

ERRATUM NOTICE
Important: There has been an erratum issued for this article. Read more …
Summary
ここでは、マウス小脳の顆粒ニューロンがシナプス構造を精緻化し、シナプスを形成して脳回路全体に集積する、生後脳の発達過程におけるシナプス形成を可視化する方法について述べる。
Abstract
神経細胞は、脳の発達中にその構造と機能が動的に変化し、他の細胞と適切な接続を形成します。げっ歯類の小脳は、単一の細胞型である小脳顆粒ニューロン(CGN)の発生と形態形成を経時的に追跡するための理想的なシステムです。ここでは、発達中のマウス小脳における顆粒ニューロン前駆細胞の in vivo エレクトロポレーションを使用して、その後の形態学的分析のために細胞をまばらに標識しました。この技術の有効性は、これらの細胞がシナプス入力の大部分を受け取る特殊な構造である樹状突起爪の形成に特に焦点を当てて、CGN成熟の主要な発達段階を示す能力で実証されています。この技術は、小脳の発達を通してCGNシナプス構造のスナップショットを提供することに加えて、細胞自律的な方法で顆粒ニューロンを遺伝的に操作して、関心のある遺伝子の役割とCGN形態、爪の発達、およびシナプス形成への影響を研究することができます。
Introduction
脳の発達は、胚発生から出生後の生活に及ぶ長期にわたるプロセスです。この間、脳は内因性刺激と外因性刺激の組み合わせを統合し、樹状突起と軸索の間のシナプスの配線を彫刻して最終的に行動を導きます。げっ歯類の小脳は、前駆細胞から成熟ニューロンに移行する際に、単一のニューロンタイプである小脳顆粒ニューロン(CGN)の発達を追跡できるため、シナプスがどのように発達するかを研究するための理想的なモデルシステムです。これは、小脳皮質の大部分が出生後に発達し、出生後の遺伝子操作と細胞標識が容易になるという事実に一部起因しています1。
哺乳類では、CGN分化は、後脳の増殖細胞のサブセットが菱形の唇の上を移動して小脳の表面に二次胚帯を形成する胚発生の終わりに始まります2,3,4。それらは顆粒ニューロン前駆細胞(GNP)の同一性に完全に関与していますが、これらの細胞は生後14日目(P14)まで外部顆粒層(EGL)の外側部分内で増殖し続けます。この層の増殖は、これらの細胞がもっぱらCGNを生じさせるため、小脳の大規模な拡張をもたらします5。生まれたばかりのCGNは、EGLの細胞周期を出ると、内顆粒層(IGL)に向かって内側に移動し、小脳の分子層を分岐して移動する軸索を残し、プルキンエ細胞にシナプスする平行繊維を形成します6。分子層内のこれらの繊維の位置は、細胞周期の出口のタイミングに依存する。
最初に分化したCGNは、分子層の下部に向かって平行な繊維を残しますが、後で分化するCGNの軸索は上部7,8にクラスター化されます。CGN細胞体がIGLに到達すると、樹状突起を精緻化し、近くの抑制性ニューロンおよび興奮性ニューロンとシナプスを形成し始めます。CGNの成熟した樹状木は、4つの主要なプロセスを持つステレオタイプアーキテクチャを示します。CGNの成熟の過程で、これらの樹状突起の末端の構造は、シナプス後タンパク質で濃縮される爪を形成します9,10。樹状突起爪と呼ばれるこれらの特殊な構造は、顆粒ニューロン上のシナプスの大部分を含み、橋に由来する苔状線維神経支配からの興奮性入力と、局所ゴルジ細胞からの抑制性入力の両方を受け取るために重要です。完全に構成されると、CGNのシナプス接続により、これらの細胞は小脳前核からの入力をプルキンエ細胞に中継し、プルキンエ細胞は小脳皮質から深部小脳核に突出します。
GNPのin vivo出生後エレクトロポレーションは、所望の構築物の発現が速いタイムラインで達成でき、この方法は細胞の小さな集団を標的とするため、ウイルス感染やトランスジェニックマウス株の生成などの他の標識ベースの方法よりも有利であり、細胞自律効果の研究に有用である。この方法は、CGNの形態学的発達を研究するための以前の研究で使用されています。ただし、これらの研究は、単一の時点または短い時間枠のいずれかに焦点を当てています9、10、11、12、13。この標識法を画像解析と組み合わせて、生後生後3週間のCGN分化の全時間経過にわたって発生するCGN形態の変化を文書化しました。これらのデータは、小脳回路の構築の根底にあるCGN樹状突起発生のダイナミクスを明らかにしています。
Subscription Required. Please recommend JoVE to your librarian.
