I - TASSER 파이프라인을 사용하여 단백질의 구조와 기능 특성화 기반 컴퓨터에 대한 지침이 설명되어 있습니다. 쿼리 단백질 시퀀스에서 시작, 3D 모델은 여러 스레딩 정렬을 사용하여 생성 및 구조 조립 시뮬레이션을 반복하고 있습니다. 기능성 inferences는 이후 알려진 구조와 기능을 가진 단백질 일치를 기반으로 그려져있다.
Method Article
컴퓨터 기반의 단백질 구조 및 기능 예측을위한 프로토콜
Opens in a new tab
In This Article
Summary
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
Abstract
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
게놈 시퀀싱 프로젝트는 생물 학적 역할에 대한 이해를 향상시키기 위해 자신의 구조와 기능에 대한 지식을 필요로 단백질 시퀀스의 수백만을 계산합니다. 실험 방법이 단백질의 작은 일부분에 대한 자세한 정보를 제공할 수 있지만, 전산 모델링을 실험적으로 uncharacterized 아르 단백질 분자의 대부분 필요합니다. I - TASSER 서버는 단백질 구조와 기능의 고해상도 모델링을위한 온라인 작업대입니다. 단백질 시퀀스 감안할 때, I - TASSER 서버에서 전형적인 출력, 보조 구조 예측을 포함 각 물질의 용매 접근성, 스레딩 및 구조 정렬에 의해 감지 동종 템플릿 단백질, 최대 다섯 전체 길이 차 구조 모델 및 구조 기반을 예측 효소 분류에 대한 주석 기능, 유전자 온톨로지 조건과 단백질 - 리간드 결합 사이트. 모든 예측은 신뢰 점수 태그가있는예측은 실험 데이터를 모르고하는 방법 정확하게 알려줍니다. 최종 사용자의 특별한 요청을 촉진하기 위해서, 서버는 사용자가 지정한 간 잔여 거리를 수락하고 대화형 I - TASSER 모델링을 변경하려면지도를 연락 채널을 제공하고, 또한 사용자가 템플릿으로 어떤 단백질을 지정할 수 있습니다, 또는 서식 파일을 제외 구조 조립 시뮬레이션 중에 단백질. 구조 정보는 I - TASSER 예측의 품질을 향상 목적으로 실험 증거 또는 생물 학적 지식을 바탕으로 사용자가 수집한 수 있습니다. 서버가 최근 사회 전체 CASP 실험에서 최고의 단백질 구조에 대한 프로그램 및 기능 예측으로 평가되었다. 온라인 I - TASSER 서버를 사용하는 100 개 이상의 나라에서> 20000 등록 과학자들은 현재이 있습니다.
Protocol
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
방법 개요
순서 - 투 - 구조 - 투 - 기능 패러다임에 따라 구조와 기능 모델링을위한 I - TASSER 절차 1-4의 연속 4 단계를 포함 : LOMETS 5 (A) 템플릿 식별, reassembly하여 복제 (B) 조각 구조 교환 몬테카를로 시뮬레이션 6, REMO 7 FG - MD 8을 사용 (C) 원자 수준의 구조 상세 및 (D) COFACTOR 9를 사용 구조 기반의 기능 해석.
템플릿 식별이 : 사용자가 제출한 쿼리 시퀀스 들어, 시퀀스가 먼저 메타 스레딩 서버를 로컬에 설치된 LOMETS에 의해 대표 PDB 구조 라이브러리를 통해 스레드입니다. 스레딩 비슷한 구조를 가지고 또는 쿼리 단백질과 비슷한 구조 모티브를 포함할 수 있습니다 템플릿 단백질을 식별에 사용되는 시퀀스 구조의 정렬 절차입니다. 동종 templ의 범위를 늘리려면먹고 탐지, LOMETS 다른 스레딩 방법을 다루는 여러 최첨단 알고리즘을 결합한 제품입니다. 다른 스레드 프로그램은 서로 다른 점수 시스템과 정렬 감성을 가지고 있기 때문에, 각 스레딩 프로그램에서 생성된 스레딩 정렬의 품질로 정의됩니다 정상화 Z - 점수로 평가됩니다 : 
Z - 점수는 프로그램에 의해 생성된 모든 정렬의 통계 의미에 대한 상대 표준 편차 단위로 점수가 어디에 있는지, 그리고 Z 0 '좋은을 차별화하는 데 대규모 스레딩 벤치 마크 테스트를 5 기준으로 결정하는 프로그램 - 특정 Z - 점수 차단됩니다 '와'나쁜 '템플릿. 높은 Z - 점수 템플릿 상단 템플릿은 일반적으로 정렬이 좋은 모델에 해당하는 것을 의미 다른 서식 파일, 대부분의보다 훨씬 높은 정렬 평가 점수를 의미합니다. 상단 스레딩 템플릿의 대부분은 안녕있다면정규 GH는 Z - 점수, 최종 I - TASSER 모델의 정확성은 일반적으로 높습니다. 그러나 단백질은 대형이고 스레딩 제휴의 범위는 쿼리 단백질의 작은 지역에 갇혀있다면, 높은 표준 Z - 점수는 반드시 전체 길이 모델 높은 모델링의 정확성을 의미하지는 않습니다. 각 스레딩 프로그램에서 상위 2 스레딩 정렬은 구조 어셈블리의 다음 단계에 대한 수집 및 사용하고 있습니다.
구조 조립 시뮬레이션을 반복 : 스레딩 절차에 따라 쿼리 시퀀스는 정렬과 unaligned 지역 스레딩로 나뉩니다. unaligned 루프 지역은 순이 론적 모델링에 의해 만들어진하는 동안 스레딩 정렬 지속 조각, 템플릿에서 excised 및 구조 어셈블리를 직접 사용됩니다. 구조 조립 절차는 복제 교환 몬테카를로 시뮬레이션 6 안내 격자 시스템에서 수행됩니다. I - TASSER 역장 수소 보를 포함상호 작용 10, PDB 11 SVMSEQ 12 시퀀스 기반의 연락처 예측 및 LOMETS 5 스레딩 템플릿에서 수집된 공간 억제의 알려진 단백질 구조에서 파생된 지식 기반 통계 에너지 조건을 nding. 시뮬레이션 동안 저온 복제에서 생성된 conformational 디코이 낮은 자유 에너지 상태의 구조를 파악하기 위해 SPICKER 13 클러스터된 수 있습니다. 상단 클러스터의 클러스터 centroids는 모든 클러스터 구조 디코의 3D 좌표를 평균하여 최종 모델 생성에 사용하여 얻을 수 있습니다. 시뮬레이션 및 클러스터링 절차는 steric 충돌을 제거하고 더욱 글로벌 토폴로지를 수정을 위해 두 번 반복됩니다.
