I - TASSERのパイプラインを使用してタンパク質の構造と機能解析に基づくコンピューターのためのガイドラインが記述されています。クエリタンパク質配列から始まる、3Dモデルは、複数のスレッドアラインメントを使用して生成され、構造組立シミュレーションを反復している。機能的な推論は、その後、既知の構造と機能を持つ蛋白質の一致に基づいて描画されます。
Method Article
コンピュータベースのタンパク質の構造と機能の予測のためのプロトコル
Opens in a new tab
In This Article
Summary
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
Abstract
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
ゲノムシーケンシングプロジェクトは、その生物学的役割についての理解を向上させるために、それらの構造と機能の知識を必要とするタンパク質の配列の何百万を、暗号化されている。実験方法は、これらの蛋白質のごく一部のために詳細な情報を提供することができますが、計算モデルは実験的に未知されているタンパク質分子の大部分のために必要です。 I - TASSERサーバは、タンパク質の構造と機能の高解像度モデリングのためのオンラインワークベンチです。タンパク質配列を考えると、I - TASSERサーバからの典型的な出力は、二次構造予測が含まれ、各残基の溶媒露出度、スレッドと構造アラインメントによって検出された相同テンプレートのタンパク質、最大5つのフルレングスの三次構造モデル、および構造ベースの予測酵素の分類のための機能のアノテーション、遺伝子オントロジーの用語と蛋白質 - リガンド結合部位。すべての予測は信頼性スコアでタグ付けされている予測は実験データを知らなくてもあるか正確に伝えます。エンドユーザーの特殊な要求を容易にするために、サーバはユーザが指定した残基間の距離を受け入れ、対話的にI - TASSERのモデリングを変更するマップを連絡するチャネルを提供し、それはまた、ユーザーがテンプレートとして任意のタンパク質を指定するには、または任意のテンプレートを除外することができます構造の組み立てシミュレーション中のタンパク質。構造情報は、I - TASSER予測の質を改善する目的で実験的証拠または生物学的洞察に基づいて、ユーザーによって収集することができます。サーバは、最近の社会全体のCASP実験でタンパク質の構造と機能の予測のための最高のプログラムとして評価した。現在ありません>オンラインI - TASSERサーバを使用している100カ国以上から20,000登録の科学者が。
Protocol
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
メソッドの概要
シーケンス間の構造から機能のパラダイムに続いて、構造と機能のモデリングのためのI - TASSERの手順1-4の4つの連続したステップが含まれます。LOMETS 5による()テンプレートの識別、再構築によってレプリカ- (b)のフラグメント構造交換モンテカルロシミュレーション6; REMO 7、FG - MD 8を使用して (C)原子レベルの構造精密化、および(d)補9を使用して構造ベースの機能の解釈。
テンプレートの識別は :ユーザーによって送信されるクエリーのシーケンスは、シーケンスは、最初にローカルにインストールさLOMETSのメタスレッドサーバで代表的なPDB構造のライブラリを介してスレッド化されている。スレッディングは、同じような構造を持っているか、クエリのタンパク質と同様の構造モチーフを含む可能性のあるテンプレートのタンパク質を同定するために使用される配列構造のアライメントの手順です。相同templのカバレッジを向上させるために食べた検出、LOMETSは別のスレッドの方法論をカバーする複数の最先端のアルゴリズムを組み合わせたもの。別のスレッドのプログラムが別のスコアリングシステムとアライメントの感度を持っているので、各スレッドプログラムから生成されたスレッドアライメントの品質は、次のように定義されている正規化されたZ -スコアで評価される。 
Z -スコアは、プログラムによって生成されたすべてのアライメントの統計的平均値からの相対標準偏差単位でのスコアです、そして、Z 0は "善を区別するために大規模なスレッドのベンチマークテスト5に基づいて決定プログラム固有のZ -スコアのカットオフです。 'と'悪い'テンプレート。高Z -スコアのテンプレートは、トップのテンプレートは、通常、アライメントが良いモデルに対応していることを意味する他のテンプレートのほとんどよりも有意に高いアラインメントスコアが、持っていることを意味します。トップスレッドテンプレートのほとんどは、ハイテクがある場合GH正規化されたZ -スコアは 、最後のI - TASSERモデルの精度は、通常、高いです。しかし、タンパク質が大きく、スレッディングアライメントの範囲は、クエリタンパク質の小さな領域に限定されている場合、高規格化Z -スコアは、必ずしもフルレングスモデルのための高いモデリングの精度を意味するものではありません。各スレッドプログラムから上位2つのスレッドのアライメントは、構造アセンブリの次のステップのために収集され使用されています。
