

Summary
Las células bacterianas se organizan espacialmente muy. Para seguir esta organización en el tiempo en lento crecimiento células de Myxococcus xanthus , se desarrolló un montaje de imágenes de células vivas de fluorescencia de alta resolución espacio-temporal durante varias generaciones. Usando este método, se pudo determinar dinámica espaciotemporal de proteínas importantes para la segregación cromosómica y la división celular.
Abstract
Imágenes de células vivas de la fluorescencia de las células bacterianas son un método clave en el análisis de la dinámica espacial y temporal de las proteínas y los cromosomas subyacen eventos del ciclo celular central. Sin embargo, la proyección de imagen de estas moléculas en bacterias de crecimiento lento representa un reto debido al fotoblanqueo de fluoróforos y fototoxicidad durante la adquisición de la imagen. Aquí, describimos un protocolo simple para evitar estas limitaciones en el caso de Myxococcus xanthus (que tiene un tiempo de generación de 4-6 h). Para ello, M. xanthus las células se cultivan en una gruesa almohadilla de agar que contiene nutrientes en un ambiente húmedo controlado por temperatura. En estas condiciones, determinamos el tiempo de duplicación de células individuales, siguiendo el crecimiento de las células. Por otra parte, celular clave procesos tales como segregación cromosómica y la división celular pueden ser reflejadas por la proyección de imagen de células vivas de la fluorescencia de celdas que contiene relevantes proteínas marcador fluorescente etiquetado como extrasede YFP, FtsZ-GFP y mCherry alcohólico sobre múltiples ciclos de la célula. Posteriormente, se procesan las imágenes adquiridas para generar montajes o películas.
Introduction
Las células bacterianas se organizan espacialmente muy con muchas proteínas localizar asimétricamente dentro de compartimentos celulares1,2,3,4. Esta localización suele ser muy dinámica y cambia con el tiempo en respuesta a señales del ciclo celular o señales externas. Igualmente, el cromosoma bacteriano es espacial altamente organizado con loci individuales se colocan a localizaciones subcelulares específicos antes y durante el proceso de segregación5. Esta organización espacial dinámica es importante para el crecimiento, división, regulación del ciclo celular, diferenciación, motilidad, transduction de la señal así como organización del cromosoma y segregación; por lo tanto, afecta esencialmente todos los aspectos de la función bacteriana.
La dinámica espacio-temporal de estos procesos celulares está siendo analizada en una variedad de especies bacterianas con Escherichia coli, Bacillus subtilis, Vibrio choleraey Caulobacter crescentus que sirve como importante organismos modelo. Sin embargo, estas cuatro especies cubren solamente un pequeño espectro de la enorme diversidad bacteriana y, tal vez como era de esperar dada la gran distancia filogenética entre estas especies, mecanismos celulares de la organización y la polarización son diferentes en estos bacterias. Esto plantea la necesidad de estudiar otras especies bacterianas para poder eventualmente extraer principios generales subyacentes a la dinámica espacio-temporal de las células bacterianas.
El delta-proteobacterium gramnegativa M. xanthus es un organismo modelo en el estudio de comportamientos sociales y la cooperación en las bacterias6. M. xanthus es un aerobio estricto y en presencia de nutrientes, forma colonias en los cuales las células separados hacia fuera en un altamente coordinada, enjambre moda y presas en otros microorganismos7. En respuesta al hambre de nutrientes, las células inician un programa de desarrollo que resulta en la formación de cuerpos de fructificación que consiste en miles de células y dentro de la cual, las células móviles en forma diferencian a esférico diploide esporas8. Ambos tipos de comportamientos, es decir, enjambre y formación de cuerpo fructífero, se ejecutan solamente por las células que se colocan sobre una superficie sólida. Por otra parte, en ambas condiciones de nutrientes, las células participan en procesos que involucran contacto directo célula incluyendo el intercambio de las lipoproteínas de la membrana externa que puede estimular la motilidad o funcionar como toxinas en el recipiente9,10 , el intercambio de LPS11, estimulación de la motilidad de exopolisacáridos en los vecinos células12y señalización proteína13,14anclado superficie intercelular señalización por una célula.
Recientemente, M. xanthus también se ha convertido en un organismo modelo para estudiar los mecanismos subyacentes a la motilidad y su Reglamento15división celular16,17,18y organización del cromosoma19 ,20,21. Pasos críticos en la M. xanthus ciclo celular han sido analizados en detalle por microscopia de fluorescencia utilizando instantánea imágenes o grabaciones time-lapse corto en cepas llevan proteínas fluorescencia de etiquetado pertinentes16, 1718,de,19,20. Idealmente, muchas células deben seguirse con resolución sola célula por célula vivo de fluorescencia de imagen para que al menos un ciclo celular completo obtener datos cuantitativos robustos en los parámetros de ciclo celular. Sin embargo, esto es un reto en el caso de M. xanthus debido a su tiempo de generación relativamente largo de 4-6 h en condiciones estándar de laboratorio y fotoblanqueo de fluoróforos y fototoxicidad durante la adquisición de la imagen.
Aquí, describimos un protocolo a seguir M. xanthus las células con una resolución de única célula por fluorescencia células vivas imágenes durante al menos 24 h y que abarcan varios ciclos celulares. Lo importante es que, durante el conjunto del Protocolo, las células se mantienen en una almohadilla de agar y en cercano contacto teniendo en cuenta las actividades dependientes de contacto esencial para el estilo de vida social de M. xanthus. El protocolo también permite a los usuarios monitor forma, tamaño, divisiones y sondas fluorescentes a una alta resolución temporal y resolución única célula y por lo tanto, permite la cuantificación de la variabilidad de célula a célula y correlaciones de los eventos del ciclo celular.
Protocol
1. preparación y crecimiento de M. xanthus cepas
Nota: Ver tabla 1 y tabla 2.
- Preparar 1% casitone caldo (CTT) crecimiento medio 1% (p/v) digerido pancreático de caseína (p. ej., casitone Bacto), 10 mM Tris-HCl pH 8.0, 1 mM KH2PO4 pH 7.6, 8 mM MgSO422, complementado con kanamicina (50 μg/mL) o oxitetraciclina (10 μg/mL). Agregar gentamicina (10 μg/mL) a todos los medios para reducir el riesgo de contaminación con otras bacterias, ya que M. xanthus células son naturalmente resistentes a él.