Protocol
注:すべての手順は、デューク大学施設動物管理および使用委員会(IACUC)によって承認されたプロトコルに基づいて実行されました。
1.インビボエレクトロポレーションまたはIVEのためのDNA調製(手術の1日前)
- 次の材料を収集します:精製DNA(動物あたり0.5〜25 μg)、3 M酢酸ナトリウム、エタノール、ファストグリーン染料、超高純度蒸留水、リン酸緩衝液(PBS)(材料の表を参照)。
注:DNAについては、ヒトユビキチンプロモーター下で緑色蛍光タンパク質(GFP)を発現する構築物をAddgene(FUGW、https://www.addgene.org/14883/)から取得しました。ユビキタスプロモーターの制御下でGFPまたは他の蛍光タンパク質を発現する任意の構築物が機能するはずである。この技術によるCGN特異的標識は、構築物に依存するのではなく、エレクトロポレーションに依存する。 - 所望の量のDNA、10容量%の3 M酢酸ナトリウムおよび250容量%の100%氷冷エタノールを混合することにより、エレクトロポレーション用のDNAを調製する。DNAはすぐに溶液から沈殿することに注意してください。
- DNA混合物を-20°Cで一晩沈殿させるか、-80°Cで1時間沈殿させ続けます。
- ペレットを卓上遠心分離機で>16,000 × g で沈殿させ、70%エタノールで2回洗浄します。
- DNAペレットを完全に乾燥させ、1x PBS + 0.02%ファストグリーン溶液で再構成します。

図1:スペーサーを使用して射出深さを1.5 mmに制限します。 (A)11.2 mmのセグメントをかみそりの刃を使用してローディングピペットから切り取ります。(B)スペーサーは、ハミルトンシリンジの先端(全長1.27cmまたは0.5インチ)に取り付けられ、接着剤またはパラフィルムのいずれかで固定されます。露出した先端の長さは1.5 mmである必要があります。この図の拡大版を表示するには、ここをクリックしてください。
2. 生後7日齢マウスにおける顆粒ニューロン前駆細胞の in vivo エレクトロポレーション
注:すべてのエレクトロポレーション手術は、無菌で換気の厳しい手術室で行われ、すべての職員は手袋、フェイスマスク、ヘアボンネット、ガウン、靴カバーなどの完全な個人用保護具を着用しました。あるいは、手術は換気された無菌フードで行うことができます。
- 次の資料を収集します:エレクトロポレーション用のDNA、小型外科用ハサミ、小型手術用ピンセット、カスタマイズされたハミルトンシリンジ、コットンチップアプリケーター、加熱パッド、ベタジン、70%エタノール、1x PBS、パラフィルム、組織接着剤(n-ブチルエステルシアノアクリレート)、イソフルラン、エレクトロポレーター、およびピンセットタイプの電極( 材料表を参照)。
- 滅菌されたローディングチップからスペーサーを切り取り、ハミルトンシリンジにフィットし、注入深さを1.5 mmに制限します(図1A、B)。スペーサーを接着剤またはパラフィルムで固定します。
- P7子犬をイソフルランチャンバーで0.8 L /分の送達速度で麻酔します。.呼吸の減少とつま先または尾のつまみ反応の欠如について動物を監視することにより、完全な麻酔を確認します(図2A)。
- 動物が完全に麻酔されたら、ノーズコーンを取り付けた台座に子犬を置き、0.8 L / minの送達速度で一定の4%イソフルランを送達します。ベタジンの滅菌綿棒で子犬の頭のてっぺんを3回洗浄し、次に70%エタノールを交互に使用して、サイトを準備します。続行する前に、溶液を乾燥させてください。
- 滅菌したハサミを使用して、耳の上部から付け根までの距離にまたがる1つのカットで小さな切開を行い、後脳を明らかにします(図2B)。
- 小脳の位置を特定し(図2C)、ハミルトンシリンジの露出した先端を頭蓋骨から脳に垂直に挿入し、シリンジのバックプランジャーをゆっくりと押して1.5μLのDNA混合物を小脳実質に注入します。DNA混合物の送達後、背中のこぼれを防ぐために針をゆっくりと引き戻し、DNA溶液を30秒間拡散させます。
- イソフルランをオフにし、子犬を37°Cの加熱パッドに置きます。両端を滅菌済みの1x PBSに浸して、エレクトロポレーション用のピンセット型電極を準備します。
注意: ピンセットタイプの電極を濡らすと、電気パルスの投与中に子犬の皮膚に接触する火傷を防ぐことができます。 - ピンセット電極を注射部位の上に向け、プラス端を下向きにし、マイナス端を動物の頭の上に向けます(図2D)。エレクトロポレーターから5つの電気パルスを、50 ms、130 V、および950 msのパルス間隔の設定で管理します。
