy enzimi e analizzare la funzione, tra cui la reazione catalizzata, co-fattore di requisiti e la via metabolica, della proteina modello in dettaglio.
9. Gene Ontology (GO) Termine previsioni
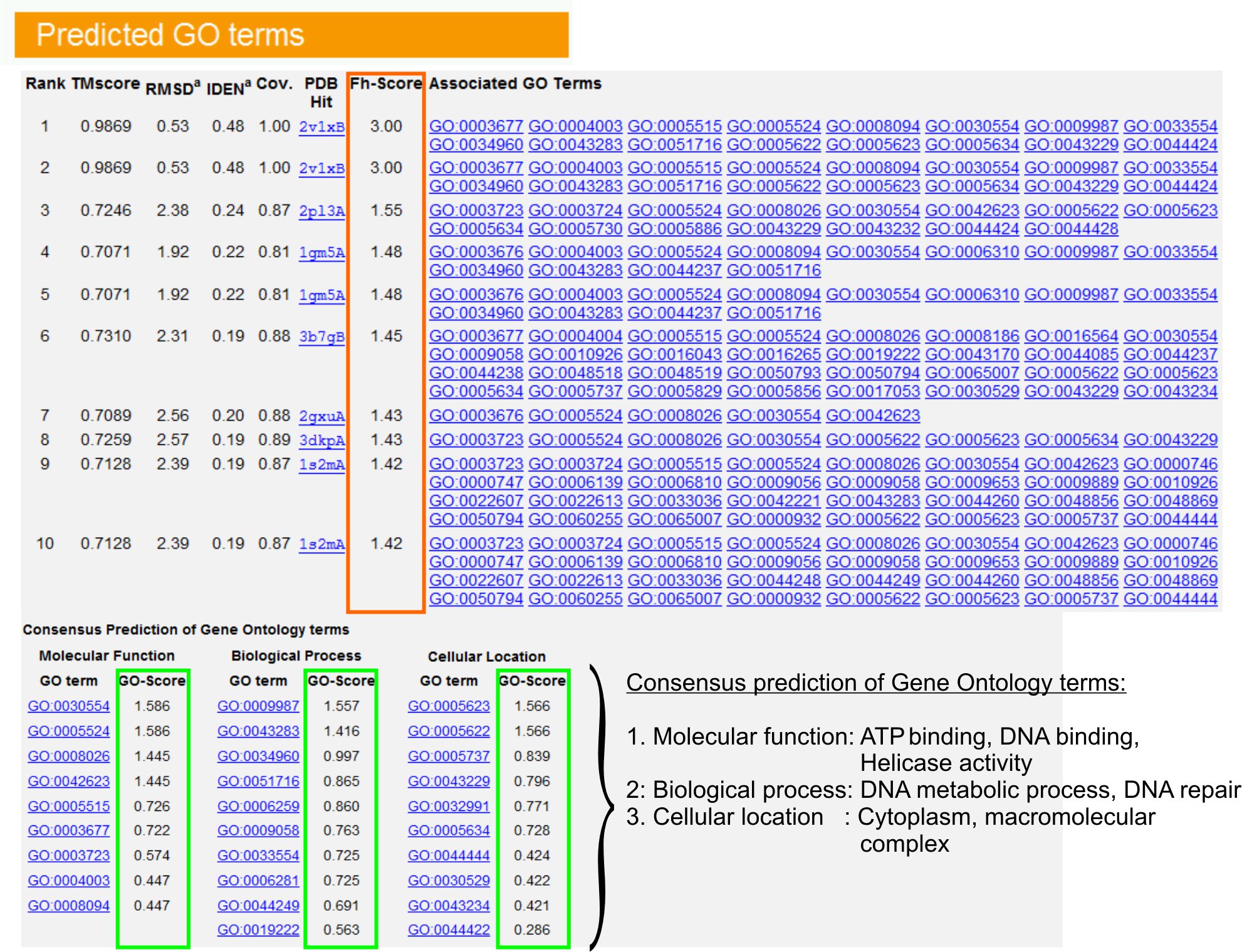
- Vista la "Predicted GO termini" tabella (Fig.URE 6) per identificare i top ten omologhi di proteine query nella biblioteca PPB, annotati con Gene Ontology (GO) termini. Ogni proteina è di solito associata a termini GO multipli, descrivendo le sue funzioni molecolare (MF), i processi biologici (BP) e la componente cellulare (CC). Clicca su ogni termine a visitare il sito Amigo e analizzare la sua definizione e lignaggio.
- Analizzare la Fh-score (punteggio di omologia funzionale) della colonna per accedere alla somiglianza funzionale tra la query e le proteine modello e stimare il livello di affidabilità del trasferimento di annotazione funzionale di queste proteine. Nel nostro studio di benchmarking 23, il 50% dei termini GO nativo potrebbe essere identificato correttamente dal primo modello identificato con un Fh-score di taglio pari a 0,8, con una precisione complessiva del 56%.
- Visualizzare la tabella "previsione di termini di consenso GO" per analizzare il concorso di funzione tra i modelli. Queste funzioni comuni sono utilizzati per prevedere i termini GO (MF, BP e CC) della queryproteine e valutare il livello di confidenza (GO-score) di GO previsioni a lungo termine. In base al test di benchmarking 23, i migliori tassi di falsi positivi e falsi negativi sono ottenuti per le previsioni con GO-score di taglio = 0,5, con la diminuzione della copertura della previsione a livelli più profondi ontologia.
10. Proteina-ligando predizioni sito di legame
- Scorrere fino alla parte inferiore della pagina per visualizzare top ten previsioni ligando sito di legame per la proteina richiesta. Previsto siti di legame sono classificati in base al numero di conformazioni ligando predetto che condividono tasca di legame. Il miglior sito individuato legame è già visualizzato nell'applet Jmol. Fare clic sul pulsanti di opzione per analizzare altre previsioni e visualizzare i residui ligando interagenti.
- Analizzare la BS-score colonna per valutare similarità locali tra il modello e sito di legame modello. Basato sul benchmark 9, BS-score> 1,1 indica la sequenza e la struttura di alta similarity vicino al luogo previsto obbligatorio in tutti i modelli e conosciuto sito di legame nel modello.
- Scarica il file in formato PDB struttura del complesso, cliccando sul link "Download". Gli utenti possono aprire questi file in qualsiasi programma di visualizzazione molecolare e interattivo visualizzare il sito predetto vincolante e ligando-proteina interazioni sul proprio computer locale.
11. Rappresentante risultati