원자 수준의 모델 구축 및 개선 : SPICKER 클러스터링 후 얻은 클러스터 centroids는 단백질 모델 (는 C의 α와 대량의 사이드 체인 센터 대표 각 물질)와 H를 감소거리를 제한 생물 학적 응용 프로그램입니다. 감소 모델의 전체 원자 모델의 건설은 두 단계로 이루어집니다. 첫 번째 단계에서, REMO 7 H - 결합 네트워크를 최적화하여 C - 알파 성분의 전체 원자 모델을 만드는 데 사용됩니다. 두 번째 단계에서, REMO 전체 원자 모델 추가로 백본 비틀림 각도, 본드 길이와 사이드 체인 rotamer 오리 엔테이션을 향상 FG - MD 14,에 의해 정제되며, 분자 동적 시뮬레이션에 의해,로에서 검색한 구조 파편에 의해 안내 에 의해 PDB 구조 TM 번 맞춥니다. FG - MD 세련된 모델은 I - TASSER로 차 구조 예측을위한 최종 모델로 사용됩니다.
생성된 모델의 품질은 LOMETS 스레딩 정렬의 Z - 점수와 I - TASSER 시뮬레이션의 융합, 수학적으로 공식화에 따라 정의된 신뢰 점수 (C - 점수)을 기준으로 예상됩니다 
어디에 13 식별 구조 클러스터의 구조 디코의 다중성이며 M 더하다는 클러스터링에 제출된 디코의 총계입니다; 클러스터 centroids에 클러스터된 디코의 평균 RMSD이며 Norm.Z - 점수는 (i) 나는 LOMETS 5 서버를 스레딩 세기에서 얻은 상단 스레딩 정렬의 표준 Z - 점수 (Eq. 1)이며 N은 LOMETS에 사용되는 서버의 개수입니다.
C - 점수는 I - TASSER 모델의 품질과 강한 상관 관계가 있습니다. C - 점수와 단백질의 길이를 결합함으로써, 최초로 I - TASSER 모델의 정확성은 TM - 점수와 RMSD 15 2에 대한 0.08의 평균 오류가 예상됩니다. 일반적으로 C - 점수>와 모델 - 1.5은 올바른 이길 것으로 예상된다. 여기 RMSD 및 TM - 점수는 모두 모델과 기본 구조 사이의 topological 유사성 잘 알려져 조치하고 있습니다. TM - 점수 valu[0, 1]에서 알면서도 범위, 높은 점수가 더 나은 구조는 16,17 일치 나타냅니다. 그러나 낮은 순위 모델 (즉, 2 차 -5 일 모델)에 대한, TM - 점수와 RMSD로 C - 점수의 상관 관계가 훨씬 약한입니다 (~ 0.5), 그리고 것은 절대 모델 품질의 신뢰성 평가를 위해 사용할 수 없습니다.
첫번째 모델은 항상 I - TASSER 시뮬레이션에서 최고의 모델인가? 이 질문에 대한 답변은 대상 유형에 따라 다릅니다. 쉽게 목표에 대한, 첫 번째 모델은 일반적으로 최고의 모델이며 C - 점수는 일반적으로 모델의 나머지 부분보다 훨씬 높다. 그러나, 스레드는 상당한 템플릿 히트가없는 하드 타겟 들어, 첫 번째 모델은 반드시 최고의 모델이 아니므로 I - TASSER 사실은 최고의 템플릿과 모델을 선택하는 어려움이있다. 그것은 따라서 하드 타겟에 대한 모든 5 모델을 분석하고 그들 실험 정보 및 생물 학적 지식을 바탕으로 선택하는 것이 좋습니다.
기능 predictions : 마지막 단계에서, FG - MD에서 발생하는 최종 3 차원 모델은 즉, 단백질 기능의 세 측면을 예측하는 데 사용됩니다 :) 효소위원회 (EC) 번호 18 (B) 유전자 온톨로지 (GO) 19 용어와 ( C) 작은 분자 리간드에 대한 바인딩 사이트. 모두 세 가지 측면 들어, 기능 해석은 알려진 구조와 기능 PDB의 템플릿 단백질에 글로벌 및 로컬 유사성에 따라 단백질의 기능을 예측하는 새로운 접근이다 COFACTOR를 사용하여 생성됩니다. 첫째, 예측 모델의 글로벌 토폴로지는 20 TM은 - 정렬 구조 정렬 프로그램을 사용하여 기능 템플릿 라이브러리에 대해 일치하는 것입니다. 다음 대상 모델에 가장 유사한 단백질의 집합들은 글로벌 구조 유사성을 기반으로 라이브러리에서 선택, 그리고 광범위한 지역 정보 검색은 바인딩 / 활성 사이트 지역 근처의 구조와 순서 유사성을 식별하는 수행됩니다. 결과 글로벌 및 로컬 유사성 점수는 순위를하는 데 사용됩니다템플릿 단백질 (기능 homologues)와 최고 점수 히트에 따라 주석을 (EC 번호와 유전자 온톨로지 19 조항) 전송. 마찬가지로, 리간드 바인딩 사이트 잔류물과 리간드 바인딩 모드는 최고 점수 함수 템플릿 9 알려진 리간드 바인딩 사이트 잔류물과 쿼리의 로컬 정렬 기반으로 유추됩니다.