構造のアセンブリのシミュレーションの反復 :スレッド手順に従って、クエリ配列は整列し、整列領域をスレッドに分割されます。アライメントされていないループ領域はのab initioモデリングによって構築されている間スレッディングアライメントの継続的なフラグメントは、テンプレートから切り出し、構造の組み立てのために直接使用されます。構造の組立手順は6レプリカ交換モンテカルロシミュレーションによって導か格子システム上で実行されます。 I - TASSER力場は、水素- BOが含まれています相互作用10、PDB 11で既知のタンパク質の構造に由来する知識ベースの統計的エネルギー項をnding、SVMSEQ 12、および空間的な制約からシーケンスベースのコンタクト予測がLOMETSから5スレッドテンプレートを収集した。シミュレーション中に低温のレプリカで生成された立体配座のデコイは、低い自由エネルギー状態の構造を識別するためにスピッカー13でクラスター化されます。トップクラスタのクラスタの重心は、クラスタ化されたすべての構造的なデコイの3次元座標を平均することによって得られ、最終的なモデルの生成に使用されます。シミュレーションおよびクラスタリングの手順は、立体の衝突を除去し、さらにグローバルなトポロジーを改良するために二度繰り返されます。
原子レベルのモデルの構築と改良 :スピッカークラスタリング後に得られたクラスタの重心は、タンパク質モデル(そのCのαと質量の側鎖の中心に代表される各残基)とhを削減しているaveの限られた生物学的アプリケーション。縮小モデルからフル原子モデルの構築は2段階で行われます。最初のステップでは、REMO 7は 、H -結合ネットワークを最適化することにより、C -αのトレースからフルアトミックモデルを構築するために使用されます。第二段階では、REMOフルアトミックモデルをさらにバックボーンのねじれ角、結合長、および側鎖の回転異性体の向きを改善FG - MD 14によって洗練され、分子動力学シミュレーションによって、などから検索した構造の断片に導かTM -揃えでPDBの構造。 FG - MD洗練されたモデルは、I - TASSERによる立体構造予測のための最終的なモデルとして使用されています。
生成されたモデルの質はLOMETSスレッドアラインメントのZ -スコアとI - TASSERシミュレーションの収束、数学的に定式化に基づいて定義されている信頼度スコア(C -スコア)、に基づいて推定されている。 
ここで、 13によって識別される構造的なクラスタ内の構造のデコイの多重度であり、M TOTは、クラスタリングに提出したデコイの合計数です。 クラスタの重心にクラスタ化されたデコイの平均RMSDです。Norm.Z -スコアは、(i)iは LOMETS 5のスレッドサーバをi番目から得られた上部スレッディングアライメントの正規化されたZ -スコア(式1)であり、Nは LOMETSで使用されているサーバーの数。
C -スコアは、I - TASSERモデルの質と強い相関を持っています。 C -スコアとタンパク質の長さを組み合わせることで、最初のI - TASSERモデルの精度は、TM -スコアと2ÅRMSDを15 0.08の平均誤差で推定することができる。一般的には、C -スコアとモデル> - 1.5は正しいフォールドを持つことが期待されます。ここでは、RMSDおよびTM -スコアは、両方のモデルとネイティブ構造との間のトポロジー類似性のよく知られている指標です。 TM -スコアVALUにおけるESの範囲[0、1]、より高いスコアはよりよい構造の一致16,17を示す。しかし、低いランクのモデル(すなわち第2 -5 番目のモデル)のために、TM -スコアとRMSDとC -スコアの相関は、(〜0.5)はるかに弱いです、そして絶対的なモデルの品質の信頼性の推定に使用することができます。
最初のモデルは、常にI - TASSERシミュレーションで最適なモデルです?この質問に対する答えは、ターゲットの種類によって異なります。簡単なターゲットの場合、最初のモデルは、通常、最良のモデルであり、そのC -スコアは、通常モデルの他の部分よりもはるかに高いです。しかし、スレッドは、重要なテンプレートのヒットを持っていないハードターゲット、のために、最初のモデルは、必ずしも最良のモデルではなく、I - TASSERは実際に最高のテンプレートとモデルを選択するのは困難です。したがって、ハードターゲット用の全5モデルを分析し、それらを実験的情報と生物学的知見に基づいて選択することを推奨します。