Figura 1 Un estratto di I-Tasser pagina dei risultati che mostrano (A) in formato FASTA sequenza query;. (B) previsto struttura secondaria e decine confidenza, e (C) prevede l'accessibilità dei residui di solvente. Regione centrale del sito analizzato e idratazione potenziale nella query sono evidenziati in azzurro e rettangoli rossi, rispettivamente.

Figura 2.

Figura 3. Un esempio di I-Tasser pagina dei risultati che mostrano top ten dei modelli individuati threading e allineamenti da LOMETS 5 programmi filettatura. La qualità degli allineamenti threading è valutato sulla base normalizzata Z-score (evidenziato in verde), dove un valore> 1 riflette un allineamento fiducioso. Residui allineati nel modello che sono identici ai residui interrogare corrispondenti sono evidenziati a colori per indicare la presenza di residui conservati / motivo, mentre la mancanza di omogeneità nella maggior parte dei modelli top indica la presenza di più domini nella proteina query e dei residui non allineati corrispondono alle regioni linker dominio. Clicca qui per visualizzare la versione full-size di figura 3.

Figura 4. Un esempio di pagina dei risultati che mostrano top ten analoghi individuati strutturali e allineamenti strutturali, identificati da TM-align 20 programma strutturale di allineamento. La classifica degli analoghi mostrato in si basa sul TM-score (evidenziata in blu) l'allineamento strutturale. Un TM-score> 0,5 indica che le due strutture hanno confrontato una topologia simile, mentre un TM-score <0,3 indica una somiglianza tra due strutture casuali. Coppie di residui strutturalmente allineati sono evidenziati in colore in base alla loro amino-acido proprietà, mentre le regioni non allineate sono indicati con "-".ove.com/files/ftp_upload/3259/3259fig4large.jpg "> Clicca qui per visualizzare la versione full-size della figura 4.

Figura 5. Un esempio di I-Tasser pagina dei risultati che mostrano omologhi enzima identificato della proteina query nella biblioteca PPB. Il livello di confidenza della previsione numero CE viene analizzato sulla base di CE-score (evidenziato in verde), dove CE-score> 1,1 indica somiglianza funzionale (stessa prime 3 cifre del numero CE) tra query e proteine modello.

Figura 6. Un esempio di I-Tasser pagina dei risultati che mostrano GO previsioni a lungo termine per la proteina richiesta. Omologhi funzionali per la proteina richiesta nella libreria modello Gene Ontology sono classificate in base al loro Fh-score (nel rettangolo arancione). Comuni caratteristiche funzionali di questi top-hit di punteggio sono derivati per generare mangiato la finale GO previsioni a lungo termine per la proteina richiesta. La qualità del GO termini previsti è stimato sulla base GO-score (in verde), dove un GO-score> 0,5 indica una previsione affidabile. Clicca qui per visualizzare la versione full-size di figura 6.

