Solidny sposób badania lawin neuronalnych, tj. niezmiennych od skali wybuchów aktywności czasoprzestrzennej, wskazujących na dynamikę stanu krytycznego w korze mózgowej. Lawiny pojawiają się spontanicznie w rozwijających się powierzchownych warstwach hodowanej kory mózgowej, co pozwala na długoterminowe pomiary aktywności za pomocą planarnych zintegrowanych układów wieloelektrodowych (MEA) w precyzyjnie kontrolowanych warunkach.
Method Article
Wieloelektrodowe zapisy lawin neuronalnych w kulturach organotypowych
Opens in a new tab
In This Article
Summary
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
Abstract
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
Kora jest spontanicznie aktywna, nawet przy braku jakiegokolwiek szczególnego sygnału wejściowego lub motorycznego. Podczas rozwoju aktywność ta jest ważna dla migracji i różnicowania typów komórek kory mózgowej oraz tworzenia połączeń neuronalnych1. U dojrzałego zwierzęcia ciągła aktywność odzwierciedla przeszły i obecny stan zwierzęcia, w którym bodźce sensoryczne są płynnie integrowane w celu obliczenia przyszłych działań. Tak więc jasne zrozumienie organizacji trwającej, tj. spontanicznej aktywności jest warunkiem wstępnym do zrozumienia funkcji kory mózgowej.
Liczne techniki rejestracji ujawniły, że trwająca aktywność w korze mózgowej składa się z wielu neuronów, których indywidualne aktywności przejściowo sumują się do większych zdarzeń, które można wykryć w lokalnym potencjale pola (LFP) za pomocą zewnątrzkomórkowych mikroelektrod lub w elektroencefalogramie (EEG), magnetoencefalogramie (MEG) i sygnale BOLD z funkcjonalnego rezonansu magnetycznego (fMRI). LFP jest obecnie metodą z wyboru przy badaniu aktywności populacji neuronalnej z wysoką rozdzielczością czasową i przestrzenną w skali mezoskopowej (kilka tysięcy neuronów). W mikroelektrodzie zewnątrzkomórkowej lokalnie zsynchronizowane działania przestrzennie sąsiadujących neuronów powodują gwałtowne odchylenia w LFP do kilkuset mikrowoltów. Korzystając z układu mikroelektrod, organizacja takich ugięć może być wygodnie monitorowana w przestrzeni i czasie.
Lawiny neuronalne opisują niezmienniczą w skali czasoprzestrzenną organizację trwającej aktywności neuronalnej w mózgu2,3. Są one specyficzne dla powierzchownych warstw kory mózgowej, jak ustalono in vitro4,5, in vivo u znieczulonego szczura 6 i u przytomnej małpy7. Co ważne, zarówno badania teoretyczne, jak i empiryczne2,8-10 sugerują, że lawiny neuronalne wskazują na znakomicie zrównoważoną dynamikę stanu krytycznego kory mózgowej, która optymalizuje przekazywanie i przetwarzanie informacji.
Aby badać mechanizmy powstawania, utrzymywania i regulacji lawin neuronalnych, preparaty in vitro są bardzo korzystne, ponieważ pozwalają na stabilne zapisy aktywności lawinowej w ściśle kontrolowanych warunkach. Obecny protokół opisuje, w jaki sposób badać lawiny neuronalne in vitro, wykorzystując rozwój warstwy powierzchownej w kulturach kory organotypowej, tj. kulturach warstwowych, hodowanych na płaskich, zintegrowanych układach mikroelektrod (MEA; patrz również 11-14).
Protocol
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
1. Sterylna, zamykana szklana komora z MEA do długoterminowych nagrań
- Gwintowane cylindry szklane z nasadką z tworzywa teflonowego (Ace Glass), wymagane do bezpiecznego i szczelnego zamknięcia komory hodowlanej, są cięte (Aceglass) (Aceglass) na około 2 mm od dołu gwintu (rys. 1A, B). Szklane pierścienie oczyścić, spłukując wodą (3x) i gotując przez 5 minut w 200-procentowym alkoholu etylowym, pozostawić do wyschnięcia.
- Roztwór krzemu o podwielokrotności wymagany do przymocowania szklanych pierścieni do powierzchni MEA. Dokładnie wymieszaj 15 ml części A i B zestawu elastomerów silikonowych Sylgard 184, Pozostawić na 15 minut w celu usunięcia pęcherzyków powietrza, przechowywać w 1 ml porcji w temperaturze -20 °C.