この関数は、predがictions:最後のステップでは、FG - MDから生成された最終的な3D -モデル、すなわち、タンパク質の機能の3つの側面を予測するために使用されています:a)の酵素委員会(EC)番号18および(b)の遺伝子オントロジー(GO)19用語と( c)の小分子リガンドの結合部位。すべての3つの側面については、機能的な解釈は、既知の構造と機能を持つPDBのテンプレートのタンパク質へのグローバルおよびローカルな類似性に基づくタンパク質の機能を予測する新しいアプローチである補因子を、使用して生成されます。最初に、予測モデルのグローバルトポロジーの構造アライメントプログラムTM -揃える20を使用して機能的なテンプレートライブラリと照合されます。次に、ターゲットモデルに最も類似したタンパク質のセットは、彼らのグローバルな構造の類似性に基づいてライブラリーから選択されており、広範囲のローカル検索がアクティブ/結合部位領域付近の構造とシーケンスの類似性を識別するために実行されます。その結果、グローバルとローカルの類似度スコアがランク付けに使用されていますテンプレートは、蛋白質(機能的ホモログ)とトップスコアのヒットに基づいて、注釈(EC番号や遺伝子オントロジー19項)を転送する。同様に、リガンド結合部位の残基とリガンドの結合モードがトップスコア関数テンプレート9の既知のリガンド結合部位の残基を持つクエリのローカルアラインメントに基づいて推測されます。
機能の品質(ECおよびGO用語は)I - TASSERの予測は、クエリとテンプレートの間で、グローバルとローカルの類似性の尺度である機能的相同性スコア(FH -スコア)に基づいて評価され、次のように定義されます。 
C -スコアは式で定義された予測モデルの品質の推定値です。 (2)、TM -スコアは、モデルとテンプレートのタンパク質間のグローバルな構造的類似性を測定します。RMSD アリは、TM -揃える20から構造的に整列地域のモデルとテンプレートの構造とのRMSDです; COVは、構造アラインメント(すなわちクエリの長さで割った構造的に整列残基の比)の範囲を表し、ID アリは、TM -揃えアラインメントにおける配列同一です。 EC番号の予測の推定信頼度のスコアはまたとして計算、定義された局所領域内のクエリとテンプレートの間の活性部位の一致(ACM)を評価するための用語が含まれています。 
N tはローカルエリア内に存在するテンプレートの残基の数を表し、N アリが整列クエリテンプレートの残基ペアの数である、D IIが整列残基のi番目のペアの間にCαの距離であり、D 0 = 3.0 Åである距離のカットオフ、M IIは整列残基のi番目のペア間のBLOSUMのスコアです。一般的には、FH -スコアの範囲は[0、5]であり、ACMのスコアは[0、2の間です。より高いスコアをより確信した機能的な割り当てを示す]、。 ACMのスコアは、局所構造とBS -スコアと呼ばれるリガンド結合部位、近くに配列類似性を評価するために使用されます。
1。タンパク質配列の提出
- でI - TASSERのWebページを参照してくださいhttp://zhanglab.ccmb.med.umich.edu/I-TASSER構造と機能のモデリングの実験を開始する。
- コピーアンドペーストしアミノ酸配列を提供する形にまたは直接、"Browse"ボタンをクリックしてコンピュータからアップロードしてください。 I - TASSERサーバは、現在、最大1500残基まででシーケンスを受け付けます。 1500残基以上のタンパク質は通常、マルチドメインタンパク質であり、そしてI - TASSERに提出する前に個々のドメインに分割することをお勧めします。
- あなたの電子メールアドレス(必須)と仕事(省略可能)の名前を入力します。
- ユーザーは、必要に応じて外部の間の解像度を指定することができます。idueコンタクト/距離の制約は、追加のテンプレートに追加したり、構造のモデリングプロセス中にいくつかのテンプレートの蛋白質を除外する。 "考察"のセクションでこれらのオプションを使用して詳細についてはこちらをご覧ください。
- シーケンスを送信するには、"I - TASSERを実行"ボタンをクリックしてください。ブラウザはユーザーが指定した情報、ジョブ識別(ジョブID)番号と結果がジョブの完了後に堆積されるWebページへのリンクを表示する確認ページが表示されます。ユーザーはこのリンクをブックマークしたり、後で参照できるようにジョブ識別番号を書き留めすることがあります。
2。結果の可用性
- でI - TASSERキューのページにアクセスして投入されたジョブのステータスを確認http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php 。検索タブをクリックし、ジョブID番号または投入されたジョブを検索するクエリシーケンスを使用してください。
- 構造と機能MO後delingが終了すると、予測構造とウェブリンクの通知電子メールが含まれているイメージがあなたに送信されます。このリンクをクリックするか、結果を表示してダウンロードするには、ステップ1.5にブックマークリンクを開きます。
3。二次構造や溶媒露出度予測
- 結果ページの上部に表示されるFASTA形式のクエリシーケンスを確認してください。追加の拘束/テンプレートは、シーケンスの提出、ユーザー指定の情報を表示するウェブページへのリンク時に指定した場合も(図1A)見ることができます。