기능의 품질 (EC와 GO 용어)는 I - TASSER의 예측은 쿼리와 템플릿 간의 세계와 지역 유사성을 측정하기위한 한 방법입니다 상동 기능 점수 (FH - 점수)에 따라 평가하고,로 정의됩니다 : 
C - 점수는 EQ에 정의된 예측 모델의 품질 추정치입니다. (2) TM - 점수는 모델과 템플릿 단백질 사이의 구조적 유사성을 글로벌 대책, RMSD 알리 TM - 정렬 20 구조적으로 정렬 영역의 모델과 템플릿 구조 간의 RMSD입니다; Cov은 구조적 배열 (즉, 쿼리의 길이로 나눈 구조적으로 정렬 잔류물의 비율)의 범위를 나타냅니다; ID 알리 TM - 정렬 정렬의 순서 정체성이다. EC 번호 예측에 대한 예상 신뢰 점수는 또한 계산 정의된 로컬 지역 내의 쿼리와 템플릿 간의 적극적인 사이트 검색 (ACM)를 평가하기위한 용어를 포함 : 
N t이 지역 내에서 현재 템플릿 잔류물의 수를 나타냅니다 여기서 N 알리가 정렬 쿼리 템플릿 잔여물 쌍 번호입니다, D II가 정렬 잔류의 I 번째 쌍 사이의 C α 거리이며, D 0 = 3.0입니다 거리의 단절은, M II는 정렬 잔류의 ith 쌍 사이의 BLOSUM 점수입니다. 일반적으로 FH - 점수의 범위에 [0, 5]와 ACM의 점수는 [사이의 0, 2] 어디에 더 자신감이 기능 할당을 나타내는 높은 점수. ACM 점수도 지역 구조와 BS - 점수로 언급됩니다 리간드 바인딩 사이트, 근처 시퀀스 유사를 평가하는 데 사용됩니다.
1. 단백질 시퀀스 제출
- 에서 I - TASSER 웹 페이지 방문 http://zhanglab.ccmb.med.umich.edu/I-TASSER 구조와 기능 모델링 실험을 시작합니다.
- 복사 제공된 양식에 아미노산 시퀀스를 붙여넣거나 직접 "찾아보기"버튼을 클릭하여 컴퓨터에서 업로드할 수 있습니다. I - TASSER 서버는 현재까지 1,500 잔류물을 가진 시퀀스를 사용할 수 있습니다. 1500 잔류물 이상 단백질은 일반적으로 다중 도메인 단백질이며, I - TASSER에 제출하기 전에 개별 도메인으로 분할하는 것이 좋습니다.
- 이메일 주소 (필수) 및 작업 (옵션)에 대한 이름을 제공합니다.
- 사용자는 선택적으로 외부 상호 해상도를 지정할 수 있습니다idue 연락처 / 거리 지지대, 추가 기능에 추가 템플릿이나 구조 모델링 과정에서 몇 가지 템플릿 단백질을 제외할 수 있습니다. "토론"섹션에서 이러한 옵션을 사용에 대해 자세히 알아보십시오.
- 순서를 제출하려면, "실행 I - TASSER '버튼을 클릭하십시오. 브라우저는 사용자 지정 정보, 직업 식별 (작업 ID) 번호와 결과를 작업의 완료 후에 입금됩니다 웹페이지에 대한 링크를 표시하는 확인 페이지로 이동합니다. 사용자는이 링크를 북마크하거나 나중에 참조할 수 있도록 작업 식별 번호를 메모 수 있습니다.
2. 결과의 가용성
- 에서 I - TASSER 대기열 페이지 방문하여 제출한 작업의 상태를 확인 http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php을 . 검색 탭을 클릭하고 작업 ID 번호 또는 제출된 작업을 검색하는 쿼리 순서를 사용합니다.
- 구조와 기능 MO 후deling가 완료되면, 예측 구조와 웹 링크의 통지 전자 우편이 포함된 이미지는 귀하에게 발송됩니다. 이 링크를 클릭하거나 결과를 확인하고 다운로드하는 단계 1.5에서 책갈피 링크를 엽니다.
3. 차 구조 및 용매 접근성 예측
- 결과 페이지 상단에 표시되는 FASTA 형식의 쿼리 순서를 확인합니다. 추가 구속 / 템플릿 시퀀스 제출, 사용자가 지정한 정보를 표시 웹 페이지에 대한 링크시 지정한 경우에는 (그림 1A) 볼 수 있습니다.
- 알파 나선 구조 (H), 베타 좌초 (S) 또는 코일 (C) 각 잔여물 예측의 신뢰 점수 (0 = 낮음, 9 = 높음)로 표시 보조 구조 예측을 검사합니다. 단백질의 코어 영역을 평가하기 위해 정기적으로 보조 구조의 길이 펼쳐진 (H 또는 S) 예측과 함께 지역보세요. 단백질의 구조 클래스도 분석 보조 구조 요소의 분포에 따라 수 있습니다. 알그래서, 단백질의 코일 요소의 긴 지역은 일반적으로 구조화 / 무질서 지역을 나타냅니다.
- 쿼리에서 확인할 묻힌 및 솔벤트 노출 지역에 대한 예측 용매 접근성 (그림 1C)을 볼 수 있습니다. 의 값은 0 (묻혀 잔여물)에서 9 (노출 잔여물)을 용매 접근 범위를 예측했다. 솔벤트 노출 및 친수성 잔류물과 지역 잠재 수화 / 기능성 사이트에있는 동안 거의 묻혀 잔류물을 포함하는 지역은, 단백질의 핵심 지역을 윤곽을 그리다하는 데 사용할 수 있습니다.
4. 차 구조 예측
- 대화형 Jmol 애플릿 (그림 2)에 표시되는 쿼리 단백질의 구조 예측 차를 보려면 아래로 스크롤합니다. 표시되는 구조의 모양을 변경할 수있는 애플릿을 클릭하여 왼쪽, 특정 지역으로 확대가 예측 모델의 특정 잔여물 유형을 선택하거나 간 잔여 거리를 계산합니다.
- 긴 구조화되지 않은 영역의 존재에 대한 모델을 분석할 수 있습니다. 이 Regions는 보통 단백질 무질서 지역에 해당하는 또는 템플릿 정렬 부족을 나타냅니다. 이 지역은 일반적으로 N & 지역 모델링의 정확도를 향상시킬 C - 말단에서 모델링하는 동안 낮은 모델링 정확성과이 지역을 제거있다.
- "다운로드 모델"링크를 클릭하여 모델의 PDB 형식의 구조 파일을 다운로드합니다. 당신은 구조 기능의 추가 분석을위한 분자 시각화 소프트웨어 (예 : Pymol, Rasmol 등)에서 해당 파일을 열 수 있습니다.