- Przyklej szklany pierścień do MEA (siatka 8x8 z wewnętrzną elektrodą uziemiającą, średnica elektrody 30 μm, odległość między elektrodami 200/100 μm dla szczura/myszy) (rys. 1A, Pobrać 1 ml silikonu (23 °C) do strzykawki z igłą o małej średnicy. Nałóż silikon na niepolerowaną powierzchnię cięcia szklanego pierścienia, środkowy szklany pierścień na MEA, nałożyć dodatkową warstwę silikonu na zewnątrz pierścienia, aby uzyskać mocniejsze uszczelnienie, pozostawić do utwardzenia na 1 - 2 godziny w temperaturze ~60 °C na gorącej płycie.
- Wysterylizuj komorę MEA i nasadki komory w okapie z przepływem laminarnym, 3x spłukując w wodzie dejonizowanej, a następnie 70% alkoholu (3 x; na ostatnie płukanie pozostaw na 10 minut w alkoholu), a następnie przez 10 minut wystawiaj wnętrze komory i nakrętki na działanie światła UV. Komorę MEA w autoklawie (120 °C; na mokro; 45 min) i pozostawić do wyschnięcia.
- Pokryj powierzchnię MEA wewnątrz komory hodowlanej poli-D-lizyną. W przypadku nowych MEA, które są raczej lipofilowe, powlekaj przez wielokrotne zasysanie kropelkami roztworu z siatki elektrod. W przypadku zużytych MEA należy zalać dno komory roztworem, odessać nadmiar płynu, pozostawić do odparowania w sterylnych warunkach wewnątrz okapu laminarnego. Przymocuj nasadkę, aby uszczelnić komorę MEA do przechowywania i wykorzystania w przyszłości.
2. Składniki niezbędne do przygotowania i wzrostu kultur organotypowych
- Sterylny agar rozpuścić w 0,9% NaCl, wlać na sterylną szalkę Petriego (Falcon, 100 x15; poziom °5 mm), ostudzić i sterylnie owinąć Parafilmem do przechowywania. Wytnij bloki 20 x 10 x 5 mm z litego agaru do użycia.
- Przechowuj super klej, np. Devcon Super Glue II, z opakowaniem przetartym 70% EtOH przed otwarciem, wewnątrz kaptura z przepływem laminarnym, aby zachować sterylność.
- Przygotuj 50% D-glukozy (SIGMA ultra, G7528), dodając 40 g glukozy do 40 ml dejonizowanej wody hodowlanej (Sigma). Przechowywać w 2 ml porcjach w temperaturze -20°C.
- Dodaj 4 ml 50% D-glukozy do 500 ml roztworu soli Gey's Balanced Salt i schłodź do błota pośniegowego (mieszanina płynu/kryształków lodu) w zamrażarce przed użyciem.
- Rozpuść osocze kurczaka w 5 ml zdejonizowanej wody hodowlanej (delikatnie wstrząsnąć, unikać tworzenia się pęcherzyków), pozostawić roztwór na 5 – 10 minut, delikatnie zamieszać i zdekantować klarowną zawartość na sterylną szalkę Petriego. Sterylny roztwór osocza z filtrem (filtr porowy 0,22 μm; białko), podaniec 350 μl do krioprobówek (NuncTM), przechowywać w temperaturze -20°C.
- Odpowiednio rozpuścić trombinę z osocza bydlęcego, sterylny filtr (filtr porowy 0,22 μm), podwielokrotność 40 μl do krioprobówek (NuncTM), przechowywać w temperaturze -20°C. Aby uzyskać roztwór roboczy, rozcieńczyć 40 μl roztworu trombiny w 375 μl zbilansowanego roztworu soli Geya z / D-glukozą.
- Przygotuj 400 ml pożywki hodowlanej, mieszając 100 ml surowicy końskiej, 200 ml Basal Medium Eagle, 100 ml buforowanego roztworu soli fizjologicznej Hanka, do którego dodaje się 4 ml 50% glukozy i 2 ml 200 mM L-glutaminy. Może być przechowywany przez 4 – 8 tygodni w 100 ml butelkach PYREX w temperaturze 4°C.
- Przygotować inhibitor mitozy przez zmieszanie 0,3 mM urydyny, 0,3 mM cytozyno-β-D-arabinofuranozydu ARA-C i 0,3 mM 5-fluoro-2'-deoksyurydyny, sterylny filtr, podwielokrotność 200 μl i przechowywać w temperaturze -20°C przez 6 – 12 miesięcy.
3. Sekcja tkanek kory mózgowej i brzusznego obszaru nakrywki (VTA) (czas: < 1 godzina)
- Procedura polega na pobraniu skrawków tkanki kory i VTA dla »12 kokultur szczurów lub myszy) i jest przygotowywana w okapie z przepływem laminarnym w sterylnych warunkach. Całkowity czas pobrania tkanek powinien wynosić niż 1 godzina.