- αヘリックス(H)、βストランド(S)またはコイル(C)と各残基の予測の信頼性スコア(0 =低、9 =ハイ):として表示される二次構造予測を調べます。蛋白質のコア領域を推定するために、定期的な二次構造(HまたはS)の予測の長いストレッチで地域を探します。タンパク質の構造クラスはまた、二次構造要素の分布に基づいて分析することができます。らので、タンパク質のコイルエレメントの長い地域では、通常、非構造化/乱れた領域を示す。
- クエリの中に埋もれと溶媒露出された領域を確認するために予測溶媒露出度(図1C)を表示します。の値は0(埋葬渣)から9(露出残基)に溶媒露出度の範囲を予測した。溶剤曝露と親水性残基を持つ地域は、潜在的な水和/機能部位である間、主に埋もれ残基を含む領域は、タンパク質のコア領域を区切るために使用することができます。
4。三次構造予測
- インタラクティブJmolのアプレット(図2)で表示されるクエリタンパク質の予測された立体構造を、確認するためにスクロール。表示された構造の外観を変更するためにアプレットを左クリックし、特定の地域に拡大、予測モデルの特定の残基の種類を選択するか、残基間の距離を計算します。
- 長い非構造化部位の存在のためのモデルを分析する。これらのRegionsは、通常、タンパク質の乱れた地域に対応したり、テンプレートのアラインメントの欠如を示している。これらの領域は一般的に低モデリングの精度を持ち、N&からモデリング中にこれらの領域を削除するC末端領域は、モデリングの精度が向上します。
- "ダウンロード型"のリンクをクリックして、モデルのPDBフォーマットされた構造のファイルをダウンロードしてください。あなたは、構造的特徴をさらに分析するための任意の分子の可視化ソフトウェア(例:pymolの、RasMolはなど)にこれらのファイルを開くことができます。
- 予測構造の品質を推定する構造のモデリングの信頼性スコア(C -スコアを)分析する。 C -スコア(式2)の値が範囲内に典型的には[-5、2]、ここでは、より高いスコアはより良い品質のモデルを反映している。推定TM -スコアと最初のモデルのRMSDは、"モデル1の推定精度"として表示されます。 TM - scoreがRMSDより位相的な変化に対してより敏感である限り、タンパク質については、それは、TM -スコアに基づいてモデルの品質を評価することをお勧めします。 < liは> C -スコアを分析するためのリンク、クラスターのサイズとすべてのモデルのクラスタの密度"C -スコアの詳細"をクリックしてください。低いランクのモデルのC -スコアが強くTM -スコアまたはRMSDと相関されていないため、推定TM -スコアとRMSDは、最初のI - TASSERモデルに対してのみ表示されます。低いランクのモデルの質は、部分的に近いネイティブ構造に平均している大きなクラスターと高い密度からモデルを特徴とする、それらのクラスタの密度と最初のモデルの相対的なクラスタサイズに基づいて評価することができます。
- 低C -スコアの予測は、通常、低精度の予測を示している。ほとんどのそのようなケースでは、クエリのタンパク質は、ライブラリで良いテンプレートを欠いているとのab initioモデリング(すなわち> 120残基)の範囲を超えた大きさを持っています。これらのケースでは、ユーザーが追加の空間的な制約のために追求し、I - TASSERモデリング(ディスカッションのセクションを参照)改善するためにそれらを使用することができます。また、(私たちのQUARKのサーバへのシーケンスを提出するよう奨励され純粋な第一原理モデリングのためのQUARK /"> http://zhanglab.ccmb.med.umich.edu/QUARK/)蛋白質のサイズは以下の200残基である場合。
5。 LOMETSのターゲットテンプレートのアラインメント
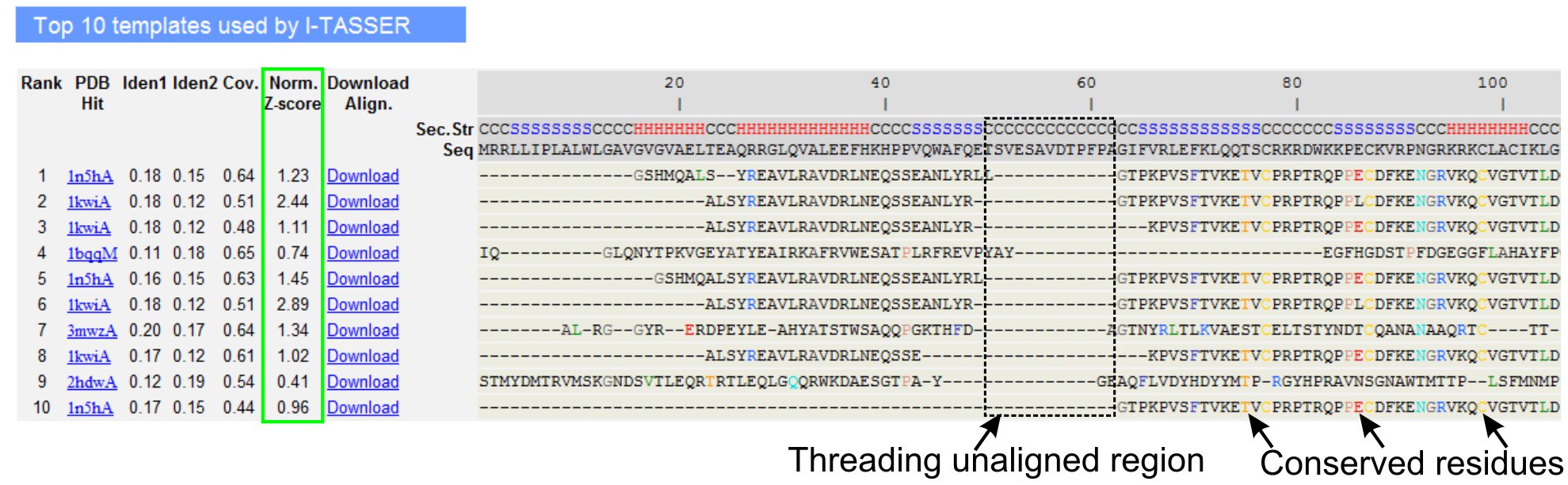
- としてLOMETSスレッドのプログラム(図3)によって識別されるクエリタンパク質のトップテンスレッドのテンプレートを、分析するためにスクロールダウンします。 "ノームに示すように正規化されたZ -スコア(式1)を、表示する。スレッディングアライメントの品質を分析するために、Z -スコア"列、。正規化されたZ -スコア> 1のアライメントは、自信を持って配置を反映しており、ほとんどは、クエリのタンパク質と同じ折りを持っている。
- スレッド揃え領域(列"アイデン。1')で、クエリおよびテンプレートのタンパク質間の相同性を評価するために、チェーン全体(列'アイデン。2')のための配列同一性を分析する。高い配列同一性は、クエリおよびテンプレートのタンパク質の間の進化的関係の指標です。
- 視覚的に短所を識別するために、色に示すように、スレッド整列残基を見るerved残基/クエリおよびテンプレートのタンパク質のモチーフ。全体の鎖のアライメントと比較してスレッド揃え地域におけるより高い配列同一性は、また、クエリ内の保存構造モチーフ/ドメインの存在を示している。
- 表示することにより、スレッドの配置の適用範囲を評価する"COVを。"列とアラインメントを検査する。トップアラインメントのカバレッジが低いと、クエリシーケンスの長いセグメントのクエリタンパク質または不在の小領域のみに限定されている場合、クエリタンパク質は通常、複数のドメインが含まれており、それが順序と、モデルを分割することを推奨します。個別のドメイン(図3)。