- 예측 구조의 품질을 평가하는 구조 모델링의 신뢰 점수 (C - 점수)를 분석할 수 있습니다. C - 점수 (Eq. 2) 값이 범위에 일반적으로 아르 [-5, 2], 궁극적는 높은 점수는 더 나은 품질의 모델을 반영합니다. 예상 TM - 점수와 첫번째 모델 RMSD는 "모델 1 예상 정확도"로 표시됩니다. TM - 점수 RMSD보다 topological 변화에 더 민감 한 단백질에 대한, 그것은, TM - 점수에 따라 모델의 품질을 평가하는 것이 좋습니다. < 리> C - 점수를 분석 링크 클러스터 크기와 모든 모델의 클러스터 밀도 "C - 점수에 대한 자세한 내용"을 클릭합니다. 낮은 순위 모델의 C - 점수가 강하게 TM - 점수 또는 RMSD와 상관 없기 때문에 TM - 점수와 RMSD은, 첫 번째 I - TASSER 모델 제공됩니다 추산했다. 낮은 순위 모델의 품질은 부분적으로 가까이 기본 구조에 평균에있는 더 큰 클러스터와 높은 밀도의 모델이 궁극적으로 자신의 클러스터 밀도와 첫 번째 모델에 비해 클러스터 크기에 따라 평가하실 수 있습니다.
- 낮은 C - 점수 예측은 일반적으로 낮은 정확도 예측을 나타냅니다. 대부분 이러한 경우에는 쿼리 단백질은 도서관에서 좋은 템플릿을 부족하고 순이 론적 모델링 (예> 120 잔류물)의 범위를 넘는 크기를하고 있습니다. 이러한 경우에는, 사용자는 추가적인 공간적 구속에 대한 추구와 I - TASSER 모델링을 (토론 섹션을 참조) 개선하기 위해 그들을 사용할 수 있습니다. 또한 (우리 QUARK 서버에 시퀀스를 제출 권장합니다QUARK /는 "> http://zhanglab.ccmb.med.umich.edu/QUARK/) 순수한 순이 론적 모델링에 대한 단백질의 크기는 아래 200 잔류물 경우.
5. LOMETS 대상 템플릿 정렬
- 로 LOMETS 스레딩 프로그램 (그림 3)에 의해 식별 쿼리 단백질의 10 스레딩 템플릿을 분석 아래로 스크롤합니다. '규격에 표시된 표준 Z - 점수 (Eq. 1), 볼 수 있습니다. 스레딩 정렬의 품질을 분석하는 Z - 점수 '항목. 정규화된 Z - 점수> 1 정렬은 자신감 정렬을 반영하고 대부분의 쿼리 단백질과 같은 접을 수 있습니다.
- 스레딩 - 정렬 영역 (열 'Iden 1.')와 쿼리와 템플릿 단백질 간의 상동을 평가하기 위해 전체 체인 (열 'Iden. 2')에 대한 시퀀스 ID를 분석합니다. 높은 시퀀스 ID는 쿼리 및 템플릿 단백질 사이의 진화 relatedness의 지표입니다.
- 시각적으로 죄수를 식별하는 색깔에 표시되는 스레딩 정렬 잔류물보기erved 잔류물 / 쿼리 및 템플릿 단백질의 모티브. 전체 체인 정렬에 비해 스레딩 - 정렬 영역에서 높은 시퀀스 ID는 또한 쿼리에 보존되어 구조적 모티프 / 도메인의 존재를 나타냅니다.
- 확인하여 스레딩 정렬의 범위를 평가 'Cov합니다.' 컬럼과 정렬을 점검. 상단 정렬의 범위가 낮은 쿼리 단백질이나 쿼리 시퀀스의 긴 구간에 대한 결석만이 작은 영역에 국한되어있다면, 쿼리 단백질은 일반적으로 하나 이상의 도메인을 포함하고 이것은 순서와 모델을 분할하는 것이 좋습니다 개별적으로 도메인 (그림 3).
- 클릭하여 PDB 포맷 시퀀스 구조 정렬 파일을 다운로드 링크를 "다운로드하면 정렬". 이러한 정렬 파일은 자료 섹션에 나열된 모든 분자 시각화 프로그램에서 열 수 있으며, 또한 구조 모델링 (단계 1.4) 기간 동안 추가적인 제한을 추가하는 데 사용할 수 있습니다.
6.PDB에 구조 analogs
- 20 TM - 정렬 구조 정렬 프로그램으로 식별 첫 번째 예측 모델의 상위 10 analogs 구조를 결정하기 위해 결과 페이지의 다음 표 (그림 4)을 참조하십시오. TM - 점수> 0.5은 TM - 점수 이들은 <0.3은 임의의 구조 유사성을 의미하는 동안 감지된 아날로그 및 모델, 유사한 토폴로지를 가지고 쿼리 단백질 16 구조 클래스 / 단백질 가족을 결정하는 데 사용할 수있는 것을 나타냅니다.
- 순서 정체성을 분석하고 구조적으로 정렬된 지역 RMSD가 표시된 'IDEN'와 컬럼 모델의 공간 작품의 보존과 구조 아날로그을 평가하는 'RMSD'을 선택하십시오. 시각이 구조적으로 보존 잔류물과 모티브를 식별하는 정렬의 컬러와 정렬된 쌍을 잔류물을 검사합니다.
- 그들의 구조적 분류 (SCOP에 대한 RCSB 웹사이트를 방문 하셔서 자세한 내용을 보려면 'PDB 조회'열에 표시된 PDB 코드를 클릭하십시오, 선배와 PFAM) 및 이용 바운드 리간드로 이동 관련된 기능 정보 (EC 번호).
7. COFACTOR를 사용하여 기능 예측
- 검색어 단백질에 대한 기능 해석을 분석할 결과 페이지에서 아래로 스크롤합니다. 효소위원회 (EC) 번호, 유전자 온톨로지 (GO) 조건, 그리고 리간드 바인딩 사이트 : 단백질 기능이 표시, 세 컨텍스트 테이블에 열거됩니다.
- 'TM - 점수를'보기 'IDEN'와 'RMSD'Cov을. ' 각 테이블의 열은 세계 구조와 유사하고 모델과 식별 기능 homologues (템플릿) 사이의 공간 패턴의 보존의 매개 변수를 분석합니다.
8. 효소위원회 번호 예측
- 에 표시된 쿼리 단백질의 5 대 잠재 효소 homologues보기는 테이블 (그림 5) "EC 번호를 예상". 이러한 템플릿을 사용하여 EC 번호 예측의 신뢰 수준은 'EC - 점수'항목에 표시됩니다. benchma에 따라분석 23, 쿼리 및 템플릿 단백질 사이의 기능적 유사성 (EC 번호의 처음 3 자리)를 rking 것은 안정 EC - 점수> 1.1을 사용 해석될 수 있습니다.