- Zdrowe, dobrze odżywione młode (wielkość miotu ̃10; obecność plamki mlecznej w jamie brzusznej) należy przyjmować w 1 – 2 dni po urodzeniu (PND). Delikatnie chwyć szczeniaka za pysk, pozwól mu swobodnie zwisać i szybko odetnij głowę u podstawy szyi ostrymi nożyczkami.
- W celu usunięcia mózgu usuń skórę (dwa boczne nacięcia nożycami), rozetnij czaszkę cienkimi nożycami do oczu (1 strzałkowe cięcie linii środkowej; 1 cięcie koronalne na połączeniu kora/móżdżek). Odwróć wszystkie 4 klapy czaszki. Za pomocą zaostrzonej szpatułki, przeciąć czołowo opuszkę węchową, wsuń szpatułkę doogonowo pod mózg. Delikatnie wyjmij mózg z czaszki i pozwól mu wsunąć się do sterylnego, schłodzonego roztworu Geya w celu szybkiego schłodzenia i tymczasowego przechowywania. Powtórz kroki od 3.2 do 3.3 dla 2 kolejnych mózgów (łączny czas: <20 min).
- Aby uzyskać tkankę VTA, przenieś mózgi na sterylną, suchą szalkę Petriego za pomocą małej szpatułki. Następnie usuń nadmiar płynu, delikatnie przesuwając każdy mózg na około 1 cm na boki. Usuń pień mózgu za pomocą koronalnego, pionowego cięcia na poziomie móżdżku za pomocą żyletki.
- Klej blok agarowy na tarczy montażowej do mechanicznej stabilizacji mózgów podczas procedury krojenia. Umieść cienką linię superglue kilka milimetrów przed blokiem agaru na dysku (unikaj dotykania agaru przez klej).
- Za pomocą małej szpatułki przenieś i zamontuj każdy mózg tyczką czołową w dół. Upewnij się, że przednie bieguny są przyklejone do tarczy montażowej i że boczne boki brzuszne dotykają agaru bez żadnych pozostałości kleju, aby uzyskać odpowiednią stabilizację mechaniczną podczas cięcia i łatwe podnoszenie pokrojonych plastrów.
- Ostrożnie zanurz i zabezpiecz dysk montażowy z zespołem mózgu w tacy wibratomicznej (np. Leica VT1000) wypełnionej sterylnym, schłodzonym roztworem Geya. Za pomocą starannie oczyszczonej żyletki (90% EtOH) wytnij wycinki koronalne śródmózgowia z najwyższą częstotliwością wibracji i stosunkowo niską prędkością naprzód o grubości 400 500 μm. Używając odwróconej pipety Pasteura z bańką ssącą, przenieść i zebrać plastry zawierające VTA na szalkach Petriego o wymiarach 35 x 10 mm wypełnionych sterylnym, schłodzonym roztworem Geya (ryc. 1C; patrz także płytka czołowa 18 – 20 w E22 w15).
- W przypadku sekcji kory powtórz kroki 3.2 – 3.6, ale zastosuj pionowe cięcie między korą a móżdżkiem i zamontuj przodomózgowie tyczką czołową do góry. Około 3 wycinki koronalne (o grubości 350 μm) zaczynające się na poziomie prążkowia są pobierane do przyszłej sekcji kory mózgowej.
- Za pomocą mikronoża wykonanego ze złamanych żyletek wypreparuj pod mikroskopem stereoskopowym przekrój czołowy kory czołowej i obszarów śródmózgowia o szerokości ̃2 mm zawierających VTA (ryc. 1C). Skrawki tkanek należy zbierać oddzielnie w małych naczyniach (np. szkiełkach samochodowych) wypełnionych schłodzonym roztworem Geya.
4. Montaż plastrów tkanki Cortex i VTA na MEA (Czas: <1 godz.)
- Umieść MEA w temperaturze pokojowej pod mikroskopem stereoskopowym z matrycą elektrod w ostrości. Wyśrodkuj kroplę osocza o wielkości 25 μl na czystej, wolnej od kurzu i sterylnej matrycy elektrod. Za pomocą małych szpatułek ostrożnie wsuń korę i wycinek VTA do kropli osocza.