注意: 必要に応じて、テスト注射を実行して、注射部位が小脳朱茎にあることを確認します(図2E)。 - 切開部をつまんで閉じ、無毒のn-ブチルエステルシアノアクリレート組織接着剤で創傷を密封します。微量の血液があると親の子殺しや共食いの可能性が高まるため、70%エタノールで傷口をきれいにします。
- 子犬をダムに戻す前に、動物が37°Cの加熱パッドで回復するのを待ちます。手術後少なくとも30時間、2分ごとに子犬を監視して、完全に回復するようにします。
注:どちらかの親による子殺しは非常に一般的です。共食いを防ぐために、エレクトロポレーションを開始する前に種馬を別のケージに収容し、常に洗浄および回収された子犬(つまり、血痕がなく、 完全に可動性)を元の寝具の元のケージに戻します。子犬は、血の臭いを最小限に抑えるために、元のケージからの糞で拭くこともできます。元のダムが彼女の子犬を共食いし続ける場合、代理ダムの使用が必要になるかもしれません。

図2:P7野生型マウス子犬における顆粒ニューロン前駆細胞のin vivo小脳エレクトロポレーション。 (A)子犬は、DNA溶液の注入を通して麻酔を確実にするために、0.8L / minの速度で送達される4%イソフルランで麻酔されます。イソフルランは0.8 L/minの速度で送達されます。 (B)マウスをベタジンと70%エタノールで3回滅菌した後、耳の距離にまたがる切開を行い、後脳を明らかにします。(C)注射部位のランドマークである頭蓋骨の白い境界の拡大画像。DNAコンストラクトは、マークから1 mm以内に注入する必要があります。点線は境界の輪郭を描き、黒い矢印は注射部位を示します。小脳虫の隆起が見える場合があり、注射部位を見つけるのに役立ちます。(D)効率的なエレクトロポレーションのためのピンセット型電極配向。プラス(+)端は、電気パルスを投与する前に、負に帯電したDNAを小脳実質に引き込むために下向きにする必要があります。(E)1μLの0.02%ファストグリーン染料の試験注射は、注射が小葉5〜7の間の小脳虫の中央に局在することを示しています。この図の拡大版を表示するには、ここをクリックしてください。
3. エレクトロポレーションCGNの免疫組織化学
- 次の材料を収集します:イソフルラン、1x PBS、4%パラホルムアルデヒド(PFA)、30%スクロース、通常のヤギ血清、非イオン性洗剤、スライドガラス、ガラスカバーガラス、マニキュア、封入剤、ヘキスト核色素、および適切な一次抗体および二次抗体(材料の表を参照)。
- 実験動物をイソフルランで麻酔し、つま先と尾のつまみで完全麻酔を確認します。
- 動物の心臓の左心室に1x PBSと4%PFAをゆっくりと注射することにより、経心灌流を実行します。大静脈を切断することによって動物から血液を排出させます。
- 脳を4°Cで4%PFAに浸して一晩固定します。 翌日、1x PBSで脳をすばやくすすぎ、少なくとも24時間凍結保護のために、脳を1x PBSの30%スクロースに移します。
- 必要に応じて、脳を吻側尾側軸に沿って半分にスライスし、直立蛍光解剖顕微鏡を使用してトランスフェクトされたレポーター構築物の発現を確認します。
注意: 脳が乾かないように、小さな皿に1x PBSに浸したままにします。 - 凍結するミクロトームに脳を取り付け、25 μm矢状切片をスライスし、1x PBSとグリセロールの1:1混合物で切片を展開させます。
注:切片は、長期保存のために-20°Cでこの凍結保護剤溶液に保存することができます。 - 切片を1x PBSでそれぞれ10分間3回洗浄して凍結保護剤を除去し、1x PBS + 10%正常ヤギ血清+ 0.2%非イオン性界面活性剤で組織をオービタルシェーカーで室温で1時間ブロックします。
- 一次抗体溶液:1x PBS、10%正常ヤギ血清、0.2%非イオン性界面活性剤、および抗GFP抗体を調製し、溶液を>16,000 × gで5分間遠心分離します。抗体溶液中の切片をオービタルシェーカーで4°Cで48時間インキュベートします。
- 一次抗体溶液を1x PBS + 0.2%非イオン性界面活性剤で15分間5回洗い流します。
- 二次抗体溶液を調製する:1x PBS、10%正常ヤギ血清、0.2%非イオン界面活性剤、およびGFPを検出するための適切な二次抗体。溶液を>16,000 × gで遠心分離します。抗体溶液中の切片をオービタルシェーカー上で室温で2〜3時間インキュベートします。漂白を防ぐために、セクションを光への露出から保護してください。
- 二次抗体溶液を1x PBS + 0.2%非イオン性界面活性剤で15分間3回洗い流します。切片を1x PBS + Hoechstで5分間インキュベートして、核を染色します。
- 1x PBS + 0.2%非イオン性洗剤でヘキスト溶液を洗い流し、スライドガラスにマウントします。セクションを封入剤で覆い、スライドを覆い、蒸発を防ぐためにマニキュアでスライドを密封します。
4. CGNの形態素解析 - 3次元(3D)再構成と表面積・細胞体積