- 검색어 단백질로 유사한 접어 (예 : TM - 점수> 0.5)이 템플릿 중에 함수의 합의 (EC 번호)를 찾아보십시오. 여러 템플릿 같은 EC 번호, EC - 점수> 1.1이있다면, 예측의 신뢰 수준은 매우 높습니다. 그러나, EC - 점수가 높은지만 확인 히트곡 가운데 공감대의 부족이있다면, 다음 예언은 덜 안정적인되고 사용자는 GO 용어 예측을 참조하시기 바랍니다.
- 자세히 템플릿 단백질의 반응 촉매, 공동 요소 요구 사항 및 신진 대사 경로를 포함하여 ExPASy 효소 데이터베이스를 방문하여 기능을 분석하기 위해 EC 번호에 제공된 링크, 클릭하십시오.
9. 유전자 온톨로지 (GO) 기간 예측
- "용어를 GO 예상"보기 테이블 (그림ure 6) 유전자 온톨로지 (GO) 조건 주석 PDB 도서관에서 쿼리 단백질의 10 대 homologues를 식별합니다. 각 단백질은 보통의 분자 기능 (MF), 생물 학적 프로세스 (BP)과 세포 구성 요소 (CC)를 설명하는 여러 GO 용어와 연결되어 있습니다. 아미고 웹 사이트를 방문하고 정의와 혈통을 분석하기 위해 각 용어를 클릭하십시오.
- 쿼리 및 템플릿 단백질 사이의 기능적 유사성에 액세스하고 이러한 단백질의 기능 주석 전송의 신뢰 수준을 추정하는 FH - 점수 (기능 상동 점수) 컬럼을 분석할 수 있습니다. 우리의 벤치마킹 연구 23, 기본 GO 용어의 50 %가 제대로 56%의 전체 정확도와 0.8의 FH - 점수 컷오프를 사용하여 최초로 확인된 템플릿에서 확인할 수 있습니다.
- 템플릿 간의 기능의 동시를 분석하기 위해 "GO 용어의 합의 예측"이 테이블을 볼 수 있습니다. 이러한 일반적인 기능은 쿼리 GO 용어 (MF, BP와 CC) 예측에 사용됩니다단백질과 장기 예측을 GO의 신뢰 수준 (GO - 점수)를 평가합니다. 벤치마킹 테스트 23을 기반으로 최고의 거짓 긍정과 거짓 부정적인 속도는 깊이 온톨로지 수준에서 예측의 범위를 감소와 함께, GO - 점수 컷오프과 예측 = 0.5에 대해 얻을 수 있습니다.
10. 단백질 - 리간드 결합 사이트 예측
- 검색어 단백질에 대한 상위 10 리간드 바인딩 사이트 예측을 볼 수있는 페이지의 하단으로 스크롤합니다. 예상 바인딩 사이트는 일반적인 바인딩 주머니를 공유 예측 리간드 conformations의 숫자에 따라 순위가 결정됩니다. 최고의 식별 바인딩 사이트는 이미 Jmol 애플릿에 표시됩니다. 다른 예측을 분석하고 리간드 상호 작용의 잔류물을 시각화하기 위해 라디오 버튼을 클릭하십시오.
- 모델과 템플릿의 구속력이 사이트 사이의 지역 유사성을 평가하기 위해 BS - 점수 컬럼을 분석합니다. 벤치 마크 9을 기반으로, BS - 점수는> 1.1 높은 순서와 구조 SIM을 나타냅니다모델에서 예측 구속력 사이트와 템플릿에 알려진 바인딩 기지 근처 ilarity.
- '다운로드'링크를 클릭하여 단지의 PDB 형식의 구조 파일을 다운로드합니다. 사용자는 어떤 분자 시각화 프로그램에서 이러한 파일을 열고 대화형 예상 구속력 사이트와 자신의 로컬 컴퓨터에 리간드 - 단백질 상호 작용을 볼 수 있습니다.
11. 대표 결과

그림 1은 I - TASSER 결과 페이지의 발췌가 게재 (A) FASTA는 쿼리 순서를 포맷;. (B) 이차 구조와 관련된 신뢰 점수를 예측하며, (C) 잔류 용매의 접근을 예측했다. 분석 핵심 지역 및 쿼리에 잠재적인 수화 사이트는 각각 시안과 붉은 사각형으로 강조 표시됩니다.

그림 2.

그림 3. LOMETS 5 스레딩 프로그램에 의해 10 대 식별 스레딩 템플릿과 제휴를 보여주는 I - TASSER 결과 페이지의 예. 스레딩 정렬의 품질은 가치> 1 자신감 정렬을 반영하는 정규화된 Z - 점수 (녹색으로 강조 표시)에 따라 평가됩니다. 해당 쿼리 잔류물에 동일 템플릿에 정렬된 잔류물은 보존 잔여물 / 모티브의 존재를 나타내기 위해 색상으로 강조하는 동안 대부분의 정렬의 부족 상단 템플릿 쿼리 단백질과 unaligned 잔류물 여러 도메인의 존재가 도메인 링커 지역에 해당 나타냅니다. 그림 3의 전체 크기 버전을 보려면 여기를 클릭하십시오.