- Inocular los 5 mL de 1% CTT que contienen los antibióticos con una sola Colonia recién crecida de salvaje tipo DK1622 (WT) 23, SA4420 (ΔmglA)24, SA4797 (ΔmglA, Δalcohólico/PpomZ mCherry-alcohólico )16, SA8241 (ΔmglA, ftsZ+/PnatftsZ-gfp), o SA4749 (ΔmglA, extrasede+/Pnatextrasede-yfp) en el mañana del día 1.
- Suspender una sola M. xanthus Colonia en 500 μl de 1% CTT suplementado con antibióticos en un tubo estéril y transferir la suspensión entera a un erlenmeyer que contenga 5 mL de 1% de 50 mL CTT.
Nota: Utilice un matraz de Erlenmeyer con 10 veces el volumen de la cultura para garantizar suficiente aeriation y un crecimiento óptimo.
- Suspender una sola M. xanthus Colonia en 500 μl de 1% CTT suplementado con antibióticos en un tubo estéril y transferir la suspensión entera a un erlenmeyer que contenga 5 mL de 1% de 50 mL CTT.
- Crecen las células para ocho generaciones (aproximadamente 40-48 h con un tiempo de generación de 4-6 h) a 32 ° C, agitación a 220 rpm, en la oscuridad. Mantener las células en la fase de crecimiento exponencial (OD550 < 1.2) y evitar que lleguen a la fase estacionaria. Si es necesario, diluir las células en medio fresco de CTT de 1% con los antibióticos pertinentes a un OD550 de 0.1 - 0.2.
Nota: Un óptimo OD550 para microscopía de una sola célula es 0.5 - 0.7. En este OD550, un número suficiente de células está presente por imagen para permitir la cuantificación y análisis estadístico de los parámetros celulares.
2. preparación de muestras de microscopia
Nota: Las células para ser visualizadas por microscopía se colocan sobre un cubreobjetos microscopio y cubiertas por un cojín de agarosa que contiene nutrientes. El cubreobjetos se pega al plástico o soporte de marco de metal para proporcionar mecánico. En preparación para la microscopia, un cojín grande de 1% agarose/TPM/0.2% CTT debe preparar con antelación como se describe en los pasos 2.1-2.3. Consulte también la Tabla de materiales para productos específicos aquí.
- Preparar 500 mL de tampón TPM (10 mM Tris-HCl pH 7,6, 1 m m KH2PO4 pH 7.6, 8 mM de MgSO4) y filtro o autoclave esterilice usando un filtro de botella.
Nota: El buffer estéril puede almacenarse durante varios meses a temperatura ambiente. - Preparar solución de microscopia 1% agarosa que contiene 0.2% CTT (mezcla de 1 g de agarosa con 80 mL de buffer TPM y 20 mL de medio CTT 1%). Calentar en el microondas hasta que la agarosa fundida.
Nota: El 0.2% CTT es suficiente para que las células crecer y evitar el hambre. Concentraciones más altas de CTT en el medio de la microscopia producirá fluorescencia de fondo alto. - Llenar un plato de Petri con la agarosa fundida en un espesor de 0,5 cm (para una plaza de 11,5 x 11,5 cm plato de Petri, aproximadamente 60 mL de agarosa fundida es necesaria) y deje que se enfríe a temperatura ambiente.
Nota: La almohadilla de agarosa puede almacenarse a 4 ° C en un ambiente húmedo durante 2 días.- Precaliente la almohadilla CTT agarose/TPM/0.2% 1% a 32 ° C al menos 15 minutos antes de usarlo.
Nota: Para preparar las células para microscopia, siga los pasos 2.4-2.8.
- Precaliente la almohadilla CTT agarose/TPM/0.2% 1% a 32 ° C al menos 15 minutos antes de usarlo.
- Coloque un cubreobjetos de vidrio estéril (60 mm x 22 mm, espesor: 0,7 mm) en un marco de plástico o metal que tiene un agujero en el medio (figura 1A); este marco sirve como soporte mecánico para el fino cubreobjetos y ayuda a reducir la deriva en la microscopia. Fije el cubreobjetos a la estructura con cinta.
- Para preparar el marco, cortar un marco de 75 x 25 mm de una placa de metal grueso de 1 mm, luego recortar un agujero de tamaño adecuado (20 x 30 mm en este experimento) en el centro.
- Añadir 10-20 μl de crecido exponencialmente M. xanthus células sobre cubreobjetos.
- Añadir microesferas de μm 0,5 fluorescentes como marcadores de referencia a las células para simplificar el seguimiento de células o proteínas en grabaciones time-lapse.
- Diluir las microesferas de 1: 100 en tampón de TPM y almacenar a 4 ° C por hasta varios meses. Agitar bien antes de usar y agregar 5-10 μl de las microesferas diluidas a las células.
Nota: Aquí las microesferas son fluorescentes en todos común azul, verde, amarillo y rojo fluorescentes canales fueron utilizados.
- Diluir las microesferas de 1: 100 en tampón de TPM y almacenar a 4 ° C por hasta varios meses. Agitar bien antes de usar y agregar 5-10 μl de las microesferas diluidas a las células.
- Cortar una almohadilla pequeña aproximadamente el tamaño del cubreobjetos de la grande precalentado 1% agarose/TPM/0.2% CTT almohadilla y coloque encima de las células (figura 1B). Coloque un cubreobjetos sobre el 1% agarose/TPM/0.2% CTT agarosa cojín para evitar la evaporación y para mantener las células en un ambiente húmedo.
Nota: El cubreobjetos solo evitará evaporación significativa durante al menos 2 h. Para más grabaciones time-lapse, el sándwich 1% agarose/TPM/0.2% CTT pad y cubreobjetos debe sellarse con la película de parafina para evitar la evaporación. - Incubar la muestra microscopía a 32 º C durante 15-20 min dejar las células fije a la parte inferior de la almohadilla de agarosa. Comienzan entonces las grabaciones time-lapse de la microscopia.
3. microscopio instalación y adquisición de Time-lapse
Nota: El protocolo descrito aquí fue desarrollado para un microscopio invertido widefield con autofocus, 100 X / NA 1.30 aceite PH3 objetivo, una X, Y motorizado etapa, una cámara sCMOS, una fuente de luz, filtros de verde fluorescente, rojo fluorescente o fluorescente amarillo proteínas y una cámara de incubación controlada de la temperatura. Esta cámara mantiene células protegidas de la luz y a temperatura constante.
- Precalentar la cámara de incubación y el microscopio a 32 ° C para 2 ~ 1 h antes de la microscopia.