- 共焦点顕微鏡で63倍の対物レンズで2倍ズームで単一のエレクトロポレーションCGNを画像化し、スタックあたり0.5μmのzスタック画像を撮影します。画像ウィンドウごとに1つのセルを画像化して、画像分析と再構成を容易にします。
- 次のリンク(https://imagej.net/Simple_Neurite_Tracer:_Basic_Instructions)を使用してFIJI用のSimple Neurite Tracerプラグインをインストールし、3次元(3D)空間でエレクトロポレーションされたCGNの構造を簡単かつ効率的にトレースします。
注: プラグイン (https://imagej.net/SNT) の更新バージョンがあります。 - 神経突起長と樹状突起爪形成を簡易神経突起トレーサーを用いて盲検化して解析します。エレクトロポレーションされたCGNのシングルチャンネルzスタック画像をFIJIにアップロードし、 プラグイン|セグメンテーション |単純な神経突起トレーサー (図3D)。
- ドロップダウン メニューにアクセスし、[新しい 3D ビューアを作成 (図 3D)] を選択します。
- 樹状突起の基部までスクロールし、そこでセルソーマに接続し、接合部をクリックしてパスを開始します。セルフィル信号が最も明るいセクションをクリックし、[y]を押してトレースを保持することにより、パスを手動でトレースします。爪が含まれていない場合は樹状突起の端まで、または爪の付け根までトレースし、[f]を押して経路を確認します(図4D)。
- 次に、構造の基部から経路を開始し、最も長い神経突起の終わりまで追跡することによって爪をたどります。二次分岐と三次分岐をトレースするには、Windows では [ctrl] を押し、Mac OS では [alt] を押したままパスをクリックします。[f]を押してパスを確認します。
- トレースの測定値が別のウィンドウに表示されていることを確認します。爪の枝(一次、二次、三次)のすべての測定値を合計して、各爪の全長を求めます。
- エレクトロポレーションCGNの表面積と細胞体積を分析するには、Imaris細胞分析ソフトウェア(https://imaris.oxinst.com/)をダウンロードしてください。
注:FIJIは、すぐに利用できる無料のプラグインを使用して、zスタック画像から3Dでセルを再構築するためにも使用できます。さらに、Simple Neurite Tracerにはボリュームレンダリング機能がありますが、以下に概説する理由でImarisが使用されました。 - エレクトロポレーションされたCGNのzスタック画像をImarisにアップロードします。3D再構成ツールキットにアクセスするには、[ 超過]を押します。
- CGN を再構築するには 、「サーフェス」を押して、イメージウィンドウ内のセル全体を含む対象領域を選択します。終了したら、[ 作成]の下の右下隅にある青い進む矢印を押します。
- 画像に異なる信号の複数のチャンネルが含まれている場合は、エレクトロポレーションされたCGNを含むチャンネルを選択し、青い進む矢印を押します。
- スライドバーを使用して、エレクトロポレーションセルの信号に最も正確に適合する所望の閾値を設定する。セルの表面に近づけると、閾値を正確に決定できます。終了したら、二重緑色の矢印を押してセルを再構築し、メタデータから表面積と体積サイズを取得します。

図3:エレクトロポレーション顆粒ニューロンの免疫組織化学的分析と3次元再構成。 P7 CD-1マウスを、GFPを発現するコンストラクトでエレクトロポレーションした。脳を採取し、免疫組織化学、共焦点顕微鏡、および形態素解析のための3D再構成にかけました。(A) エレクトロポレーションから10DPIマウスの画像処理までのタイムライン。(B)エレクトロポレーション小脳10-DPIの矢状断面の最大投影像;白い線は小脳層を画定し、スケールバーは25μmです。 (C)単一のエレクトロポレーションされた顆粒ニューロン10-DPIおよび対応する3Dトレースの最大投影画像、スケールバーは10μmである。 (D)3D再構成は、FIJIプラグインのシンプルニューライトトレーサーを使用して生成されました。すべての測定値は、セルフィル信号に従ってzスタックを介して追跡されました。シャフトと爪の測定は、樹状突起ごとに別々に追跡されました。点線は、現在の平面内の樹状突起の部分を示します。略語:3D =3次元。GFP = 緑色蛍光タンパク質;DPI =注入後の日数。PSD-95 =シナプス後密度タンパク質95;GNP =顆粒ニューロン前駆細胞;PFA = パラホルムアルデヒド。 この図の拡大版を表示するには、ここをクリックしてください。
Subscription Required. Please recommend JoVE to your librarian.