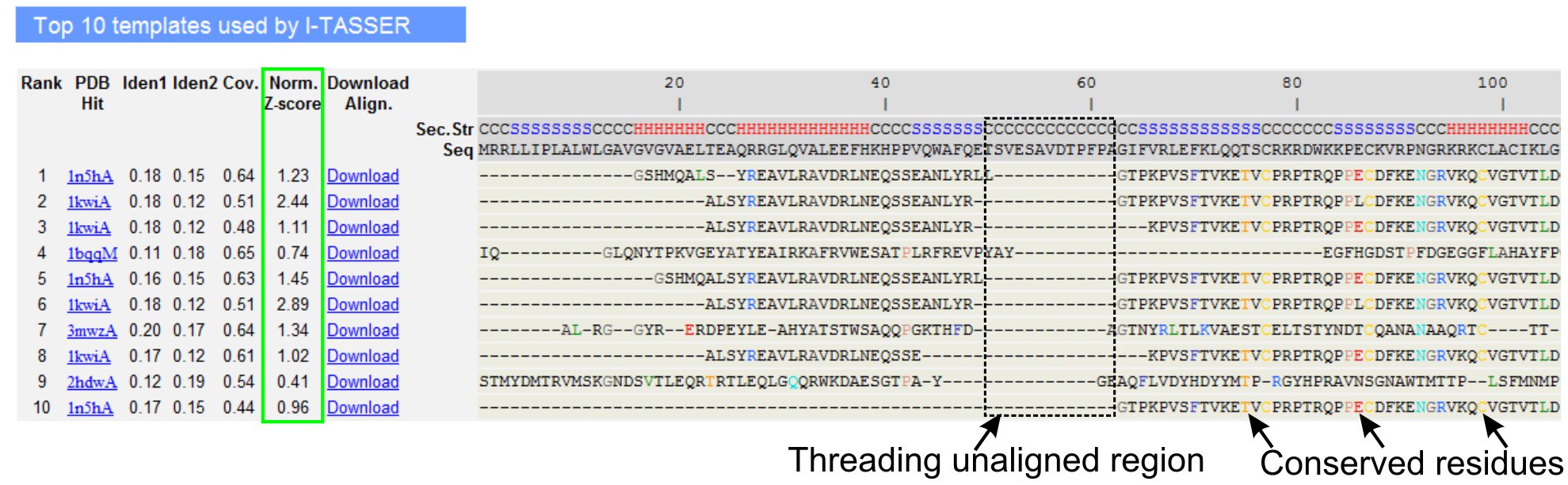{kind=link}

그림 4. 식별 10 대 식별 구조 analogs 및 구조 정렬, 보여주는 결과 페이지의 예를 들면 20 구조적 정렬 프로그램을 TM 번 맞춥니다. 에 표시된 analogs의 순위는 구조 정렬의 TM - 점수 (파란색으로 강조 표시)을 기반으로합니다. TM - 점수 <0.3 두 개의 임의의 구조 사이의 유사성을 의미하는 동안 TM - 점수> 0.5은 두 비해 구조가 유사한 토폴로지를 나타냅니다. "-"unaligned 지역에 의해 표시되는 동안 구조적으로 정렬된 잔기 쌍은 그들의 아미노 산 속성에 따라 색상으로 강조 표시됩니다.ove.com/files/ftp_upload/3259/3259fig4large.jpg "> 그림 4의 전체 크기 버전을 보려면 여기를 클릭하십시오.

그림 5. PDB 도서관에서 검색어 단백질의 식별 효소 homologues를 보여주는 I - TASSER 결과 페이지의 예. EC 번호 예측의 신뢰 수준은 EC - 점수> 1.1 쿼리 템플릿 단백질 사이의 기능적 유사성 (EC 번호의 동일한 처음 3 자리)를 나타냅니다 EC - 점수 (녹색으로 강조 표시)를 기반으로 분석됩니다.

그림 6. 보여주는 I - TASSER 결과 페이지의 예제는 쿼리 단백질에 대한 장기 예측을 이동합니다. 유전자 온톨로지 템플릿 라이브러리에서 쿼리 단백질에 대한 기능 homologues들은 FH - 점수 (오렌지 사각형)에 따라 순위가 결정됩니다. 이러한 최고 점수 조회에서 공통 기능 기능은 gener으로 파생됩니다 검색어 단백질의 최종 GO 용어 예측을 먹었어요. 예측 GO 용어의 품질은 예상 GO - 점수 (녹색 표시), GO - 점수> 0.5은 신뢰할 수있는 예측을 나타냅니다.을 기반으로 그림 6의 전체 크기 버전을 보려면 여기를 클릭하십시오.
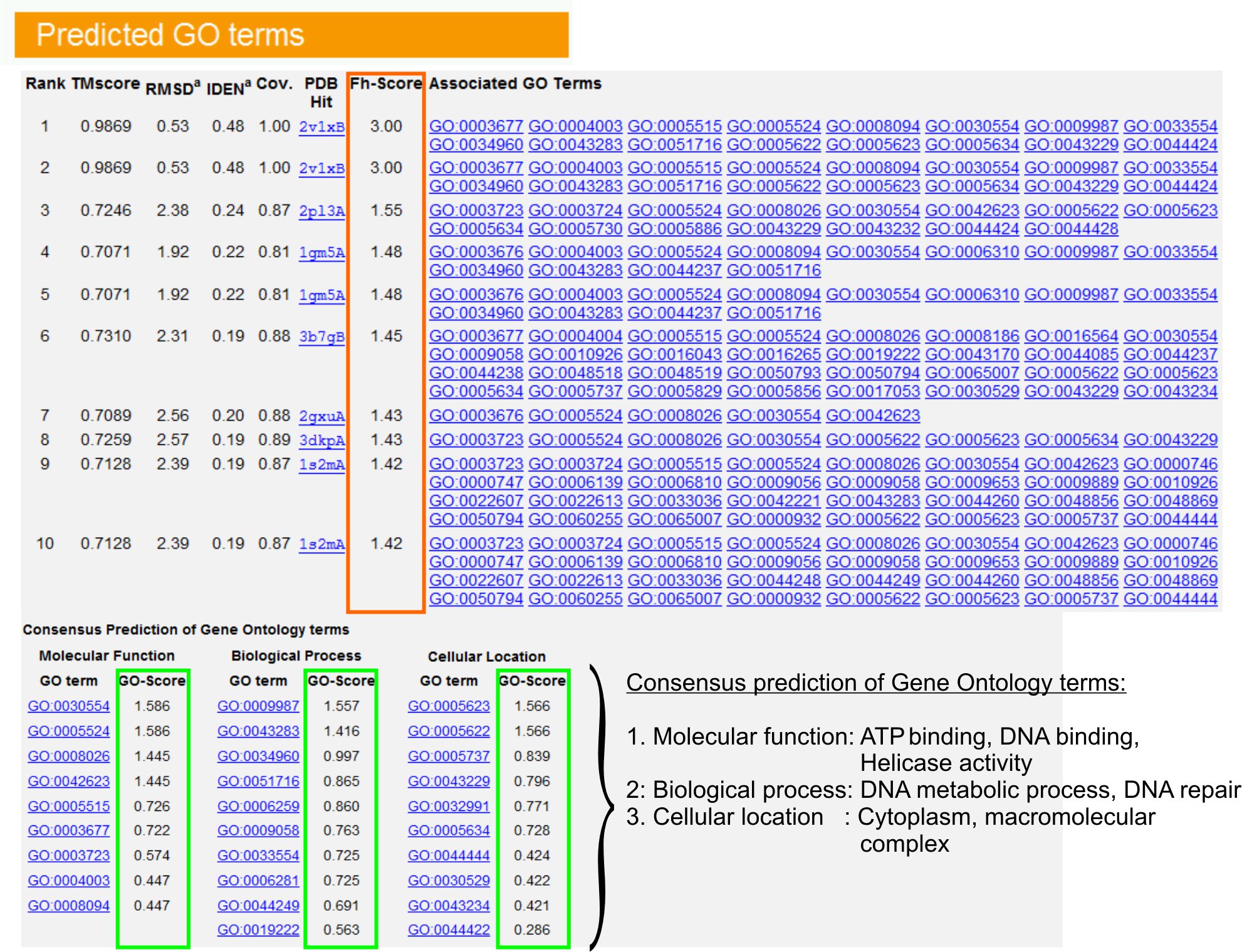{kind=link}

그림 7. COFACTOR 9 알고리즘을 사용하여 상위 10 단백질 리간드 바인딩 사이트 예측을 보여주는 I - TASSER 결과 페이지의 예. 예측 바인딩 사이트의 순위는 쿼리의 일반적인 구속력 주머니를 공유 예측 리간드 conformations의 숫자에 따라 달라집니다. BS - 점수 (빨간색으로 강조 표시) 지역의 순서와 예측하고 템플릿의 구속력 사이트 간의 구조 유사성을 측정하기위한 한 방법입니다, 그리고 바인딩 사이트 주머니의 절약을 분석하는 데 유용합니다.