Nota: Dependiendo de la configuración del microscopio, el calentamiento puede tardar más. Precalentamiento es esencial para reducir la deriva y estabiliza el sistema de control de enfoque automático. - Encienda el microscopio e inicie el software de control de microscopio. Seleccionar el objetivo correcto y el correcto espejos y filtros para adquirir fase imágenes de contraste así como imágenes de proteínas verde fluorescente, rojo fluorescente o fluorescente amarillo.
Nota: Un microscopio normalmente se suministra con un software preferido para la adquisición de control y la imagen del microscopio. Aquí un software comercialmente disponible (véase la Tabla de materiales) fue utilizado para el control de la adquisición de microscopio y de la imagen. - Añadir una gota de aceite de inmersión de alta calidad sobre la lente del objetivo y en la parte inferior de la muestra previamente incubada a 32 ° C. Coloque el objetivo en la posición más baja Z-para no dañar la lente del objetivo cuando la muestra se coloca sobre la platina del microscopio. Coloque el marco de metal con la muestra sobre la platina del microscopio y con el "lado de agujero" hacia el objetivo. Fijar la muestra con seguridad en el soporte de la etapa.
- Se centran en las células mediante el movimiento de la etapa en la dirección Z más cerca al objetivo. Mover la etapa más lenta cuando el aceite en la parte de la muestra y el objetivo hacer contacto. Mover el escenario en el X dirección Y hasta varias celdas individuales son visibles en la región del punto de vista, cuando las células están en el plano focal. Asegúrese de que al menos una microesfera fluorescente es en la región de vista para luego alinear las imágenes adquiridas.
Nota: En condiciones óptimas, debe alcanzarse una densidad celular de las células de 15-30 por región de vista (2.048 x 2.048 píxeles o 133.1 x 133.1 μm). - Abrir al asistente de Adquisición multidimensional del software de control de microscopio para establecer un experimento de lapso de tiempo que permite el microscopio adquirir imágenes en múltiples longitudes de onda y las posiciones de etapa si es necesario.
- En la pestaña principal activar Timelapse y Varias longitudes de onda. Fichas adicionales aparecerá en el lado izquierdo de la ventana.
- Haga clic en la ficha Guardar y Seleccionar directorio para seleccionar una carpeta vacía en el disco duro del ordenador para guardar las imágenes adquiridas. Activar el nombre de base de incremento si el archivo existe para asegurarse de que los datasets consecutivos no sobrescribir los anteriores. Luego dar el experimento un nombre con la fecha y el nombre de la cepa o el título del experimento.
- Haga clic en la ficha de lapso de tiempo para ajustar los parámetros Time-lapse. Definir la duración a las 24 h y establecer el Intervalo de tiempo a 20 minutos. El Número de puntos del tiempo cambiará automáticamente.
Nota: El intervalo de tiempo óptimo depende de la experiencia y la función celular a analizar. Adquisiciones de imagen frecuente pueden causar fotoblanqueo. Por lo tanto, una relación inversa entre la resolución temporal y photobleaching debe encontrarse empíricamente. En un tiempo de duplicación de 4-6 h, imágenes pueden ser fácilmente adquiridas en un intervalo de 5 minutos (o incluso más pequeños intervalos si se desea) para microscopía de contraste de fase. Si la microscopía de fluorescencia sobre un curso de tiempo de 24 h se desea imágenes deben grabarse en un intervalo de unos 15-30 min. - Haga clic en la ficha de longitudes de onda Seleccione el número de longitudes de onda para la adquisición de cada imagen en cada momento cambiando el número.
Nota: para cada longitud de onda, aparecerá una nueva pestaña en el lado izquierdo de la adquisición de multidimensional " mago y longitudes de onda se adquirirán en el orden de arriba a abajo. Para cada longitud de onda, puede modificarse la configuración de la adquisición por separado. - Haga clic en la primera pestaña de la longitud de onda de la parte superior. Seleccione contraste de la fase en la lista desplegable de iluminación . Seleccione 100 ms para la exposición y Cada momento en la lista desplegable de adquisición . Desactivar Auto exponer seleccionando nunca en la lista desplegable.
- Repita el paso 3.5.5 para cada longitud de onda que necesita adquirir en cada momento. Para el montaje experimental y fluorescencia etiquetadas proteínas descritas aquí, utilice los siguientes parámetros para exposición: 250 ms para proteínas de fusión mCherry, 200 ms para proteínas de fusión YFP y 1.000 ms para las proteínas de la fusión de GFP.
Nota: La configuración de iluminación óptimo para cada cepa y proteína fluorescente debe determinarse con antelación cambiando la intensidad de la lámpara y el tiempo de adquisición de imagen para cada longitud de onda. Tiempos de adquisición de imágenes demasiado aumentará el efecto fototóxico y en última instancia conducir a crecimiento detención y muerte celular. Por lo tanto, debe lograrse un equilibrio entre viabilidad de célula y la calidad de imagen. - Adquisición de imágenes desde múltiples posiciones de etapa para aumentar el número de células en el mismo experimento.
- Para adquirir imágenes desde múltiples posiciones de la etapa, activar Múltiples posiciones de la etapa en la ficha principal . Luego haga clic en el escenario y pulse el botón de Live para mirar el campo de visión.
- Mover el escenario en la dirección X/Y hasta una región de interés (ROI) en el campo de visión. Guardar las coordenadas X e Y-haciendo clic en el "+" en la ficha de etapa mueva la etapa otra vez en el X/Y-dirección hasta encontrar un retorno de la inversión nueva y guardar las coordenadas nuevamente haciendo clic en el "+". Siga hasta el número deseado de regiones está guardado.
Nota: En caso de adquisición de imágenes de fluorescencia, asegúrese de que las regiones de interés (ROIs) no están muy cerca entre sí para minimizar la fototoxicidad.
- Compruebe una vez más que las células están en foco haciendo clic en la guardada X - y Y-posiciones e iniciar el enfoque automático de hardware haciendo clic en AFC mantener para mantener la posición de Z guardada constante a lo largo del experimento.
- Comienzan las grabaciones time-lapse en el software de control de microscopio haciendo clic en adquirir en el Asistente para la Adquisición de multidimensional .
Nota: Aparecerá una ventana para cada longitud de onda que es adquirido y aparecerá una ventana adicional que muestra el número de puntos adquiridos tiempo y el tiempo hasta la próxima adquisición de imagen. - Comprobar que las células todavía están en foco después de los primeros puntos de tiempo en las grabaciones time-lapse para maximizar la calidad de las imágenes y reorientar si es necesario.