- リンクを"整列のダウンロード"をクリックして、PDBフォーマットされた配列構造体のアラインメントファイルをダウンロードしてください。これらのアライメントファイルは、マテリアルのセクションに記載されている任意の分子可視化プログラムで開くことができ、また構造のモデリング(ステップ1.4)の間に追加の制約を追加するために使用することができます。
6。PDBの構造類似体
- 20 TM -揃える構造アライメントプログラムによりとして同定された最初の予測モデルのトップテン構造類似体を、決定するために、結果のページの次の表(図4)を表示します。 TM -スコア> 0.5は、TM -スコアとのそれらは、<0.3はランダム構造の類似性を意味している間に検出アナログとモデルが、同じようなトポロジーを持っていると、クエリの蛋白質16の構造クラス/タンパク質ファミリーを決定するために使用できることを示します。
- 配列同一性を分析し、モデルの空間的なモチーフの保全及び構造類似体を評価するために"IDEN"と"RMSD"列に示すように構造的に整列領域でRMSD。視覚的にこれらの構造的に保存された残基とモチーフを識別するためのアライメントに着色し、整列残基のペアを点検。
- RCSBのウェブサイトを訪問し、それらの構造的分類(SCOPについての詳細は、"PDBヒット"列に示されているPDBコードをクリックしてください、CATHとPFAM)と機能的な情報(EC番号、用語及び結合リガンドのGOに関連する)。
7。補因子を用いて機能予測
- クエリのタンパク質の機能的な解釈を分析するために、結果のページでスクロールダウンします。酵素委員会(EC)番号、ジーンオントロジー(GO)の用語、およびリガンド結合部位を:タンパク質の機能を表示、次の3つのコンテキストの表に列挙されます。
- "TM -スコアを"表示する、、"IDEN"と"RMSD'COVを。"モデルと識別された機能ホモログ(テンプレート)との間の空間パターンのグローバルな構造の類似性と保全のパラメータを分析するために、各テーブル内の列。
8。酵素委員会番号の予測
- に示すように、クエリタンパク質の上位5潜在的な酵素のホモログを表示するテーブル(図5)"EC番号が予測"。これらのテンプレートを使用してEC番号の予測の信頼度は、"EC -スコア]列に表示されます。 benchmaに基づいて解析23、クエリおよびテンプレートのタンパク質間の機能的類似性(EC番号の最初の3桁)をrkingする信頼性の高いEC -スコア> 1.1を使用して解釈することができます。
- クエリタンパク質として同様のフォールド(すなわちTM -スコア> 0.5)を持つテンプレートが、間で機能のコンセンサス(EC番号)を探します。複数のテンプレートが同じEC番号とEC -スコア> 1.1をお持ちの場合、予測の信頼度は非常に高いです。しかし、EC -スコアが高いですが、同定されたヒットの間でコンセンサスの欠如がある場合、その後の予測は信頼性が低くなり、ユーザはGO的な予測に相談することをお勧めします。
- 詳細テンプレートのタンパク質の反応の触媒、補因子の要件と代謝経路を含むExPASy酵素のデータベースを訪問し、機能を解析するためのEC番号の上のリンク、、をクリックしてください。
9。遺伝子オントロジー(GO)長期予測
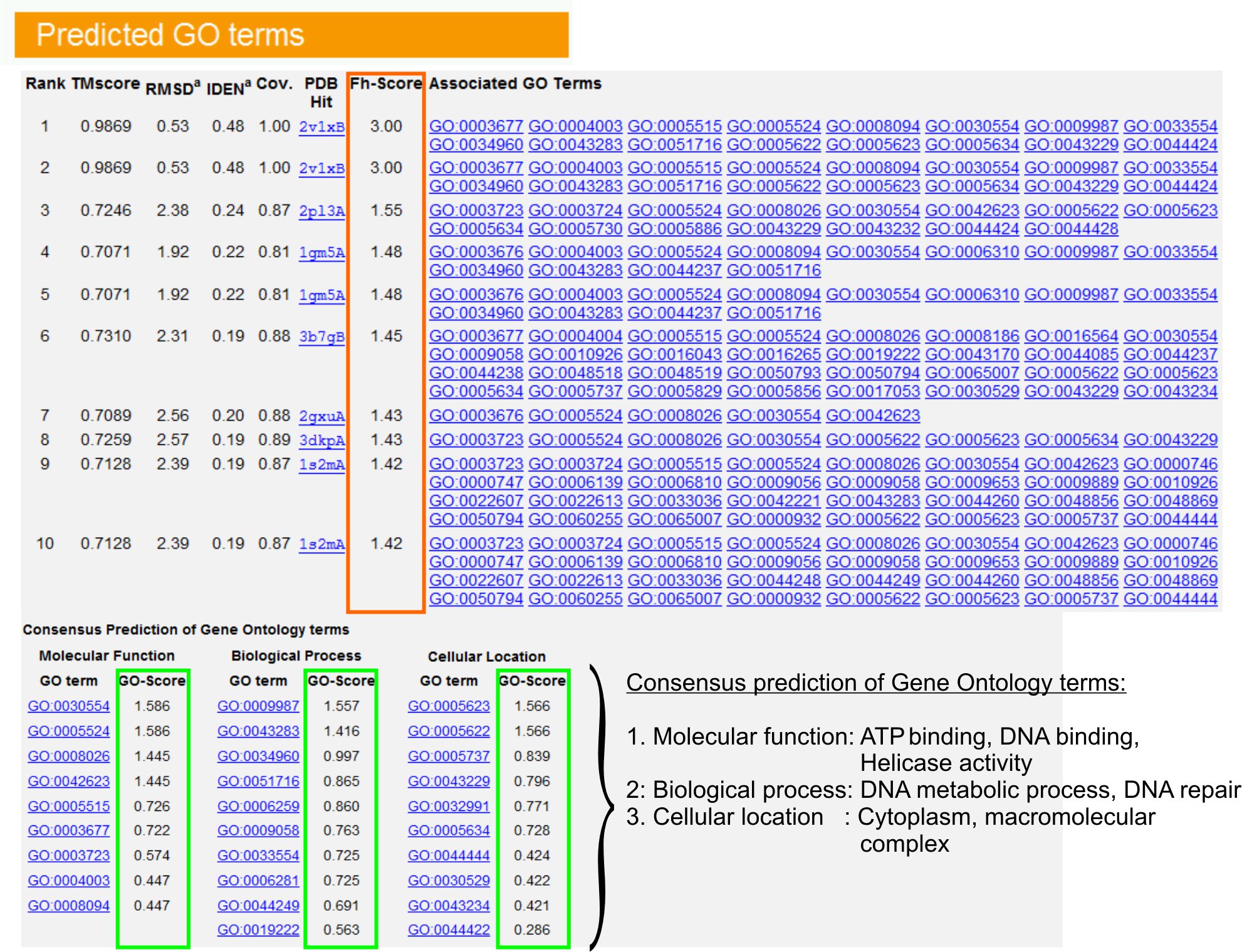
- "GO用語を予測"テーブル(図を見る遺伝子オントロジー(GO)の用語で注釈PDBライブラリのクエリタンパク質のトップテンホモログを、識別するために、URE 6)。それぞれのタンパク質は、通常、その分子機能(MF)、生物学的プロセス(BP)と細胞成分(CC)を記述する、複数のGO用語に関連付けられています。アミーゴのウェブサイトを訪問し、その定義と血統を分析するために各用語をクリックしてください。
- クエリおよびテンプレートのタンパク質間の機能的類似性にアクセスし、これらのタンパク質からの機能注釈を転送するの信頼度を推定するFH -スコア(機能的相同性のスコア)の列を分析する。私たちのベンチマーク調査23では、ネイティブのGO用語の50%は正しく56%の全体的な精度で、0.8のFH -スコアのカットオフを使用して最初に同定されたテンプレートから識別することができる。
- テンプレート間の機能の同意を分析するために"GO用語のコンセンサス予測は、"テーブルを表示します。これらの共通機能は、クエリのGO用語を(MF、BPやCC)を予測するために使用されています蛋白質と長期的な予測を行くの信頼水準(GO -スコア)を評価する。ベンチマークテスト23日に基づいて、最高の偽陽性と偽陰性率は、より深いオントロジーのレベルでの予測の範囲を小さくすると、GO -スコアのカットオフと予測= 0.5のために取得されます。
10。タンパク質 - リガンド結合部位の予測
- クエリのタンパク質のためのトップテンリガンド結合部位の予測を表示するページの一番下までスクロールダウンします。予測結合部位は、一般的な結合ポケットを共有する予測リガンドの立体配座の数に基づいてランク付けされます。最高の同定された結合部位は、既にJmolのアプレットに表示されます。他の予測を分析し、リガンドの相互作用残基を可視化するためにラジオボタンをクリックしてください。