- Umieścić MEA na płycie chłodzącej, ponownie ustawić ostrość widoku, pozostawić do ostygnięcia na 15 s, a następnie dodać 25 μl trombiny do kropli osocza. Za pomocą końcówki pipety trombinowej ostrożnie rozprowadzić mieszaninę osocza/trombiny małymi okrężnymi ruchami w poprzek MEA. Nie dotykaj bezpośrednio kruchego układu elektrod. Delikatnie umieść korę na matrycy z brzegiem grzbietowym wzdłuż drugiego rzędu elektrod matrycy. W ten sposób rozwijające się warstwy powierzchowne ostatecznie zakryją przypomnienie tablicy. VTA umieszcza się w sąsiedztwie brzusznej granicy przekroju kory (ryc. 1D).
- Zakryj i luźno zamknij komorę MEA, aby utrzymać wysoką wilgotność, podczas gdy zespół MEA/kultura siedzi przez 5 minut w okapie w temperaturze pokojowej, aby umożliwić koagulację osocza/trombiny. W międzyczasie powtórz kroki 4.1 – 4.3 dla 3 kolejnych kultur.
- Ostrożnie dodać 600 μl pożywki hodowlanej w małych kropelkach do komory hodowlanej za pomocą strzykawki o pojemności 1 ml z igłą 25 x 5/8.
- Szczelnie zamknij komorę MEA i umieść zestaw MEA/kultur na kołyszącej się tacy do przechowywania wewnątrz inkubatora (rys. 1B). Aby przyspieszyć procedurę, można złożyć 3 – 4 MEA w nakładających się na siebie sekwencjach. Czas montażu dla 12 MEA powinien wynosić <1 godzina.
- Po 2 dniach in vitro (DIV) dodać 10 μl inhibitora mitozy. Odświeżać pożywkę hodowlaną o 60% w 4 działkach, a następnie co 4 dni.
5. Zapis elektrofizjologiczny i generowanie bodźców
- Aby ustalić związek między znacznymi odchyleniami potencjału pola lokalnego (LFP) a tendencją neuronów do wystrzeliwania potencjałów czynnościowych, po ̃1 tygodniu 5,6 rejestruj spontaniczną aktywność przy 24 kHz przez ̃10 min z każdej elektrody MEA (Sprzęt: MEA1060 z obwodem wygaszającym, wzmocnienie x1200, 12 bit A/D, zakres 0–4096 mV, Systemy wielokanałowe; Oprogramowanie: MC_Rack). Uziemienie jest dostarczane albo przez wewnętrzną elektrodę uziemiającą, albo zewnętrznie przez dodanie ogniwa połówkowego Ag / AgCl.
- Oddziel LFP za pomocą filtra pasmowo-przepustowego 1 – 200 Hz od zewnątrzkomórkowej aktywności szczytowej (pasmo przepustowe 300 – 3,000 Hz). Aktywność spajków można dalej podzielić na jedno- i wielojednostkowe za pomocą sortowników spajków off-line (np. Plexon Inc.). Oblicz średnie wartości wyzwalane przez skoki dla każdej elektrody. W przypadku kultur kory mózgowej większość średnich zidentyfikuje ujemne odchylenia LFP (nLFP) jako preferowany czas impulsu neuronalnego w kulturze.
- Obliczyć dla każdej elektrody próg -3 odchyleń standardowych szumu (SD) od ścieżek LFP (rys. 2), wyznaczyć czasy szczytów i amplitudy nLFP, które przekraczają próg (rys. 2B, C). Należy wybrać przedział czasowy Δt (np. od 2 do 8 ms) i zidentyfikować czasoprzestrzenne klastry nLFP na tablicy, łącząc nLFP ze wszystkich elektrod, które znajdują się w tym samym z następujących po sobie przedziałów czasowych o długości Δt (rys. 2D; szczegóły patrz 2,4,5).
- Aby zidentyfikować lawiny neuronalne, oblicz wielkość każdego klastra nLFP, np. liczbę aktywnych elektrod lub sumę amplitud nLFP, skonstruuj histogram wielkości i wykreśl współrzędne dwulogarytmiczne. W przypadku lawin neuronalnych rozkład wielkości jest zgodny z prawem potęgowym przybliżonym linią prostą we współrzędnych dwulogarytmicznych2 (ryc. 2E,F). Patrz 16, aby zapoznać się z testami statystycznymi praw potęgowych.
- Wywołaj reakcje w tkance poprzez wybór elektrody, przez którą podawane są bodźce sterowane prądem o amplitudzie S (Generator bodźców STG 1008, systemy wielokanałowe). Aby zmniejszyć uszkodzenia elektrod, należy zastosować ograniczoną w zakresie, neutralną pod względem ładunku stymulację pojedynczych wstrząsów o bipolarnym kształcie fali prostokątnej: 50 μs o amplitudzie -S, a następnie 100 μs przy amplitudzie +S/2 i S między 10 – 200 μA. Więcej informacji można znaleźć w instrukcji obsługi.