4. generación de películas de Time-lapse y alineación de la imagen
Nota: Varios paquetes de software comercial y libre están disponibles para la adquisición de imágenes y análisis de imagen. Se utiliza un software comercialmente disponible (véase la Tabla de materiales) con múltiples plugins preinstalados y herramientas adicionales.
- Guardar las imágenes individuales de grabaciones time-lapse en un equipo que tiene instalado el software de análisis y procesamiento de imagen.
- Inicie el software y abrir imágenes como una pila haciendo clic en Informe multidimensional datos | Seleccione Archivo de Base | Seleccione el directorio. Abra la carpeta con los datos multidimensionales. Compruebe el conjunto de datos y haga clic en vista; el conjunto de datos se mostrarán como imágenes desde uno hasta el final. Activar la longitud de onda (para crear una pila), seleccione todas las imágenes que deben ser de la pila y haga clic en la Imagen de carga. Repita este paso para todas las longitudes de onda y ahorrar pilas completadas.
- (Opcional) Abrir todas las imágenes necesarias para la película con archivo | Abierto.
Nota: Se recomienda abrir imágenes por una longitud de onda adquirida a la vez para no ralentizar el equipo si poder computacional es limitada. Si deben omitirse ciertas partes de las grabaciones time-lapse, por ejemplo, el comienzo, al final o varios puntos del tiempo, entonces esto puede ajustarse en la película terminada. - Activar la pila de imágenes que debe ser corregido para la deriva. Aplicaciones para abrir la herramienta de alineación | Auto alineación... Check Stack como la fuente de las imágenes y el primer plano/tiempo punto como plano de referencia. Seleccione la pila con el botón de la pila de la fuente y haga clic en aplicar.
Nota: La alineación automática tomará algo de tiempo y poder computacional pero es una buena manera para corregir grandes pilas para la deriva de la configuración del microscopio. Esta alineación automática funciona bien si las microesferas están incluidas pero también podrían funcionar sin ellos. - Guarde la pila alineada.
- Uso de ROIs.
Nota: Microscopia fluorescente de Time-lapse fácilmente crea conjuntos grandes de archivos de datos que ocupan mucho de poder computacional y ralentizan el proceso aguas abajo de estas películas. Por lo tanto recomendamos identificar ROIs y aislar células para trabajar con archivos más pequeños.- Seleccione la herramienta de la Región Rectangular . Crear un ROI alrededor de las células de interés dibujando manualmente un ROI en la imagen de contraste de fase. Asegúrese de que las células de interés son visibles y en foco a través de la película todo Time-lapse.
- Abra la película Time-lapse de la segunda longitud de onda del mismo conjunto de datos. Para transferir el ROI de las imágenes de contraste de fase a la fluorescencia imágenes de la segunda longitud de onda utilizan la herramienta de Transferencia de regiones con regiones | Transferencia de las regiones. Seleccione el conjunto de datos de contraste de fase como Imagen de la fuente y el segundo conjunto de datos de longitud de onda como Imagen de destino. Seleccionar Todas las regiones y pulse OK.
- Repita el paso 4.6.2 para cada longitud de onda adquirida para el mismo conjunto de datos.
- Seleccione el ROI y duplicar como una pila con edición | Duplicar | Pila de... o presione las teclas Shift + Ctrl + D . Guarde la pila duplicada con archivo | Guardar en la misma carpeta que los datos originales.
- Repita el paso 4.6.4 para cada ROI de cada longitud de onda adquirida para el mismo conjunto de datos
- Para generar una película en formato MOV o AVI, abrir la función de Hacer películas mediante pila | Hacer película. Seleccione las grabaciones time-lapse con el botón de la Pila de la fuente . Seleccione el formato de salida, la velocidad de fotogramas, el número de fotogramas y haga clic en Guardar.
Representative Results
M. xanthus es una bacteria de crecimiento lento que se mueve sobre superficies sólidas. Para probar nuestro montaje experimental, se realizó un experimento de Time-lapse con células móviles de DK1622 WT. Se adquirieron imágenes de contraste de fase a intervalos de 5 min durante 24 h (figura 2A, B). La mayoría de células alineadas en grupos. Como se esperaba, las células muestran motilidad y predominante se trasladó en grupos. Además observamos que las células de vez en cuando invertir dirección del movimiento. Estos resultados sugieren que las células de la WT en las condiciones probadas se comportan normalmente en términos de motilidad celular. Sin embargo, incluso cuando las células se registran cada 5 min, la identificación de células individuales es difícil. Por otra parte, porque las células son móviles, muchas células escaparan o entrar en el campo de visión lo que hace difícil seguir las células durante períodos prolongados.
Con el fin de trazar el mismo M. xanthus células durante varias rondas del ciclo celular por proyección de imagen de células vivas, se pueden eliminar para el gen mglA , que es esencial para la motilidad25cepas individuales. Esto evita que las células mover fuera del campo de visión durante el protocolo de imagen. Eliminaciones en-marco se generan según lo descrito por Shi et al. 26
Como era de esperar, en imágenes de células vivas de contraste de fase con las células no-motile ΔmglA (figura 3), las células no se mostraban movimiento activo. Hemos sido capaces de seguir el crecimiento y la división de células durante la formación de microcolony. Basado en las grabaciones time-lapse en el que se adquirieron imágenes a intervalos de 5 min durante 24 h, fue posible cuantificar el tiempo interdivisión (el tiempo entre dos eventos de la división de célula) con una resolución de única célula. Las células del mutante ΔmglA tenían un división entre tiempo de 235 ± 50 min (n = 97 células). Con aproximadamente 4 h, el tiempo interdivisión es similar al tiempo de duplicación en las culturas de la suspensión de células de peso. Esto proporciona evidencia de que M. xanthus las células crecen de forma óptima bajo estas condiciones experimentales.
Para investigar si nuestra configuración permite a las células crecer normalmente proteínas YFP-etiqueta de seguimiento durante largos períodos, se realizó un time-lapse con la proyección de imagen de la fluorescencia M. xanthus las células que expresan una proteína YFP-marcados con etiqueta. Para ello, hemos seguido extrasede YFP como marcador para el origen de replicación (ori). Extrasede es como componente del sistema ParABS M. xanthus y se une a los sitios de parS proximales a la ori; por lo tanto, la segregación de cromosomas y duplicación de origen puede ser seguido19,20,21. Con imagen de adquisición (contraste de fases y fluorescencia, tiempo de la adquisición de 200 ms en canal YFP) cada 20 minutos, las células crecian, dividieron y muestran crecimiento incluso a las 24 h (figura 4A). En el inicio de las grabaciones, extrasede YFP formó dos grupos en las regiones subpolares en la mayoría de las células (figura 4A). Poco antes o después de la división celular, el subpolar extrasede-YFP se agrupan en el Viejo Polo celular duplicado. Uno de los dos grupos permanecieron en el antiguo polo de célula mientras que la segunda copia trasladada al nuevo polo celular, alcanzando su posición final subpolar después de aproximadamente 40-60 min (figura 4A, B). Estas observaciones están de acuerdo con los datos anteriores generados a partir de grabaciones time-lapse corto usando agar delgada almohadillas19. Concluimos que este montaje experimental permite que Time-lapse de microscopía de fluorescencia rastrear la segregación cromosómica durante varios ciclos de la célula en crecimiento lento M. xanthus células, sin perturbación el crecimiento celular o la maquinaria de segregación del cromosoma.