Representative Results

図4:小脳発達過程における顆粒ニューロン形態の解析。 (A)3-DPIから14-DPI(出生後年齢P10〜P21)、核(青)およびGFP(緑)のエレクトロポレーションCGNの最大投影画像。矢印は個々の樹状突起を示し、スケールバーは10μmです。 (B)樹状突起の平均数。(C)体細胞の基部から樹状突起の先端まで測定された平均樹状突起長。(D)爪を含む樹状突起の割合。1.00の値は100%であり、つまりすべての樹状突起には爪があります。(E)樹枝状爪の全長。N>条件ごとに少なくとも4匹の動物から収集された、条件ごとに30個の細胞;すべての測定値は一元配置分散分析によって分析され、ダネットの多重比較検定(B、C、および D)またはテューキーの多重比較検定(E)のいずれかで分析され、****は時間経過に伴うp <0.0001の有意性を示します。エラーバーはS.E.M.略語:GFP =緑色蛍光タンパク質。DPI =注入後の日数。PSD-95 =シナプス後密度タンパク質95;CGN =小脳顆粒ニューロン;ANOVA = 分散分析;S.E.M. = 平均の標準誤差。 この図の拡大版を表示するには、ここをクリックしてください。
in vivoでの顆粒ニューロン形態の発達を研究するために、ヒトユビキチンプロモーター(FUGW)の制御下でGFPを発現する構築物をCD-1マウスの発達中の小脳にエレクトロポレーションし、注射後3、5、7、10、および14日目に脳を採取しました(DPI)。エレクトロポレーションと共焦点顕微鏡法を組み合わせた細胞のスパース標識は、樹状突起の剪定、成長、成熟の期間中のCGNのスナップショットをキャプチャします。CGNシナプス構造の成長を定量的に分析および追跡するために、各樹状突起は、FIJIプラグインの単純神経突起トレーサー(SNT)を使用して追跡されました。SNTは、3次元(3D)空間で神経突起と爪の長さを測定するための簡単、高速、効率的ですぐに利用できる方法です。逆に、Imarisは、プログラムが各細胞の高速で正確なレンダリングを提供し、その閾値化機能が近くの標識された細胞破片から標識された細胞を分離できるため、CGNの3D再構成に使用され、表面積と体積の測定値を取得しました。
生まれたばかりのCGNは、樹状突起の成長の活発な段階を経て、P10からP14(3〜7-DPI)に精製され、過剰な樹状突起の50%以上が剪定されます(図4B)。この出来事は、残りのアーバーの漸進的な延長(図4C)と各樹状突起の端部での爪のような構造の形成(図4D)と一致しており、これらの発生過程が同時に起こっていることを示しています。しかし、爪はP14(7-DPI)によって樹状突起の約75%に見られますが、これらの構造はP21(14-DPI)までサイズが大きくなり続けます(図4E)。
樹状突起と爪の形態の変化は、総細胞サイズの全体的な変化または細胞膜の再分布のいずれかを反映している可能性があります。この問題に対処するために、各標識された顆粒ニューロンをImarisで再構築し、総体性樹状突起表面積と体積を定量化しました。CGNのサイズは開発全体で比較的一定でしたが(図5A、B)、P14では、CGNはP10、P12、およびP17(3、5、および10-DPI)と比較して体積が20%大幅に減少しました(図5B)。これらのデータは、樹状突起の爪への拡大を可能にするために、収縮した樹状突起からの膜リサイクルが特に重要である可能性があることを示唆しており、剪定からシナプス発達への移行における重要な時点としてP14(7-DPI)を示しています。

図5:小脳発達中の顆粒ニューロンサイズの解析。エレクトロポレーションされたCGNをImarisで再構築し、細胞サイズを決定しました。(A-B)小脳発達中の顆粒ニューロン(すなわち、細胞体細胞および樹状突起)の表面積および体積分析。N>条件ごとに30個の細胞を、条件ごとに少なくとも4匹の動物から採取する。すべての測定値は、一元配置分散分析とダネットの多重比較検定によって分析され、**はp <0.005の有意性を示します。エラーバーはSEMの略語です:DPI =インジェクション後の日数。CGN =小脳顆粒ニューロン;ANOVA = 分散分析;S.E.M. = 平均の標準誤差。この図の拡大版を表示するには、ここをクリックしてください。
Subscription Required. Please recommend JoVE to your librarian.