les/ftp_upload/3259/3259fig8.jpg "/>
그림 8. 잔류 - 잔류 문의 / 거리 제한을 지정하는 데 사용되는 외부 구속 파일의 예입니다.

그림 9. I - TASSER 서버 템플릿 단백질을 지정에 사용되는 구속 파일의 예. 또는 (B) 3D 형식, 사용자 (A) FASTA 형식 중 쿼리 템플릿 정렬을 지정할 수 있습니다.

그림 10. I - TASSER 구조 모델링 절차 중에 템플릿을 제외하고 사용 예제 파일입니다. 첫 번째 열에는 제외하는 템플릿 단백질의 PDB ID를 포함하고 있습니다. 두 번째 열은 템플릿 라이브러리에있는 다른 유사한 템플릿에 사용되는 시퀀스 신분 컷오프를 지정하는 데 사용됩니다.
Discussion
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
위에 제시 프로토콜 구조와 I - TASSER 서버를 사용하여 함수 모델링을위한 일반적인 지침입니다. 이 자동화된 절차는 단백질의 대부분 잘 작동하지만, 인간의 개입은 종종 특히 PDB 라이브러리에 가까운 템플릿 부족한 단백질을 위해, 크게 모델링의 정확도를 개선하는 데 도움이됩니다. (B) 구조 어셈블리를 개선하기 위해 외부 구속을 제공,, 멀티 도메인 단백질 (A) 분리 및 (C) 모델링하는 동안 템플릿을 제거 : 사용자는 다음과 같은 방법으로 I - TASSER 모델링하는 동안 개입하실 수 있습니다.
분할 다중 도메인 단백질 :
오랜 단백질 시퀀스가 자주 실험과 전산 두 기술을 사용하여 구조 해설 어렵게 만드는 유연 링커 지역에 의해 곁에 여러 도메인을 포함합니다. 도메인 독립적으로 엔티티를 접는하고 독특한 분자 기능을 수행할 수있는 그럼에도 불구하고, 그것은이다별도로 각 도메인 긴 다중 도메인 단백질과 모델을 분할하는 것이 바람직하다. 모델링 도메인은 개별적에만 예측 프로세스 속도를하지 않을뿐만 아니라, 쿼리 템플릿 정렬의 품질보다 신뢰할 수있는 구조와 기능 예측의 결과로, 증가합니다.
단백질 시퀀스에서 도메인 경계는 그러한 NCBI CDD 24 PFAM 25 InterProScan 26로 자유롭게 사용할 외부 프로그램을 사용하여 온라인으로 예측할 수 있습니다. LOMETS 스레딩 정렬이 쿼리 단백질에 사용할 수있는 경우 또한, 도메인 경계는 시각 (단계 5.4 참조) 상단 스레딩 템플릿 unaligned 잔류 긴 펼쳐진을 식별하여 찾을 수 있습니다. 이러한 unaligned 지역은 대부분 도메인 링커 지역에 해당합니다. 다중 도메인 템플릿 정렬 모든 쿼리 도메인으로 템플릿 PDB 라이브러리에 이미있는 경우 다음 쿼리 단백질은 전체 길이로 모델 수 있습니다.
외부 구속을 제공
I - TASSER의 구조 조립 시뮬레이션은 주로 템플릿을 스레딩 LOMETS에서 수집한 공간적 구속하여 안내하고 있습니다. 템플릿 라이브러리에 좋은 스레딩 히트 (Norm. Z - 점수> 1)이 쿼리 단백질 들어, 파생된 공간 억제는 높은 정확도의 대부분이며, I - TASSER이 단백질에 대한 고해상도 구조 모델을 생성합니다. Contrarily, 약한가 쿼리 단백질 또는 전혀 스레딩 히트 (Norm. Z - 점수 <1), 수집 공간 억제는 종종 때문에 템플릿과 정렬의 불확실성의 오류가 포함되어 있습니다. 이러한 단백질 타겟 들어, 사용자가 지정한 공간 정보는 예측 모델의 품질을 향상시키기 위해 매우 도움이 될 수 있습니다. 사용자는 두 가지 방법으로 I - TASSER 서버로 외부 구속을 제공할 수 있습니다 :A. 연락처 / 거리 제한을 지정
실험적 특징 상호 잔여물 연락처 / 거리, NMR의 예 또는가교 실험을 구속 파일을 업로드하여 지정할 수 있습니다. 예를 들어 파일을 열 1 규제의 종류를 지정합니다 그림 8에 표시됩니다 "DIST"또는 "접촉"즉. 거리 구속에 대한 (DIST), 컬럼 2, 4 잔류 위치를 (I, J)를 포함, 컬럼 3, 5 두 지정한 원자 사이의 거리를 지정 잔여물 및 열 6 원자 유형을 포함합니다. 문의 묶기 위해 (연락처), 컬럼 2, 3 연락해야 잔류물의 위치를 (I, J)가 포함되어 있습니다. 이러한 접촉 잔여 쌍 측면 체인 센터 사이의 거리는 PDB 알려진 구조에서 관찰 거리에 따라 결정됩니다. I - TASSER의 구조 상세 시뮬레이션 동안 지정된 거리에 가까운 이러한 원자 쌍을 그릴려고합니다.