En un experimento similar, intentamos seguir marcadores para la división celular por microscopia de fluorescencia Time-lapse. Similar a casi todas las bacterias, M. xanthus requiere FtsZ, una GTPasa como tubulina bacteriana, para la división celular16,17,18. FtsZ forma a anillo-como la estructura en midcell, el supuesto anillo de Z, que ayuda a reclutar todas otras proteínas necesarias para la división celular27,28. En M. xanthus, la formación de los anillos Z y su posicionamiento en midcell es estimulada por la PomXYZ tres proteínas16,17. Estas tres proteínas forman un complejo asociado a cromosoma que transfiere en el nucleoide del sitio de la división celular en la célula "madre" a la mitad del nucleoide en las dos células hijas. El centro del nucleoid coincide con midcell, antes de la segregación cromosómica y aquí los reclutas complejo PomXYZ FtsZ y estimula la formación del anillo Z.
Aquí, seguimos en primer lugar las células no-motile con ftsZ-gfp. Porque FtsZ-GFP general muestra una fluorescencia débil señal que extrasede-YFP, aumentamos el tiempo de exposición 5 veces a 1 s en el canal GFP. Como era de esperar, fuerte acumulación de FtsZ-GFP se observó sólo en midcell y esta localización dicta la posición de la constricción de la división celular (figura 5A). FtsZ-GFP predominante había formado un cluster en midcell en la celda más larga. También fue evidente que este grupo aumentó en intensidad con el tiempo. Después de la división celular, observamos que FtsZ-GFP a acumulado en midcell en la hija de dos células de aproximadamente 2 h más tarde (figura 5B). Esto es consistente con el hallazgo de que aproximadamente el 50% de las células en una población de mostrar la localización de FtsZ en midcell basado en análisis snap-shot16,17.
En un segundo experimento, seguimos Δ no-motilemglA las células durante 24 h que expresan mCherry alcohólico. Como parte del sistema PomXYZ, alcohólico ayuda a guiar la formación del anillo Z y posicionamiento, así estimulando la división celular en el midcell16. La señal de fluorescencia de mCherry alcohólico es fuerte y permite un tiempo de exposición en el canal de la fluorescencia de la Sra. 250 importante, todas las células crecieron en tamaño y muestran un evento de división celular en el transcurso del experimento, formando microcolonias después de 24 h ( Figura 6A). Como previamente divulgados16, casi todas las células contienen un clúster mCherry alcohólico. La mayoría de ellos localizados en el midcell y los racimos de midcell trasladadas a midcell durante el curso del experimento. Durante las divisiones celulares, mCherry alcohólico racimos se dividieron, con cada célula hija recibe un clúster. A diferencia de FtsZ-GFP, mCherry alcohólico había localizada en midcell 80-90% del ciclo celular y llegó a este cargo poco después de la división celular (figura 6B).
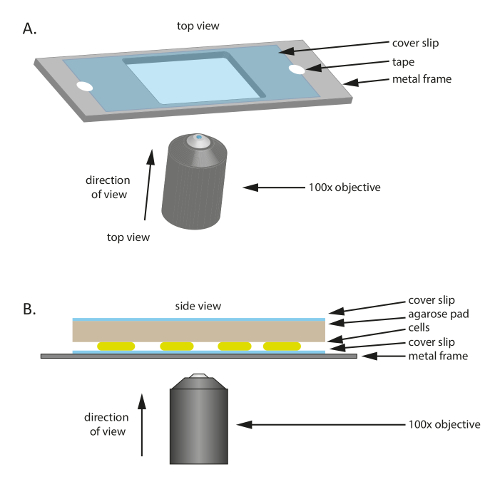
Figura 1 : Esquema de la instalación experimental utilizada en este estudio. (A) A metal o plástico marco sirve como un soporte para la muestra. Un cubreobjetos se fija a la estructura metálica con cinta para reducir el movimiento de la muestra. Ve el lado (B) de la configuración de la muestra experimental. Las células se montan en el cubreobjetos que se muestra en (A). Se coloca la almohadilla de agarosa que suministra nutrientes y humedad a las células sobre las células. La almohadilla de agarosa está cubierta por un cubreobjetos adicional para reducir la evaporación. Para imágenes de alta calidad, se utiliza un 100 X aceite fase contraste objetivo de inmersión. Haga clic aquí para ver una versión más grande de esta figura.

Figura 2 : Contraste de fase microscopio Time-lapse de WT M. xanthus células. Las células fueron seguidas durante 24 h y se adquirieron imágenes cada 5 minutos (A) se muestran imágenes representativas del mismo campo de vista cada 5 min. Flechas color indican la direccionalidad del movimiento de las células individuales. Del mismo color marca la misma celda con el tiempo. Números indican el tiempo en minutos. Barra de escala: 5 μm. (B) las imágenes del mismo campo de visión después de cada hora se muestran. Observe que se muestra el mismo campo de visión, sino porque las células son móviles, las células están constantemente que entran y salen del campo de visión. Números indican el tiempo en horas. Barra de escala: 5 μm. PH: contraste de fase. Haga clic aquí para ver una versión más grande de esta figura.

Figura 3 : Contraste de fase microscopio Time-lapse de no-motile M. xanthus células. Células ΔmglA siguieron 24 h. imágenes fueron adquiridas cada 5 min y se muestran imágenes representativas después de cada hora. División celular las constricciones están marcados con las flechas naranja. Números indican el tiempo en horas. PH: contraste de fases. Haga clic aquí para ver una versión más grande de esta figura.