- モデルとテンプレートの結合部位の間に局所的に類似性を評価するためにBS -スコアの列を分析する。ベンチマーク9、BS -スコアに基づいて、> 1.1は高い配列と構造SIMを示していますモデルで予測された結合部位およびテンプレート内の既知の結合部位の近くにilarity。
- "ダウンロード"リンクをクリックして複合体のPDBフォーマットされた構造のファイルをダウンロードしてください。ユーザーは、任意の分子可視化プログラムでこれらのファイルを開いて、インタラクティブに自分のローカルコンピュータ上で予測された結合部位とリガンド - タンパク質相互作用を見ることができます。
11。代表的な結果

図1:()FASTAフォーマットのクエリシーケンスを示すI - TASSER結果ページの抜粋を、(B)二次構造および関連する信頼度を予測、および(C)残基の溶媒露出度を予測した。分析されたコア領域と、クエリ内の潜在的な水和サイトは、それぞれ、シアンと赤の長方形で強調表示されます。

図2。

図3。LOMETS 5スレッドのプログラムでトップテン識別スレッドテンプレートとアラインメントを示すI - TASSER結果ページの例。スレッディングアライメントの品質は、値> 1は自信を持って配置を反映して正規化されたZ -スコア(緑色で強調表示)、に基づいて評価されます。対応するクエリの残基と同一のテンプレート内の整列残基は、保存残基/モチーフの存在を示すために色で強調表示されている間のほとんどの整合性の欠如トップテンプレートは、クエリの蛋白質とアラインされていない残基に複数のドメインの存在は、ドメインリンカー領域に対応することを示します。 図3のフルサイズバージョンを表示するにはここをクリック。
{kind=link}

TM -揃え20構造アライメントプログラムで識別されるトップテン構造的な類似と構造的なアラインメントを示す結果ページの4例、 図 。に示すように類似の順位は、構造整列のTM -スコア(青で強調表示)に基づいています。 TM -スコア<0.3つのランダムな構造間の類似性を意味しながらTM -スコア> 0.5は、2つの比較構造が似てトポロジーを持っていることを示しています。 " - "アライメントされていない領域がで示されている間、構造的に整列残基のペアは、それらのアミノ酸のプロパティに基づいて、色で強調表示されます。ove.com/files/ftp_upload/3259/3259fig4large.jpg">図4のフルサイズバージョンを表示するにはここをクリック。

図5。PDBライブラリのクエリタンパク質の同定された酵素のホモログを示すI - TASSER結果ページの例。 EC番号の予測の信頼水準EC -スコア(緑色で強調表示)に基づいて分析され、EC -スコア> 1.1は、クエリおよびテンプレートのタンパク質間の機能的類似性(EC番号の同じ最初の3桁)を示します。

図6。クエリタンパク質のための長期的な予測を行く示したI - TASSER結果ページの例。 Gene Ontologyのテンプレートライブラリ内のクエリタンパク質の機能的なホモログは、彼らのFH -スコア(オレンジ色の長方形で)に基づいてランク付けされます。これらのトップスコアのヒットから共通の機能的特徴は、GENERに導出されていますクエリの蛋白質の最終的なGOの長期予測を食べた。予測されたGO用語の品質は、GO -スコア> 0.5 GO -スコア(緑で表示)に基づいて推定されている信頼性の高い予測を示している。 図6のフルサイズバージョンを表示するにはここをクリック。
{kind=link}

図7。補因子9アルゴリズムを使用してトップテンタンパク質のリガンド結合部位の予測を示すI - TASSER結果ページの例。予測結合部位の順位は、クエリ内の共通の結合ポケットを共有する予測リガンドの立体配座の数に基づいています。 BS -スコアは(赤で強調表示)の予測とテンプレートの結合部位の間にローカルの配列と構造類似性の尺度であり、結合部位のポケットの保全を分析するために有用です。
les/ftp_upload/3259/3259fig8.jpg"/>
図8。残残基の接触/距離の制約を指定するために使用される外部拘束ファイルの例。

図9。I - TASSERサーバにテンプレートのタンパク質を指定するために使用される拘束ファイルの例。または(B)3Dフォーマット、ユーザーはどちらか(A)FASTA形式でクエリテンプレートのアラインメントを指定することができます。

図10。I - TASSER構造のモデリング手順の実行中にテンプレートを除くために使用するサンプルファイル。最初の列は除外されるテンプレートのタンパク質のPDB IDが含まれています。 2番目の列は、テンプレートライブラリ内の他の同様のテンプレートに使用される配列同一のカットオフを指定するために使用されます。
Discussion
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