B.는 단백질 구조 템플릿을 지정
LOMETS 스레딩 프로그램은 쿼리 제자에 대한 그럴듯한 주름을 찾을 대표 PDB 라이브러리를 사용EIN. 대표적인 구조 라이브러리를 사용하면 시퀀스 구조의 정렬을 계산하는 데 필요한 시간을 줄이기 위해 도움이되지만 그것이하더라도, 그것은 좋은 템플릿 단백질이 도서관에서보고 있거나 템플릿 LOMETS 스레딩 프로그램에서 확인되지 않았을 수도 있습니다 도서관에 존재. 이러한 경우에는 사용자는 템플릿으로 원하는 단백질 구조를 지정해야합니다.
추가 템플릿으로 단백질 구조를 지정하려면, 사용자는 하나 PDB 형식의 구조 파일을 업로드하거나 PDB 라이브러리에 쌓인 단백질 구조의 PDB ID를 지정할 수 있습니다. I - TASSER이 소집 프로그램 23을 사용하여 쿼리 템플릿 정렬을 생성하고 지정된 사용자 템플릿과 구조 조립 시뮬레이션을 안내 LOMETS 템플릿 모두에서 공간적 구속을 수집합니다. LOMETS 묶기의 정확성이 다른 대상에 대해 서로 다른이기 때문에 LOMETS 묶기의 무게는 쉬운 (상동) 타에 강해체계적으로 우리의 벤치 마크 훈련 조정되었습니다 하드 (비 상동) 목표에보다 rgets.
사용자는 자신의 쿼리 템플릿 정렬을 지정할 수 있습니다. FASTA 형식 (그림 9A)와 3D 형식 (그림 9B) : 서버는 두 형식의 정렬을 허용합니다. FASTA 형식은 표준에서 설명 http://zhanglab. ccmb.med.umich.edu / FASTA / . 3D 형식은 표준 PDB 포맷 (유사합니다 http://www.wwpdb.org/documentation/format32/sect9.html )하지만 템플릿에서 파생된 두 개의 추가 열이 원자 기록 (그림 9B 참조)에 추가됩니다 :
열 10-30 : 아톰 (단 C - 알파)와 잔여물 질의 순서에 대한 이름입니다.
열 31-54 : 템플릿에 해당 원자에서 복사한 쿼리의 C - 알파 원자 좌표.
열 55-59 : 대응 잔여물 번호를 템플릿에이 배열을 기반으로
열 60-64 : 대응 잔여물 이름 템플릿에
템플릿 단백질을 제외
단백질은 유연한 분자 및 생물 학적 활동을 변경하는 여러 conformational 상태를 채택할 수 있습니다. 예를 들어, 많은 단백질 kinases 및 막 단백질의 구조 활성 및 비활성 두 형태에서 해결되었습니다. 또한 행 리간드의 존재 또는 부재는 큰 구조적 움직임을 일으킬 수 있습니다. 템플릿의 모든 conformational 상태는 스레딩 프로그램에 똑같지만, 그것은 하나의 특정 상태에있는 템플릿을 사용하여 쿼리를 모델하는 것이 바람직하다. 서버에 새로운 옵션은 사용자가 구조를 모델링하는 동안 템플릿 단백질을 제외 수 있습니다. 이 기능은 또한 사용자가 모델링에 사용되는 템플릿의 상동 수준을 선택할 수 있도록합니다. 사용자는 템플릿 단백질 천을 제외시킬 수 있습니다톰 I - TASSER 도서관으로 :
A.이 시퀀스 신분 컷오프 지정
사용자는 I - TASSER 템플릿 라이브러리에서 상동 단백질을 제외하려면이 옵션을 사용할 수 있습니다. 상동 수준이 시퀀스 신분 컷오프에 따라 설정, 쿼리 및 쿼리 순서의 시퀀스 길이로 나눈 템플릿 단백질 사이의 동일 물질의 수를 즉. 예를 들어, 제공되는 양식에 "70%"의 사용자 유형,> 70 %를 쿼리 단백질 내가 - 것입니다 I - TASSER 템플릿 라이브러리에서 제외 될 수있는 시퀀스 ID를 가진 모든 템플릿 단백질.
B. 특정 템플릿 단백질을 제외
특정 템플릿 단백질은 제외하는 구조의 PDB ID를 포함하는 목록을 업로드하여 I - TASSER 템플릿 라이브러리에서 제외시킬 수 있습니다. 예제 파일은 그림 10에 표시됩니다. 같은 단백질은 PDB 라이브러리에서 여러 항목, I - TASSER SE로 존재 수 있듯이rver는 기본적으로 지정된 템플릿 (COLUMN1에서)뿐만 아니라 평범한 생활을 도서관에서 다른 모든 템플릿> 지정된 템플릿 90 %를 제외합니다. 사용자는 ID를 모든 템플릿> 지정된 템플릿 단백질 70 %가 제외됩니다 다른 신분 컷오프, 예를 들어 70 %, 지정할 수 있습니다.
Disclosures
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
관심 없음 충돌 선언하지 않습니다.
Acknowledgements
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
이 프로젝트는 알프레드 P. 슬로안 재단, NSF 경력 수상 (DBI 1,027,394), 그리고 일반 의료 과학 국립 연구소 (GM083107, GM084222)에 의해 부분적으로 지원됩니다.
Materials
- FASTA는 (참조 모델로 단백질의 아미노산 서열 포맷 http://www.ncbi.nlm.nih.gov/BLAST/fasta.shtml을 ).
- 인터넷에 접속하고 웹 브라우저와 함께 개인용 컴퓨터.
- 예측 차 구조와 기능 사이트를 분석하는 분자 시각화 소프트웨어, 예를 들어 RASMOL 또는 PYMOL.
References
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H. CD-Search: protein domain annotations on the fly. Nucleic. Acids. Res. 32, W327-W331 (2004).
- Finn, R. D. The Pfam protein families database. Nucleic. Acids. Res. 38, D211-D222 (2010).
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request Permission