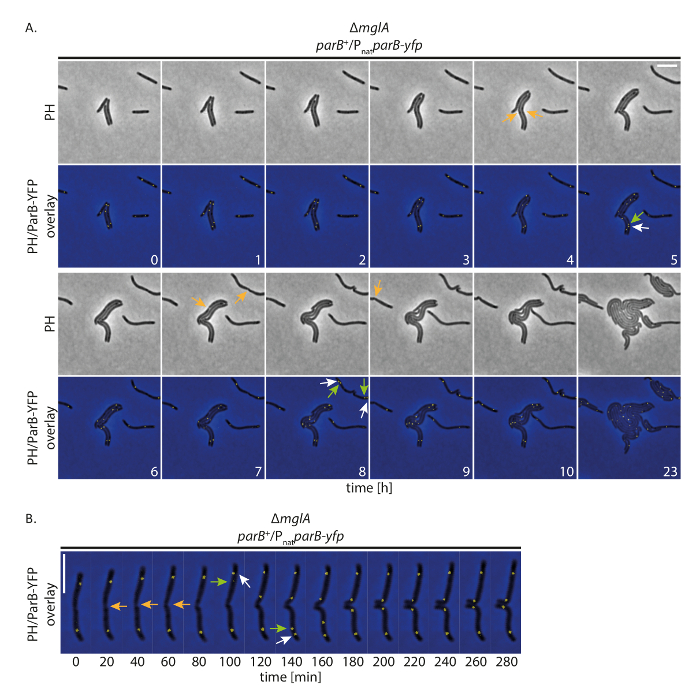
Figura 4 : Time-lapse microscopía de fluorescencia de extrasede-YFP en no-motile M. xanthus células. Células de un ΔmglA mutanteexpresando extrasede-yfp en presencia nativa extrasede (SA4749; ΔmglA; extrasede +/Pnatextrasede-yfp) fueron seguidos durante 24 h por fase contraste y fluorescencia microscopía. (A) imágenes se adquirieron cada 20 min y representante imágenes cada hora hasta las 10 h se muestran, junto con las mismas células después de 24 h. imágenes aparecen en contraste de fases (PH) y como recubrimiento de contraste de fases y la YFP de señal. Las divisiones de la célula están marcadas con flechas naranja. Blancas y verdes las flechas indican eventos de duplicación de YFP extrasede racimo, con las flechas verdes marca el clúster translocan. Números indican el tiempo en horas. Barra de escala: 5 μm. (B) las imágenes fueron adquiridas como en (A) pero se muestran a mayor resolución temporal. Números indican el tiempo en minutos. Las flechas son como en (A). Barra de escala: 5 μm. haga clic aquí para ver una versión más grande de esta figura.

Figura 5 : Time-lapse microscopía de fluorescencia de GFP de FtsZ en no-motile M. xanthus células. Células de un mutante demglA Δ expresando gfp de ftsZ en la presencia de nativos ftsZ (SA8241; ΔmglA; ftsZ +/PnatftsZ-gfp) fueron seguidos durante 24 h por microscopia de contraste y de la fluorescencia de fase. (A) imágenes se adquirieron cada 20 min y se muestran imágenes representativas cada hora hasta las 10 h, junto con las mismas células después de 24 h. se muestran imágenes en contraste de fases (PH) y como recubrimiento de GFP señal y contraste de fases. Las divisiones de la célula están marcadas con flechas naranja. Flechas blancas indican grupos de FtsZ-GFP en midcell. Números indican el tiempo en horas. Barra de escala: 5 μm. (B) las imágenes fueron adquiridas como en (A) pero se muestran a mayor resolución temporal. Números indican el tiempo en minutos. Las flechas verdes y blancas marcan racimos de FtsZ-GFP en las células de izquierda y derecha, respectivamente. Las flechas naranjas indican las divisiones de célula. Barra de escala: 5 μm. haga clic aquí para ver una versión más grande de esta figura.

Figura 6 : Time-lapse de microscopía de fluorescencia de mCherry alcohólico en no-motile M. xanthus células. Las células dealcohólico no-motile Δ acumulando mCherry-alcohólico (SA4797; ΔmglAΔalcohólico/PpomZ mCherry-alcohólico) fueron seguidas durante 24 h por microscopia de contraste y de la fluorescencia de fase cada 20 minutos (A) representante imágenes cada hora hasta las 10 h se muestran, junto con las mismas células después de 24 h. se muestran imágenes de contraste de fases (PH) y como recubrimiento de contraste de fases y mCherry señal. Las divisiones de la célula están marcadas con flechas naranja. Blancas y verdes las flechas indican mCherry alcohólico grupos antes y después de dividir eventos, respectivamente. Números indican el tiempo en horas. Barra de escala: 5 μm. (B) las imágenes fueron adquiridas como en (A) y se muestran a mayor resolución temporal. Las flechas son como en (A). Barra de escala: 5 μm. haga clic aquí para ver una versión más grande de esta figura.
| Cepa bacteriana | Correspondiente genotipo1 | Referencia |
| DK1622 | Tipo salvaje | 23 |
| SA4420 | ΔmglA | 24 |
| SA4749 | ΔmglA; extrasede+/attB:: Pnatextrasede yfp (pAH7) | Este estudio |
| SA4797 | ΔmglA; Δalcohólico / attB::PpomZ mCherry-alcohólico (pAH53) | 16 |
| SA8241 | ΔmglA; ftsZ+/ mxan18-19::PnatftsZ-gfp (pDS150) | Este estudio |
| Plásmidos en los soportes contienen fusiones del gen indicado y fueron intergated en los sitios indicados en el genoma. Plásmidos integran en el sitio de attB o la región intergénica de mxan18-19 se expresa de su promotor nativo P (nat) o el promotor nativo de pomZ (PpomZ). |
||
Tabla 1: Lista de las cepas bacterianas utilizadas en este estudio.
| Plásmidos | Características relevantes | Referencia |
| pAH7 | Pnatyfp extrasede; AttP Mx8; TTER | 19 |
| pAH53 | PpomZ mCherry alcohólico; AttP Mx8 ; KmR | 16 |
| pDS150 1 | PnatftsZ-gfp ; mxan18-19 ; TTER | Este estudio |
| pMR3691 | Plásmido de expresión génica inducible vanillate | 18 |
| pKA51 | PnatftsZ-gfp ; AttP Mx8; TTER | 17 |
| 1 pDS150: pDS150 es un derivado de la pKA51 en que fue reemplazado el sitio attP Mx8 con la región intergénica de mxan18-19 . Para esto fue amplificada la región intergénica de mxan18-19 de pMR3691 con las cartillas Mxan18 19 fwd BsdRI (GCGATCATTGCGCGCCAGACGATAACAGGC) y Mxan18-19 rev BlpI (GCGGCTGAGCCCGCGCCGACAACCGCAACC) y reproducido en pKA51. |
||
Tabla 2: Lista de los plásmidos utilizados en este estudio.