- Aby zarejestrować zakres dynamiki9, zapisz bodziec zarejestrował odpowiedzi LFP przy częstotliwości próbkowania 4 kHz na wszystkich elektrodach po 500 ms po stymulacji. Użyj obwodów wygaszających (systemy wielokanałowe), które odłączają wzmacniacze stopnia głównego podczas stymulacji, aby zredukować artefakty bodźca i zapobiec nasyceniu przedwzmacniacza.
6. Reprezentatywne wyniki:
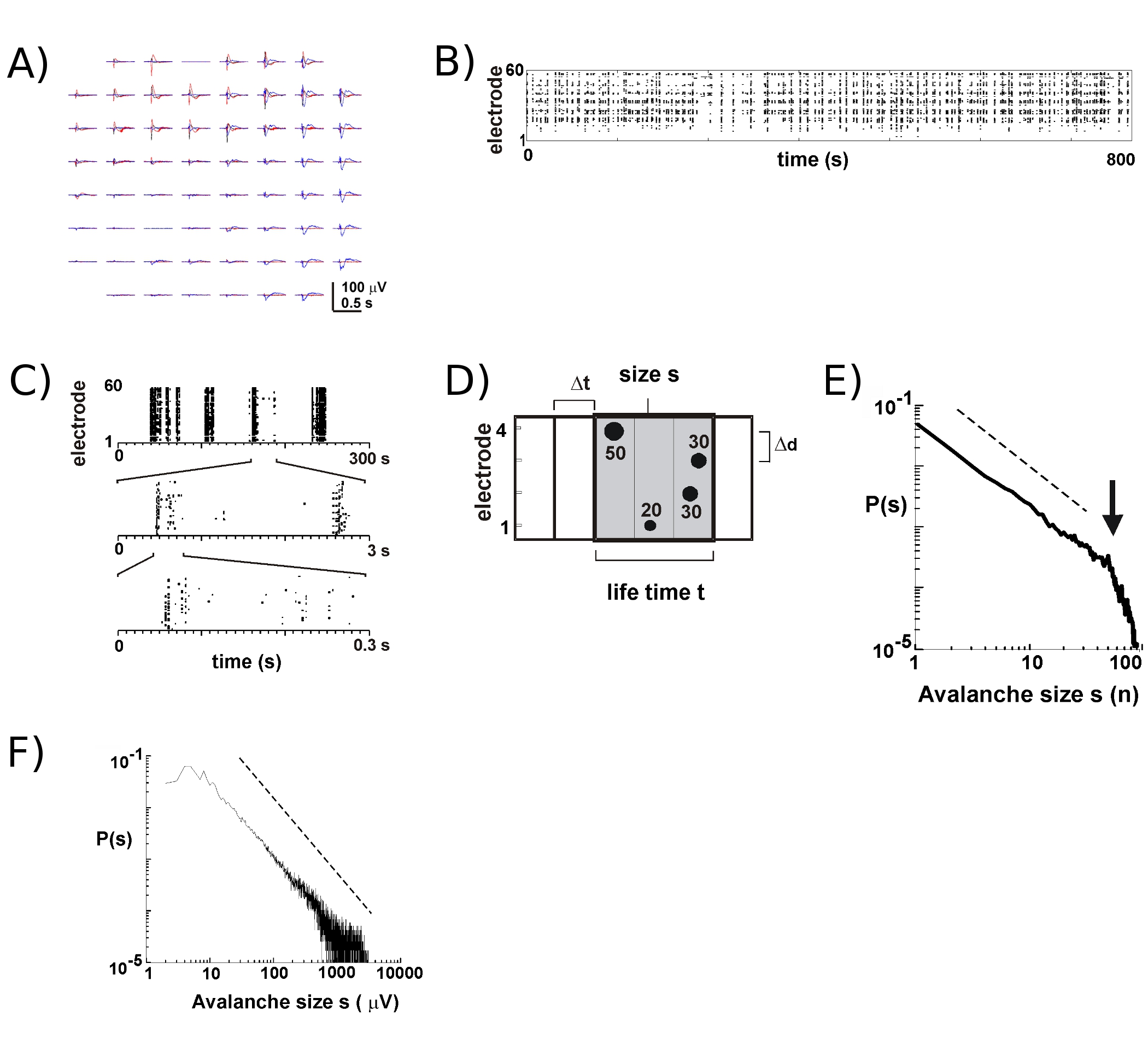
Z nowymi MEA około 8 – 9 z 10 kultur przetrwa wiele tygodni. Większość naszych długoterminowych nagrań odbywa się wewnątrz inkubatora w pożywce hodowlanej, co pozwala nam śledzić rozwój poszczególnych kultur na przestrzeni wielu tygodni5. Na podstawie naszych eksperymentów, nagrania LFP można wiarygodnie uzyskać przy użyciu MEA używanych przez ponad 100 dni hodowlanych. W przeciwieństwie do tego, zewnątrzkomórkowa aktywność kolców jest bardziej wiarygodnie mierzona za pomocą stosunkowo nowych MEA (<40 dni hodowli). W typowym eksperymencie przenosimy MEA z tacki do przechowywania (rys. 1B, po prawej) na tacę z przymocowanym stopniem głównym (rys. 1B, po lewej), utrzymując szczelną komorę hodowlaną. W przypadku kory5, kokultur kory i VTA6, a także u znieczulonego szczura6 i przytomnej małpy in vivo7, wypalanie neuronów podczas lawin neuronalnych w warstwach powierzchownych zachodzi głównie w pobliżu szczytowego ujemnego odchylenia LFP (nLFP). W ten sposób czasoprzestrzenną organizację lokalnie zsynchronizowanych grup neuronów można oszacować, mierząc występowanie nLFP w przestrzeni i czasie na tablicy 17.