上記のプロトコルは、I - TASSERサーバを使用して構造と機能のモデリングのための一般的なガイドラインです。 、この自動化された手順は、タンパク質の大部分を非常にうまく動作しますが、人間の介入は、しばしば特にPDBライブラリに近いテンプレートを欠いているタンパク質のため、大幅にモデリングの精度を向上させるのに役立ちます。 (b)の構造のアセンブリを改善するための外部制約を提供する;マルチドメインタンパク質の(A)の分割及び(c)のモデル化の際にテンプレートを削除:ユーザーは、次の方法でI - TASSERのモデリング中に介入することができます。
分割するマルチドメイン蛋白質:
多くの長いタンパク質配列は、頻繁に実験と計算の両方の技術を使用して、その構造の解明を困難にする柔軟なリンカー領域によってつなが複数のドメインが含まれています。それにもかかわらず、ドメインは独立して折り畳み式の実体であり、明確な分子機能を実行することができますように、それは個別に各ドメインの長いマルチドメインタンパク質とモデルを分割することが望ましい。モデリングのドメインは、個別にのみ予測のプロセスをスピードアップしませんが、またクエリテンプレートのアラインメントの質はより信頼性の高い構造と機能の予測の結果、増加する。
タンパク質配列のドメイン境界は、NCBI CDD 24、PFAM 25またはInterProScanの26として自由に利用できる外部のオンラインプログラムを使用して予測することができます。また、アラインメントのスレッドLOMETS、クエリタンパク質のために使用可能な場合は、ドメインの境界を視覚的に(ステップ5.4を参照)上のスレッドのテンプレートに整列されていない残基の長いストレッチを識別することによって見つけることができます。これらのアライメントされていない領域は、主にドメインリンカー領域に対応しています。すべてのクエリーのドメインが整列してマルチドメインのテンプレートがテンプレートPDBライブラリですでに使用可能な場合は、クエリのタンパク質は、全長としてモデル化することができます。
外部制約を提供する
I - TASSERの構造のアセンブリのシミュレーションは、主にテンプレートをスレッドLOMETSから収集された空間的な制約によって導かれる。テンプレートライブラリに優れたスレッドヒット(Norm. Z -スコア > 1)を持っているクエリタンパク質については、派生空間的制約は、高精度であることが多い。とI - TASSERは、これらのタンパク質の高分解能構造モデルを生成します。逆に、弱い、またはまったくスレッディングヒット(Norm. Z -スコア <1)を持つクエリのタンパク質で、収集された空間的な制約は、しばしばテンプレートとアライメントの不確実性のエラーが含まれている。これらのタンパク質のターゲットでは、ユーザーが指定した空間情報は、予測モデルの品質を向上させるために非常に役立ちます。ユーザーは、2つの方法でI - TASSERサーバーへの外部制約を提供することができます。A.は、接触/距離制約を指定します。
NMRから、例えば実験的に特徴づけられる残基間コンタクト/距離、または架橋実験を、拘束のファイルをアップロードして指定することができます。例のファイルは図8に示すように、カラム1が拘束の種類を指定する場所、"DIST"または"CONTACT"すなわち。距離拘束(DIST)の場合は、列が2と4は、残基位置(i、j)を含む、列3と5は、残基と6列目の原子の種類を含む2つの指定された原子間の距離を指定します。接触拘束(CONTACT)の場合は、列2と3は接触にあるべき残基の位置を(i、j)を含んでいます。これらの連絡残基ペアの側鎖の中心間の距離は、PDBで知られている構造で観測された距離に基づいて決定されます。 I - TASSERは、構造精密化のシミュレーション中に指定された距離に近いこれらの原子のペアを描画しようとします。
B.は、タンパク質の構造のテンプレートを指定します。
LOMETSスレッドプログラムでは、クエリのprotのためのもっともらしい襞を見つけるために代表的なPDBライブラリを使用してくださいアイン。代表的な構造のライブラリを使用すると、配列構造のアラインメントを計算するために必要な時間を減らすために役立ちますが、それは良いテンプレートのタンパク質がライブラリ内に見落とされた場合、またはテンプレートが、それがであっても、LOMETSスレッドプログラムで識別されていない可能性がありますライブラリ内に存在する。これらのケースでは、ユーザーがテンプレートとして所望のタンパク質の構造を指定する必要があります。
追加のテンプレートとしてタンパク質の構造を指定するには、ユーザーがいずれかのPDBフォーマットされた構造のファイルをアップロードするか、PDB、ライブラリ内の堆積タンパク質の構造のPDB IDを指定することができます。 I - TASSERは召集プログラム23を使用してクエリテンプレートのアラインメントを生成し、指定されたユーザーテンプレートと構造の組立シミュレーションを導くためにLOMETSテンプレートの両方から空間的な制約を収集します。 LOMETS拘束具の精度が異なるターゲットが異なるため、LOMETSの拘束の重みは、容易に(相同)TAに強力です。体系的に私たちのベンチマークトレーニングにチューニングされているハード(非相同)ターゲット、のそれよりもrgets。