Discussion
Imágenes de células vivas de fluorescencia se ha convertido en una poderosa herramienta para estudiar la dinámica espacio-temporal de las células bacterianas. Microscopía de fluorescencia Time-lapse de bacterias móviles y lento como M. xanthus, sin embargo, ha sido difícil y sólo se realizó durante períodos de tiempo corto. Aquí, presentamos un método fácil de utilizar y robusto para la proyección de imagen de células vivas de la M. xanthus por microscopía de fluorescencia Time-lapse. Este método permite al usuario seguir las células y proteínas fluorescencia marcadas por varias rondas del ciclo celular con una resolución de única célula.
Hay varios requisitos previos que influyen en el éxito de la proyección de imagen de células vivas de crecimiento lento M. xanthus células incluyendo: 1) una superficie sólida para la fijación de la célula; 2) la disponibilidad de nutrientes y oxígeno; 3) constante de humedad y temperatura; y 4) la optimización de las condiciones experimentales tales como frecuencia de adquisición de imagen y tiempo de exposición.
En nuestro montaje experimental, usamos agarosa gruesas almohadillas complementadas con nutrientes. Usando teclas de agarosa grueso en comparación con dispositivos microfluídicos para seguir las células tiene algunos beneficios fundamentales pero también algunos inconvenientes. En primer lugar, la almohadilla de agarosa no sólo proporciona una superficie de M. xanthus celular accesorio y movimiento pero también suficientes nutrientes para el crecimiento durante al menos 24 h. En segundo lugar, encaje el tiros análisis comúnmente utilizados para estudiar la localización intracelular de proteínas fluorescencia marcadas anteriormente se realizó en el mismo tipo de agarosa cojines16,17,29. Por lo tanto, datos de análisis tiro rápido pueden ser directamente comparados con datos obtenidos con el método descrito aquí. En tercer lugar, agarosa puede ser fácilmente modificado y suplementado con antibióticos u otros suplementos como el CuSO4 y vanillate que se utilizan comúnmente para gene expresión inducción18,30. Finalmente, porque las células se les permite forma microcolonias en el transcurso de un experimento, esta configuración también permite estudiar el efecto de las interacciones célula directa sobre el parámetro en particular analizado. Este aspecto es particularmente importante en el caso de M. xanthus ya que esta bacteria muestra varias de las interacciones dependientes de contacto. El principal inconveniente de este método es que las condiciones experimentales son prefijadas por la duración de un experimento. Por el contrario, dispositivos microfluídicos generalmente permiten cambiar las condiciones experimentales durante el curso de un experimento añadiendo por ejemplo antibióticos31.
Paquetes de software libre (p. ej., MicrobeJ, Oufti) están disponibles para analizar automáticamente el crecimiento de las células y la localización de proteínas dentro de células individuales. Sin embargo, estos programas sólo son adecuadas para el análisis de células individuales o pequeños grupos de células. Por lo tanto, sigue siendo un desafío para analizar automáticamente los datos generados por las grabaciones de 24 h que se describe aquí.
En Resumen, hemos descrito un protocolo reproducible y fácil de usar para realizar la proyección de imagen de células vivas con crecimiento lento M. xanthus bacterias. Demostramos que la agarosa suplido de nutrientes simples pastillas son suficientes para sostener el crecimiento durante al menos 24 h y permiten observar y analizar la localización de la proteína y el crecimiento con una resolución de única célula durante varias generaciones.
Disclosures
Los autores declaran que no tienen intereses financieros que compiten.
Acknowledgments
Este trabajo fue financiado por el Consejo de investigación alemán (DFG) en el marco de la Transregio 174 "espaciotemporal dinámica de las células bacterianas" y la sociedad Max Planck.
Materials
| Name | Company | Catalog Number | Comments |
| DMI6000B with AFC | Leica microsystems | 11888945 | Automated inverted widefield fluorescence microscope with adaptive focus control |
| Universal mounting frame | Leica microsystems | 11532338 | Stage holder for different sample sizes |
| HCX PL FLUOTAR 100x/1.30 oil PH3 | Leica microsystems | 11506197 | Phase contrast objective |
| Orca Flash 4.0 camera | Hamamatsu | 11532952 | 4.0 megapixel sCMOS camera for picture aquisition |
| Filter set TXR ET, k | Leica microsystems | 11504170 | Fluorescence filter set, Ex: 560/40 Em: 645/75 |
| Filter set L5 ET, k | Leica microsystems | 11504166 | Fluorescence filter set, Ex: 480/40 Em: 527/30 |
| Filter set YFP ET, k | Leica microsystems | 11504165 | Fluorescence filter set, Ex: 500/20 Em: 535/30 |
| ProScan III | Prior | H117N1, V31XYZEF, PS3J100 | Microscope automation controller with interactive control center |
| EL 6000 light source | Leica microsystems | 11504115 | External fluorescence light source |
| Incubator BLX Black | Pecon | 11532830 | Black incubation chamber surrounding the microscope |
| Tempcontrol 37-2 digital | Leica microsystems | 11521719 | Automated temperature control for incubation chamber |
| Gentmycin sulphate | Carl Roth | 0233.4 | Gentamycin |
| Oxytetracylin dihydrate | Sigma Aldrich | 201-212-8 | Oxytetracyclin |
| Kanamycin sulphate | Carl Roth | T832.3 | Kanamycin |
| Filtropur BT25 0.2 bottle top filter | Sarstedt | 831,822,101 | Bottle top filter for sterilization of buffers |
| Deckgläser | VWR | 630-1592 | Glass cover slip (60 x 22 mm, thickness: 0.7 mm) |
| Seakem LE agarose | Lonza | 50004 | Agarose for microscopy slides |
| Leica Metamorph AF | Leica microsystems | 11640901 | Microscope control software and software for picture analysis |
| Tetraspeck Microsperes, 0.5 µm | ThermoFisher | T7281 | Fluorescent microspheres |
| petri dish | Greiner Bio-one | 688102 | 120 mm x 120 mm x 17 mm squared petri dish for agarose pads |
| BD Bacto Casitone | Becton Dickinson | 225930 | Casitone |
| Parafilm M | VWR | 291-1213 | Parafilm |
| Tris(hydroxymethyl)-aminomethane | Carl Roth | AE15.2 | Tris |
| Magnesium sulphate heptahydrate | Carl Roth | P027.2 | Magnesium sulphate |
| Potassium dihydrogen phosphate p.a. | Carl Roth | 3904.1 | Potassium dihydrogen phosphate |
| 1% CTT medium: 1 % (w/v) BD Bacto™ casitone, 10 mM Tris-HCl ph 8.0, 1 mM potassium phosphate buffer pH 7.6, 8 mM MgSO4 | Cultivation medium for M.xanthus | ||
| TPM buffer: 10 mM Tris-HCl ph 8.0, 1 mM potassium phosphate buffer pH 7.6, 8 mM MgSO4 | Buffer for preparation of microscopy slides for M.xanthus |
References
- Shapiro, L., McAdams, H. H., Losick, R. Why and how bacteria localize proteins. Science. 326 (5957), 1225-1228 (2009).