Figura 7. Un esempio di I-Tasser pagina dei risultati che mostrano top ten delle proteine previsioni sito di legame ligando utilizzando l'algoritmo di 9 cofattore. La classifica dei siti predetto legame si basa sul numero di conformazioni ligando predetto che condividono tasca di legame nella query. BS-score (evidenziato in rosso) è una misura della sequenza locali e somiglianza tra la struttura prevista e sito di legame template, ed è utile per analizzare la conservazione delle sacche sito di legame.
les/ftp_upload/3259/3259fig8.jpg "/>
Figura 8. Un esempio di file di contenimento esterni utilizzati per per la specifica di residui di sansa di contatto / restrizioni distanza.

Figura 9. Esempio di file di contenzione utilizzati per specificare una proteina modello per l'I-Tasser server. L'utente può specificare la query modello dell'allineamento sia in (A) formato FASTA, o (B) in formato 3D.

Figura 10. Un esempio di file utilizzato per escludere modello durante la I-Tasser procedura struttura di modellazione. La prima colonna contiene l'ID PPB delle proteine modello da escludere. La seconda colonna è utilizzata per specificare l'identità di sequenza di taglio che sarà utilizzato per altri modelli simili in biblioteca modello.
Discussion
Il protocollo di cui sopra è una linea guida generale per la struttura e la modellazione funzione utilizzando l'I-Tasser server. Anche se, questa procedura automatizzata funziona molto bene per la maggior parte delle proteine, interventi umani spesso aiutano a migliorare significativamente la precisione di modellazione, soprattutto per le proteine che mancano di modelli chiudere in biblioteca PPB. Gli utenti possono intervenire durante la I-Tasser di modellazione nei seguenti modi: (a) scissione della multi-dominio proteine, (b) fornire vincoli esterni per migliorare la struttura di assemblaggio, e (c) la rimozione dei modelli durante la modellazione.
Dividere multi-dominio proteico:
Molte sequenze proteiche lunghe spesso contengono più domini legati da regioni linker flessibile, il che rende loro difficile delucidazione struttura utilizzando tecniche sia sperimentali e computazionali. Tuttavia, come i domini sono pieghevoli entità indipendente e può eseguire diverse funzioni molecolare, èauspicabile per dividere lungo multi-dominio proteine e il modello ogni dominio separatamente. I domini di modellazione individualmente non solo accelerare il processo di previsione, ma aumenta anche la qualità della query modello dell'allineamento, con conseguente maggiore struttura affidabile e previsioni funzione.
I limiti del dominio a sequenze proteiche possono essere previsti con programmi esterni liberamente disponibile online, come NCBI CDD 24, 25 o PFAM InterProScan 26. Inoltre, se allineamenti LOMETS threading sono disponibili per la proteina query, i limiti del dominio può essere individuato da individuare visivamente lunghi tratti di residui non allineati nei modelli top filettatura (vedi punto 5.4). Queste regioni non allineati per lo più corrispondono alle regioni linker dominio. Se il multi-dominio modelli sono già disponibili nella libreria di modelli PDB con tutti i domini domanda allineati, allora la proteina query può essere modellato come lunghezza.
Fornire vincoli esterni
A. Specificare contatto / distanza restrizioni
Sperimentalmente caratterizzata inter-residuo contatti / distanze, ad esempio da NMR ocross-linking esperimenti, può essere specificato il caricamento di un file di moderazione. Un file di esempio è illustrato nella figura 8, dove Colonna 1 specifica il tipo di restrizione, cioè "DIST" o "CONTATTI". Alla moderazione distanza (DIST), colonne 2 e 4 contengono residui di posizioni (i, j), colonne 3 e 5 contengono l'atomo-tipo nel residuo e colonna 6 specifica la distanza tra i due atomi specificato. Per le restrizioni di contatto (contatto), colonne 2 e 3 contengono le posizioni (i, j) di residui che devono essere a contatto. La distanza tra il centro catene laterali di queste coppie di residui di contatto è deciso sulla base di distanze osservati in strutture note nel PPB. I-Tasser cercherò di tracciare queste coppie atomo vicino alla distanza specificata durante le simulazioni raffinatezza della struttura.
B. Specificare un modello di struttura delle proteine
LOMETS programmi threading utilizzare una libreria rappresentante PDB per trovare pieghe plausibile per la richiesta protein. Sebbene l'utilizzo di una libreria struttura rappresentativa aiuta a ridurre il tempo necessario per calcolare la sequenza-struttura allineamenti, è possibile che una proteina modello buono è mancato in biblioteca o il modello non sono stati identificati dai programmi di LOMETS filettatura, anche se è presenti nella biblioteca. In questi casi, l'utente deve specificare la struttura della proteina desiderata come il modello.