Aktywność na MEA ma tendencję do pojawiania się w klastrach czasowych, tak że aktywności na jednej elektrodzie towarzyszy aktywność w innych miejscach. Typowe przebiegi LFP w takich okresach aktywności są pokazane na rysunku 2A, poprzez przekreślenie 3 klastrów występujących w odstępie kilku sekund. Dla każdego klastra można zaobserwować ujemne odchylenia pola na kilku elektrodach w oknie 1 s. Podczas wyodrębniania pików nLFP, które przekraczają próg wielokrotnego ujemnego SD, aktywność w postaci czasów szczytowych nLFP jest wygodnie wizualizowana w rastrze, w którym "kolumny" kropek reprezentują prawie zbieżne nLFP na różnych elektrodach (rys. 2B). Czasoprzestrzenna organizacja tej działalności jest dość złożona; "kolumny", które wydają się mniej lub bardziej jednorodne przy niskiej rozdzielczości czasowej, składają się z oddzielnych klastrów przy wyższej rozdzielczości czasowej i tak dalej (rys. 2C). W rzeczywistości powstawanie czasoprzestrzennych klastrów nLFP jest silnie zorganizowane w sieci korowe. Mówiąc dokładniej, organizacja jest niezmienna w skali dla lawin neuronalnych. Wykazano to, obliczając prawdopodobieństwo rozmiarów klastrów przy danej rozdzielczości czasowej Δt. W tym przypadku klastry składają się z nLFP, które występują w tych samych lub kolejnych przedziałach czasowych (rys. 2D). Gdy wielkość takiego klastra jest wyrażona w całkowitej liczbie nLFP na klaster lub zintegrowanych amplitudach nLFP na klaster, rozkłady wielkości klastra ujawniają prawo potęgowe, którego nachylenie wykazało -1,5 2,4,5,7 (rys. 2E,F). Należy zauważyć, że ten rozkład identyfikuje niezmiennicze uporządkowanie rozmiarów klastrów, czyli stosunek rozmiarów s do k x s , gdzie k jest czynnikiem stałym, wynosi k-1,5 , co jest niezależne od s. Ta organizacja prawa potęgowego jest niezależna od rozmiaru tablicy 2, rozdzielczości czasowej Δt 2 i progu używanego do identyfikacji znacznych ugięć nLFP 7. Ponieważ amplituda nLFP skaluje się z rozmiarem grupy neuronalnej 7, niezmiennicza organizacja nLFP odzwierciedla niezmienniczą skalę, tj. fraktalną, kolejność lokalnie zsynchronizowanych grup neuronów, które obejmują wszystkie rozmiary.