Discussion
小脳顆粒ニューロンは、哺乳類の脳で最も豊富なニューロンであり、げっ歯類の脳の全ニューロン集団のほぼ60〜70%を占めています1,14。小脳は、細胞の増殖、遊走、樹状突起形成、シナプス発生のメカニズムの解明に広く利用されています6,9,10,11,15,16,17,18,19,20 .さらに、何十年にもわたる電気生理学的研究は、学習された運動行動への回路の関与の根底にある、長期小脳可塑性における顆粒ニューロンの役割を確立するのに役立ちました21,22,23。したがって、顆粒ニューロンは、シナプスと回路の発達に関する重要な質問を調査するための優れたモデルシステムです。
この研究の目的は、小脳を利用してin vivoで単一ニューロンタイプの形態を追跡する技術を強調することです。小脳のエレクトロポレーションは、分裂細胞を標的とする。したがって、技術的には顆粒ニューロン前駆細胞とグリア細胞の両方を標識することができますが、顆粒ニューロン前駆細胞は非常に多いため、エレクトロポレーションされた集団は、細胞型特異的プロモーターを必要とせずに、大部分がニューロンです。さらに、この技術は、in vivoで遺伝子を遺伝子操作して、CGN発生における遺伝子の役割を研究するために適応させることができます。これは、ショートヘアピンRNAまたは低分子干渉RNAのいずれかを発現するプラスミドをトランスフェクションして遺伝子をノックダウンするか、Creリコンビナーゼを発現するプラスミドをトランスジェニックマウス系統にトランスフェクションしてLoxP部位に隣接する領域を切除し、目的の遺伝子をノックアウトすることによって達成できます10,24。
エレクトロポレーションには、遺伝子研究の長所と短所の両方があります。従来のトランスジェニック戦略と比較して遺伝子を操作するためのより高速な方法を提供しますが、エレクトロポレーションの予備性は、効果を見るために多数の遺伝子組み換え細胞を必要とする行動研究への有用性を制限します。第2の制限は、小葉5と7の間の顆粒ニューロンのみを確実に標的とすることであり、これは、部分的には、小脳が注射部位でどのように配向しているかに起因する。しかし、エレクトロポレーション領域内では、この方法は比較的多数の個別に標識された細胞を生成し、統計的に堅牢な測定の可能性を与えます。たとえば、樹状突起の発達を経時的に追跡できるだけでなく、樹状突起と爪の間の膜の微妙な再分布も測定でき、以前に公開されたデータ20を拡張しました。CGN爪は、これらの樹状構造の拡大が、顆粒ニューロンが苔状線維末端および単極性ブラシ細胞との興奮性結合および近くのゴルジ細胞との抑制性結合の両方を形成するための追加の領域を提供するため、研究するのに特に興味深い。したがって、これらの樹状突起測定と特定のシナプス前およびシナプス後タンパク質の免疫組織化学的標識を組み合わせることは、小脳回路形成およびシナプス成熟の研究を進めるのに役立つ可能性があります。
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
著者は利益相反を宣言しません。
Acknowledgments
この研究は、NIH助成金R01NS098804(A.E.W.)、F31NS113394(UC)、およびデューク大学のSummer Neuroscience Program(D.G.)の支援を受けました。
Materials
| Name | Company | Catalog Number | Comments |
| Betadine | Purdue Production | 67618-150-17 | |
| Cemented 10 µL needle | Hamilton | 1701SN (80008) | 33 gauge, 1.27 cm (0.5 in), 4 point style |
| Chicken anti-GFP | Millipore Sigma | AB16901 | Our lab uses this antibody at a 1:1000 concentration |
| Cotton-tip applicator | |||
| Donkey anti-chicken Cy2 | Jackson ImmunoResearch | 703-225-155 | Our lab uses this antibody at a 1:500 concentration |
| Ethanol (200 proof) | Koptec | V1016 | |
| Electroporator ECM 830 | BTX Harvard Apparatus | 45-0052 | |
| Fast Green FCF | Sigma | F7252-5G | |
| FUGW plasmid | Addgene | 14883 | |
| Glass slides | VWR | 48311-703 | Superfrost plus |
| Glycerol | Sigma-Aldrich | G5516 | |
| Heating pad | Softheat | ||
| Hoescht 33342 fluorescent dye | Invitrogen | 62249 | |
| Imaris | Bitplane | ||
| Isoflurane | Patterson Veterinary | 07-893-1389 | |
| Micro cover glass | VWR | 48382-138 | |
| Nail polish | Sally Hansen | Color 109 | |
| Normal goat serum | Gibco | 16210064 | |
| O.C.T. embedding compound | Tissue-Tek | 4583 | |
| Olympus MVX10 Dissecting Scope | Olympus | MVX10 | |
| P200 pipette reach tip | Fisherbrand | 02-707-138 | Used for needle spacer |
| Parafilm | Bemis | PM-996 | |
| PBS pH 7.4 (10x) | Gibco | 70011-044 | |
| Simple Neurite Tracer | FIJI | https://imagej.net/Simple_Neurite_Tracer:_Basic_ Instructions |
|
| Sucrose | Sigma | S0389 | |
| Surgical tools | RWD Life Science | Small scissors and tweezers | |
| Triton X-100 | Roche | 11332481001 | non-ionic detergent |
| Tweezertrodes | BTX Harvard Apparatus | 45-0489 | 5 mm, platinum plated tweezer-type electrodes |
| Ultrapure distilled water | Invitrogen | 10977-015 | |
| Vectashield mounting media | Vectashield | H1000 | |
| Vetbond tissue adhesive | 3M | 1469SB | |
| Zeiss 780 Upright Confocal | Zeiss | 780 |
References
- Altman, J., Bayer, S. A. Development of the cerebellar system : in relation to its evolution, structure, and functions. , CRC Press. (1997).