- Treuner-Lange, A., Søgaard-Andersen, L. Regulation of cell polarity in bacteria. J Cell Biol. 206 (1), 7-17 (2014).
- Laloux, G., Jacobs-Wagner, C. Spatiotemporal control of PopZ localization through cell cycle-coupled multimerization. J Cell Biol. 201, 827-841 (2013).
- Rudner, D. Z., Losick, R.
- Badrinarayanan, A., Le, T. B. K., Laub, M. T. Bacterial chromosome organization and segregation. Annu Rev Cell Dev Biol. 31 (1), 171-199 (2015).
- Munoz-Dorado, J., Marcos-Torres, F. J., Garcia-Bravo, E., Moraleda-Munoz, A., Perez, J. Myxobacteria: Moving, Killing, Feeding, and Surviving Together. Front Microbiol. 7, 781 (2016).
- Berleman, J. E., Kirby, J. R. Deciphering the hunting strategy of a bacterial wolfpack. FEMS Microbiol Rev. 33 (5), 942-957 (2009).
- Konovalova, A., Petters, T., Søgaard-Andersen, L.
- Nudleman, E., Wall, D., Kaiser, D. Cell-to-cell transfer of bacterial outer membrane lipoproteins. Science. 309, 125-127 (2005).
- Vassallo, C. N., et al. Infectious polymorphic toxins delivered by outer membrane exchange discriminate kin in myxobacteria. eLife. 6, 29397 (2017).
- Vassallo, C., et al. Cell rejuvenation and social behaviors promoted by LPS exchange in myxobacteria. Proc Natl Acad Sci USA. 112 (22), 2939-2946 (2015).
- Li, Y., et al. Extracellular polysaccharides mediate pilus retraction during social motility of Myxococcus xanthus. Proc. Natl. Acad. Sci. USA. 100, 5443-5448 (2003).
- Kim, S. K., Kaiser, D. Cell alignment required in differentiation of Myxococcus xanthus. Science. 249, 926-928 (1990).
- Lobedanz, S., Søgaard-Andersen, L. Identification of the C-signal, a contact dependent morphogen coordinating multiple developmental responses in Myxococcus xanthus. Genes Dev. 17, 2151-2161 (2003).
- Schumacher, D., Søgaard-Andersen, L. Regulation of cell polarity in motility and cell division in Myxococcus xanthus. Annu Rev Microbiol. 71 (1), 61-78 (2017).
- Schumacher, D., et al. The PomXYZ proteins self-organize on the bacterial nucleoid to stimulate cell division. Dev Cell. 41 (3), 299-314 (2017).
- Treuner-Lange, A., et al. PomZ, a ParA-like protein, regulates Z-ring formation and cell division in Myxococcus xanthus. Mol Microbiol. 87 (2), 235-253 (2013).
- Iniesta, A. A., Garcia-Heras, F., Abellon-Ruiz, J., Gallego-Garcia, A., Elias-Arnanz, M. Two systems for conditional gene expression in Myxococcus xanthus inducible by isopropyl-beta-D-thiogalactopyranoside or vanillate. J Bacteriol. 194 (21), 5875-5885 (2012).
- Harms, A., Treuner-Lange, A., Schumacher, D., Søgaard-Andersen, L. Tracking of chromosome and replisome dynamics in Myxococcus xanthus. reveals a novel chromosome arrangement. PLoS Genet. 9 (9), 1003802 (2013).
- Iniesta, A. A. ParABS system in chromosome partitioning in the bacterium Myxococcus xanthus. PLoS One. 9 (1), 86897 (2014).
- Lin, L., Osorio Valeriano, M., Harms, A., Søgaard-Andersen, L., Thanbichler, M. Bactofilin-mediated organization of the ParABS chromosome segregation system in Myxococcus xanthus. Nat Commun. 8 (1), 1817 (2017).
- Hodgkin, J., Kaiser, D. Cell-to-cell stimulation of movement in nonmotile mutants of Myxococcus. Proc Natl Acad Sci U S A. 74 (7), 2938-2942 (1977).
- Kaiser, D. Social gliding is correlated with the presence of pili in Myxococcus xanthus. Proc Natl Acad Sci USA. 76 (11), 5952-5956 (1979).
- Miertzschke, M., et al. Structural analysis of the Ras-like G protein MglA and its cognate GAP MglB and implications for bacterial polarity. EMBO J. 30 (20), 4185-4197 (2011).
- Hodgkin, J., Kaiser, D. Genetics of gliding motility in Myxococcus xanthus. (Myxobacterales): Two gene systems control movement. Mol Gen Genet. 171, 177-191 (1979).
- Shi, X., et al. Bioinformatics and experimental analysis of proteins of two-component systems in Myxococcus xanthus. J Bacteriol. 190 (2), 613-624 (2008).
- Bi, E. F., Lutkenhaus, J. FtsZ ring structure associated with division in Escherichia coli. Nature. 354 (6349), 161-164 (1991).
- Lutkenhaus, J., Pichoff, S., Du, S. Bacterial cytokinesis: From Z ring to divisome. Cytoskeleton. 69 (10), 778-790 (2012).
- McLoon, A. L., et al. MglC, a Paralog of Myxococcus xanthus GTPase-Activating Protein MglB, Plays a Divergent Role in Motility Regulation. J Bacteriol. 198 (3), 510-520 (2015).
- Gomez-Santos, N., et al. Comprehensive set of integrative plasmid vectors for copper-inducible gene expression in Myxococcus xanthus. Appl Environ Microbiol. 78 (8), 2515-2521 (2012).
- Treuner-Lange, A., et al. The small G-protein MglA connects to the MreB actin cytoskeleton at bacterial focal adhesions. J Cell Biol. 210 (2), 243-256 (2015).