Rysunek 1. (A) Widok z boku i z góry MEA z zamontowanym gwintowanym szklanym pierścieniem i odpowiednią pokrywą. (B) Widok z wnętrza inkubatora. Po lewej: mocowanie do montażu na scenie głównej pozwalające na nagrywanie z pojedynczej kultury w warunkach inkubatora. Po prawej: Taca z licznymi MEA do wzrostu kultury. Koła boczne: sterowane silnikiem krokowym urządzenie kołyszące do naprzemiennej fazy zanurzenia i wystawienia na działanie atmosfery, wymaganej do wzrostu kultury. (C) Schematyczny rysunek wycinków koronalnych szczurów używanych do kokultur kory mózgowej i VTA. Wycinki kory mózgowia (po lewej) i odcinki śródmózgowia (środkowe, prawe) zawierające brzuszny obszar nakrywki VTA (vta; szary) uzyskuje się przez cięcie wzdłuż linii przerywanych. Ctx: kora mózgowa; WM: Istota biała; procesor: prążkowie (ang. striatum); VTA: Pons: Obszar Pontine. Patrz także odpowiednie płytki koronalne 8, 18 i 20 na 15 . (D) Umiejscowienie i wzrost pojedynczych kokultur kora-VTA na MEA i jego rozwój w ciągu pierwszych 9 DIV w kulturze. Zwróć uwagę na spłaszczenie kultury i jej stopniową ekspansję na tablicy. Odbijające światło części tkanek wskazują na zdegenerowane komórki i szczątki tkankowe. Zdrowa tkanka jest nieprzezroczysta i szarawa pod wpływem oświetlenia światłem widzialnym.

Rysunek 2. Lawiny neuronalne w korowych kulturach organotypowych. (A) Przekreślenie trzech okresów spontanicznej aktywności na tablicy, oddalonych od siebie o kilka sekund. Należy pamiętać, że każdy okres aktywności składa się z ujemnych odchyleń LFP na wielu elektrodach na matrycy (każdy kolor oznacza jeden okres aktywności). (B) Ujemne czasy szczytów nLFP z każdej elektrody są łączone w raster aktywności. Struktury przypominające "kolumny" wskazywały okresy aktywności niemal synchronicznej. (C) Zauważ, że kolumny, które wydają się wysoce zsynchronizowane na jednej skali czasu, składają się z wielu kolumn na wyższych skalach czasowych (pokazano 3 skale czasowe). (D) Schematyczne przedstawienie algorytmu lawiny neuronalnej. Na matrycy elektrod 2 x 2 identyfikowany jest czas szczytu i amplituda ujemnych odchyleń LFP (nLFP) przekraczających próg –x SD szumu. Organizacja czasoprzestrzenna nLFP jest zgrupowana w kolejno aktywne przedziały czasowe o szerokości Δt. Wielkość klastra określa się albo na podstawie liczby miejsc aktywnych, tj. elektrod o nLFP (s = 4), albo na podstawie zintegrowanej sumy amplitud nLFP (s = 130 μV). Czas życia mierzy się w wielokrotnościach Δt. (E, F) Prawo potęgowe w rozkładzie wielkości klastrów identyfikuje klastry jako lawiny neuronalne. Należy zauważyć, że wybór konkretnych odległości międzyelektrodowych Δd dla matrycy (tutaj 200 μm) wprowadza określone Δt, przy którym należy obserwować dynamikę. Dokładniej rzecz ujmując, stosunek, w którym Δd/Δt przybliża średnią prędkość propagacji w sieci, przy której α nachylenia prawa potęgowego wynosi -1,5 dla lawin neuronalnych 2,4,5. Proszę kliknij tutaj, aby zobaczyć większą wersję rysunku 2.
{kind=link}
Discussion
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
1. Kwestie techniczne:
- Technika sterylna. Przygotowanie MEA i przygotowanie hodowli odbywa się w okapie z przepływem laminarnym w sterylnych warunkach. Antybiotyki, które wpływają na aktywność neuronów, nie są stosowane w żadnym momencie procesu przygotowania i hodowli.
- Koagulacja osocza/trombiny i przyleganie tkanek do MEA. Przeżywalność tkanek w MEA wymaga starannej równowagi między czasem wymaganym do krzepnięcia osocza/trombiny a czasem ekspozycji tkanek na działanie atmosfery. Krótki czas krzepnięcia grozi przedwczesnym oderwaniem skrawków od MEA, podczas gdy długotrwałe wystawienie na działanie atmosfery powoduje degenerację tkanek. Ponieważ moc roztworu trombiny decyduje o szybkości procesu krzepnięcia, jest to bardzo ważny parametr dla pomyślnego przyczepiania zdrowych kultur do powierzchni MEA. Najlepsze wyniki uzyskujemy stosując 1000 jednostek (1 KU; 1 jednostka NIH = 0,324 ± 0,073 mg). Co ważne, niepełne wymieszanie roztworu osocza/trombiny powoduje niejednorodną przestrzennie koagulację sprzyjającą pękaniu osocza podczas hodowli. Te "" plazmowe poważnie wpływają na zdrowie hodowli i odłączają hodowlę od mikroelektrod, pogarszając w ten sposób jakość zapisu elektrofizjologicznego. Praca z płytkami chłodzącymi podczas składania MEA/tkanki spowalnia koagulację i pozwala na jednorodne wymieszanie roztworu osocza/trombiny oraz prawidłowe ułożenie skrawków tkanki.