Per specificare la struttura della proteina come un ulteriore modello, gli utenti possono caricare un file PDB struttura formattata o specificare l'ID PPB di una struttura proteica depositato in biblioteca PPB. L'I-Tasser genererà la query-modello di allineamento con il programma MUSTER 23 e raccoglierà vincoli spaziali sia l'utente specificato modello e modelli LOMETS per guidare la simulazione di assemblaggio della struttura. Perché l'accuratezza dei vincoli LOMETS è differente per target diversi, il peso dei vincoli LOMETS è più forte in facile (omologo) targets da quello rigido (non omologhe) obiettivi, che sono state sistematicamente sintonizzato nella nostra formazione di riferimento.
Gli utenti possono anche specificare il proprio modello di query-allineamenti. Il server accetta l'allineamento in due formati: il formato FASTA (Figura 9A) e il formato 3D (Figura 9B). Il formato FASTA è standard e descritto in http://zhanglab. ccmb.med.umich.edu / FASTA / . Il formato 3D è simile al formato standard PDB ( http://www.wwpdb.org/documentation/format32/sect9.html ), ma due colonne aggiuntive derivate dai modelli vengono aggiunti ai record ATOM (vedi Figura 9B):
Colonne 1-30: Atom (C-alfa solo) e residui di nomi per la sequenza query.
Colonne 31-54: Coordinate di C-alfa atomi della query copiato dal atomi corrispondente nel modello.
Colonne 55-59: numero di residuo corrispondente nel modello sulla base di allineamento
Colonne 60-64: nome di residuo corrispondente nel modello
Escludi proteine modelli
Le proteine sono molecole flessibili e possono adottare diversi stati conformazionali di cambiare la loro attività biologica. Per esempio, le strutture di molte proteine chinasi e proteine di membrana sono stati risolti in conformazione sia attivi e inattivi. Anche la presenza o l'assenza di ligando legato possono causare grandi movimenti strutturali. Mentre tutti gli stati conformazionali del modello sono simili per i programmi di threading, è auspicabile per modellare la query utilizzando i modelli in un solo stato. Una nuova opzione sul server consente all'utente di escludere le proteine modello durante la modellazione della struttura. Questa caratteristica permetterebbe inoltre agli utenti di scegliere il livello di omologia di modelli da utilizzare per la modellazione. Gli utenti possono escludere le proteine modello from I-Tasser libreria:
A. Definizione di un taglio identità di sequenza
Gli utenti possono utilizzare questa opzione per escludere proteine omologhe da I-Tasser libreria di modelli. Il livello di omologia è impostato in base al taglio identità di sequenza, cioè il numero di residui identici tra la query e la proteina modello di divisa per la lunghezza della sequenza della sequenza query. Per esempio, se l'utente digita nel "70%" nella forma prevista, tutte le proteine modelli che hanno una identità di sequenza> 70% per la proteina richiesta I-saranno esclusi dalla I-Tasser libreria di modelli.
B. Escludi proteine modello specifico
Proteine modello specifico può essere escluso dalla I-Tasser libreria di modelli caricando un elenco contenente gli ID PPB delle strutture da escludere. Un file di esempio è illustrato nella figura 10. Come la stessa proteina può esistere come più voci nella libreria PPB, I-Tasser séfiume Eno per default escludere i modelli specificato (in Colonna1), così come tutti gli altri modelli della libreria che hanno una identità> 90% per i modelli specificati. Gli utenti possono anche specificare una soglia diversa identità, ad esempio, il 70%, dove tutti i modelli con l'identità> 70% di proteine modello specificato saranno escluse.
Disclosures
Nessun conflitto di interessi dichiarati.
Acknowledgments
Il progetto è sostenuto in parte dalla Fondazione Alfred P. Sloan, Premio alla Carriera NSF (DBI 1027394), e l'Istituto Nazionale di General Medical Sciences (GM083107, GM084222).
Materials
| Name | Company | Catalog Number | Comments |
| Material Name | Type | Company | Catalogue Number |
| FASTA formatted amino acid sequence of the protein to be modeled (see, http://www.ncbi.nlm.nih.gov/BLAST/fasta.shtml). | |||
| A personal computer with access to the internet and a web browser. | |||
| Molecular visualizing software, e.g. RASMOL or PYMOL, for analyzing the predicted tertiary structure and functional sites. |
References
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H.
- Finn, R. D.
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).

{kind=link}
{kind=link}