- Rahimi-Balaei, M., Bergen, H., Kong, J., Marzban, H. Neuronal migration during development of the cerebellum. Frontiers in Cellular Neuroscience. 12, 484 (2018).
- Alder, J., Cho, N. K., Hatten, M. E. Embryonic precursor cells from the rhombic lip are specified to a cerebellar granule neuron identity. Neuron. 17 (3), 389-399 (1996).
- Hatten, M. E., Heintz, N. Mechanisms of neural patterning and specification in the developing cerebellum. Annual Review of Neuroscience. 18, 385-408 (1995).
- Ben-Arie, N., et al. Math1 is essential for genesis of cerebellar granule neurons. Nature. 390 (6656), 169-172 (1997).
- Borghesani, P. R., et al. BDNF stimulates migration of cerebellar granule cells. Development. 129 (6), 1435-1442 (2002).
- Espinosa, J. S., Luo, L. Timing neurogenesis and differentiation: insights from quantitative clonal analyses of cerebellar granule cells. Journal of Neuroscience. 28 (10), 2301-2312 (2008).
- Markwalter, K. H., Yang, Y., Holy, T. E., Bonni, A. Sensorimotor coding of vermal granule neurons in the developing mammalian cerebellum. Journal of Neuroscience. 39 (34), 6626-6643 (2019).
- Shalizi, A., et al. PIASx is a MEF2 SUMO E3 ligase that promotes postsynaptic dendritic morphogenesis. Journal of Neuroscience. 27 (37), 10037-10046 (2007).
- Shalizi, A., et al. A Calcium-regulated MEF2 sumoylation switch controls poststynaptic differentiation. Science. 311 (5763), 1012-1017 (2006).
- Konishi, Y., Stegmuller, J., Matsuda, T., Bonni, S., Bonni, A. Cdh1-APC controls axonal growth and patterning in the mammalian brain. Science. 303 (5660), 1026-1030 (2004).
- Holubowska, A., Mukherjee, C., Vadhvani, M., Stegmuller, J. Genetic manipulation of cerebellar granule neurons in vitro and in vivo to study neuronal morphology and migration. Journal of Visualized Experiments: JoVE. (85), e51070 (2014).
- Yang, Y., et al. Chromatin remodeling inactivates activity genes and regulates neural coding. Science. 353 (6296), 300-305 (2016).
- Herculano-Houzel, S. Coordinated scaling of cortical and cerebellar numbers of neurons. Frontiers in Neuroanatomy. 4, 12 (2010).
- Wilson, P. M., Fryer, R. H., Fang, Y., Hatten, M. E. Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial-guided neuronal migration. Journal of Neuroscience. 30 (25), 8529-8540 (2010).
- Kokubo, M., et al. BDNF-mediated cerebellar granule cell development is impaired in mice null for CaMKK2 or CaMKIV. Journal of Neuroscience. 29 (28), 8901-8913 (2009).
- Schwartz, P. M., Borghesani, P. R., Levy, R. L., Pomeroy, S. L., Segal, R. A. Abnormal cerebellar development and foliation in BDNF-/- mice reveals a role for neurotrophins in CNS patterning. Neuron. 19 (2), 269-281 (1997).
- Segal, R. A., Pomeroy, S. L., Stiles, C. D. Axonal growth and fasciculation linked to differential expression of BDNF and NT3 receptors in developing cerebellar granule cells. Journal of Neuroscience. 15 (7), Pt 1 4970-4981 (1995).
- Zhou, P., et al. Polarized signaling endosomes coordinate BDNF-induced chemotaxis of cerebellar precursors. Neuron. 55 (1), 53-68 (2007).
- Dhar, M., Hantman, A. W., Nishiyama, H. Developmental pattern and structural factors of dendritic survival in cerebellar granule cells in vivo. Scientific Reports. 8 (1), 17561 (2018).
- Ito, M. Synaptic plasticity in the cerebellar cortex and its role in motor learning. Canadian Journal of Neurological Sciences. 20, Suppl 3 70-74 (1993).
- Jorntell, H., Hansel, C. Synaptic memories upside down: bidirectional plasticity at cerebellar parallel fiber-Purkinje cell synapses. Neuron. 52 (2), 227-238 (2006).
- Nakanishi, S. Genetic manipulation study of information processing in the cerebellum. Neuroscience. 162 (3), 723-731 (2009).
- Chang, C. H., et al. Atoh1 controls primary cilia formation to allow for SHH-triggered granule neuron progenitor proliferation. Developmental Cell. 48 (2), 184-199 (2019).
Tags
神経科学 172号 神経発生 小脳 樹状突起 シナプス 顆粒ニューロン エレクトロポレーションErratum
Formal Correction: Erratum: Utilizing In Vivo Postnatal Electroporation to Study Cerebellar Granule Neuron Morphology and Synapse Development
Posted by JoVE Editors on 04/06/2023.