またユーザが独自のクエリテンプレートのアラインメントを指定することができます。 FASTA形式(図9A)及び3Dフォーマット(図9B):サーバーには2つの形式で位置合わせを受け入れます。 FASTA形式は標準とで説明されていますhttp://zhanglab。 ccmb.med.umich.edu / FASTA / 。 3Dフォーマットは、標準的なPDB形式(に似ていますhttp://www.wwpdb.org/documentation/format32/sect9.html )が、テンプレートから派生した2つの追加列はATOMの記録(図9B参照)に追加されます。
列1-30:アトム(C -αのみ)と残クエリシーケンスの名前。
列31から54:テンプレートの対応する原子からコピーされたクエリのC -α原子の座標。
列55から59:アライメントに基づいて、テンプレート内の対応する残基の数
列60から64:テンプレートの対応する残基名
テンプレートの蛋白質を除外する
タンパク質は、柔軟な分子であり、それらの生物学的活性を変更するには、複数の構造状態を採用することができます。例えば、多くのプロテインキナーゼと細胞膜タンパク質の構造は、 アクティブおよび非アクティブの両方コンフォメーションで解決されている。また、結合したリガンドの有無は、大きな構造的な動きを引き起こす可能性があります。テンプレートのすべての構造状態は、スレッド化プログラムのために似ているが、それは1つだけ特定の状態でテンプレートを使用してクエリをモデル化することが望ましい。サーバ上の新しいオプションは、ユーザが構造のモデリング時にテンプレートの蛋白質を除外することができます。また、この機能は、ユーザーがモデリングに使用するテンプレートの相同性レベルを選択できるようになります。ユーザーがテンプレートタンパク質frを除外することができます。OM I - TASSERライブラリ替え:
A.は、配列同一性のカットオフを指定する
ユーザーは、I - TASSERのテンプレートライブラリから相同蛋白質を除外するには、このオプションを使用することができます。相同性のレベルが配列同一のカットオフに基づいて設定されて、クエリとクエリ配列のシーケンスの長さで割ったテンプレートのタンパク質間の同一の残基の数、すなわち。提供する形で"70%"、配列同一性を持っているすべてのテンプレートのタンパク質> 70%のユーザーがクエリタンパク質にたとえば、私は、予定I - TASSERのテンプレートライブラリから除外される。
B.特定のテンプレートの蛋白質を除外
特定のテンプレートのタンパク質が除外される構造のPDB IDを含むリストをアップロードすることによってI - TASSERのテンプレートライブラリから除外することができます。サンプルファイルは、図10に示します。同じタンパク質としてI - TASSER SE、PDBライブラリに複数のエントリとして存在することができますrverはデフォルトで指定されたテンプレート(列1の)だけでなく、アイデンティティーを持つライブラリから他のすべてのテンプレート>指定したテンプレート〜90%が除外されます。また、ユーザーは、アイデンティティーを持つすべてのテンプレート> 70%、指定されたテンプレートのタンパク質には除外されるなどの70%を、別のアイデンティティのカットオフを指定することができます。
Disclosures
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
利害の衝突は宣言されません。
Acknowledgements
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
プロジェクトは、アルフレッドP.スローン財団、NSFのキャリア賞(DBI 1027394)、および米国立総合医科学研究所(GM083107、GM084222)によって部分的にサポートされています。
Materials
- FASTAは、モデル化されるタンパク質のアミノ酸配列(参照、フォーマットhttp://www.ncbi.nlm.nih.gov/BLAST/fasta.shtmlを )。
- インターネットへのアクセスや、Webブラウザでパーソナルコンピュータ。
- 予測三次構造と機能部位を分析するための分子可視化ソフトウエア、たとえばRasMolはまたはpymolの、。
References
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H. CD-Search: protein domain annotations on the fly. Nucleic. Acids. Res. 32, W327-W331 (2004).
- Finn, R. D. The Pfam protein families database. Nucleic. Acids. Res. 38, D211-D222 (2010).
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request Permission