Citeable Link.
An erratum was issued for: Utilizing In Vivo Postnatal Electroporation to Study Cerebellar Granule Neuron Morphology and Synapse Development. A figure was updated.
Figure 2 was updated from:
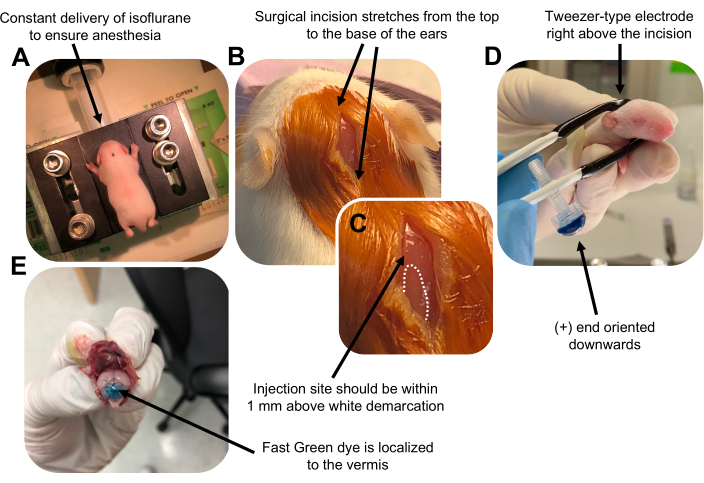
Figure 2: In vivo cerebellar electroporation of granule neuron progenitors in P7 wildtype mouse pups. (A) Pups are anesthetized with 4% isoflurane delivered at a rate of 0.8L/min to ensure anesthesia throughout the injection of the DNA solution. Isoflurane is delivered at a rate of 0.8 L/min. (B) After sterilizing the mouse 3 times with betadine and 70% ethanol, an incision is made that spans the distance of the ears, revealing the hindbrain. (C) A magnified image of a white demarcation on the cranium, a landmark for the injection site. DNA construct should be injected within 1 mm above the mark; dotted lines outline the demarcation, and black arrow denotes the injection site. The ridges of the cerebellar vermis may be visible and can be useful for finding the injection site. (D) Tweezer-type electrode orientation for efficient electroporation. Plus (+) end must be oriented downwards to pull negatively charged DNA into the cerebellar parenchyma prior to administration of electrical pulses. (E) Test injection of 1 µL of a 0.02% Fast Green dye shows injection is localized to the middle of the cerebellar vermis between lobules 5-7. Please click here to view a larger version of this figure.
to:
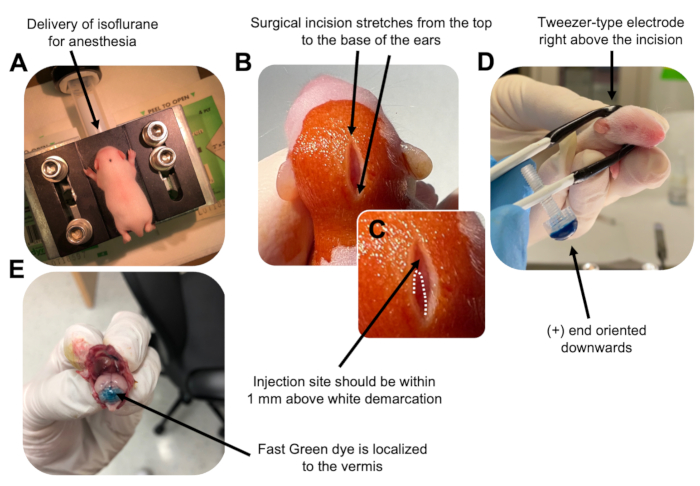
Figure 2: In vivo cerebellar electroporation of granule neuron progenitors in P7 wildtype mouse pups. (A) Pups are anesthetized with 4% isoflurane delivered at a rate of 0.8L/min to ensure anesthesia throughout the injection of the DNA solution. Isoflurane is delivered at a rate of 0.8 L/min. (B) After sterilizing the mouse 3 times with betadine and 70% ethanol, an incision is made that spans the distance of the ears, revealing the hindbrain. (C) A magnified image of a white demarcation on the cranium, a landmark for the injection site. DNA construct should be injected within 1 mm above the mark; dotted lines outline the demarcation, and black arrow denotes the injection site. The ridges of the cerebellar vermis may be visible and can be useful for finding the injection site. (D) Tweezer-type electrode orientation for efficient electroporation. Plus (+) end must be oriented downwards to pull negatively charged DNA into the cerebellar parenchyma prior to administration of electrical pulses. (E) Test injection of 1 µL of a 0.02% Fast Green dye shows injection is localized to the middle of the cerebellar vermis between lobules 5-7. Please click here to view a larger version of this figure.