Podobnie, dodanie pożywki w pojedynczych kropelkach do komory hodowlanej w celu zanurzenia kultury już po 5 minutach koagulacji, znacznie zmniejsza ryzyko oderwania tkanek z powodu napięć powierzchniowych. Udana hodowla spłaszczy się na MEA i nieznacznie się rozszerzy, wykazując zdrowy wzrost przez tygodnie, bez żadnych poważnych oznak niepełnego kontaktu tkanka-MEA lub zwyrodnienia tkanki (np. Ryc. 1D). - Rozwarstwienie tkanek. Mikro noże znacznie usprawniły nasz proces preparacji. Używamy obosiecznych żyletek (Fine Science Tools – łamliwe ostrza skalpela – 100050-00), z których za pomocą szczypiec rozłupujemy "ostrza" o szerokości ̃2 mm i przytrzymujemy je uchwytem na skalpel. Próbki tkanek wypreparowane z wycinka koronalnego za pomocą płynnego, pionowego, zdecydowanego ruchu ostrza, co znacznie zmniejsza naprężenia mechaniczne spowodowane ciągnięciem tkanki, ogólnie poprawiając zdrowie hodowli.
- Chłodzenie tkanek. Właściwe chłodzenie plastrów i skrawków tkanek podczas przygotowywania ma zasadnicze znaczenie dla powodzenia hodowli. Stosujemy wykonane na zamówienie zimne płyty zbudowane z elementów Peltiera przymocowanych pod spodem do metalowego krążka. Ciepło wytwarzane przez element Peltiera jest usuwane przez perfuzję zimnej wody. To znacznie skraca czas przygotowania i standaryzuje chłodzenie na każdym etapie przygotowania (Dold Labs & Engineering. 131 Plantation Dr. Seguin, TX 78155; (830) 560-1471)).
- Stan inkubatora. Wykonany na zamówienie inkubator z precyzyjnymi wewnętrznymi warunkami kołysania miał kluczowe znaczenie dla naszego sukcesu w uprawie plastrów na MEA. Oparte na oryginalnym projekcie firmy Multichannelsystems (obecnie niedostępnym na rynku), wewnętrzne urządzenie bujające składa się z tac, które są przymocowane do dwóch dużych bocznych kółek. Silniki krokowe i skomputeryzowane elementy sterujące pozwalają na precyzyjną trajektorię kołysania (kąt kołysania, prędkość kołysania i przerywane przerwy). Ostatecznie kultury plastrów muszą być wystawione na działanie atmosfery i pożywki hodowlanej w powolnej przemianie. Tradycyjne podejście polega na umieszczeniu kultur w wąskich rurkach, które powoli obracają się wzdłuż najdłuższej osi. W tym przypadku powolny obrót nie powoduje naprężeń mechanicznych spowodowanych samym obrotem, a prędkość obrotowa jest wystarczająco wysoka, aby uzyskać optymalny cykl "karmienia/oddychania" trwający około 5 – 10 minut. Bardziej zwarte wnętrze komory MEA, jej całkowita objętość ̃2 cm³ oraz mała objętość pożywki wymagana do kondycjonowania pożywki przez samą tkankę, stanowią poważne wyzwanie. Kołysanie MEA pod kątem ± ̃70° (czas cyklu: ̃200 s), spowolnienie prędkości kołysania w miarę przechodzenia kultury między fazą ciekłą a atmosferą oraz zatrzymanie kołysania pod ekstremalnymi kątami w celu przedłużonej ekspozycji na atmosferę było niezbędne do przetrwania kultury.
2. Wiek rozwojowy kultur kory mózgowej do badania lawin neuronalnych
Ostre wycinki z kory szczura są powszechnie pobierane w PND 0 – 1 i hodowane przez wiele tygodni na MEA. Wczesne badania wyraźnie wykazały, że hodowle z pojedynczym wycinkiem kory mózgowej, po kilku tygodniach in vitro, utrzymują warstwową strukturę z możliwymi do zidentyfikowania typami komórek, które można łatwo porównać z klasami komórek in vivo 18,18-21. Warstwowa organizacja w tym systemie in vitro została wygodnie wykorzystana do badania unerwienia wzgórza kory podczas rozwoju 22-24, a także do sterowania strukturami podkorowymi, takimi jak prążkowie 25,26. W rzeczywistości specyfika w tworzeniu połączeń neuronalnych w obrębie i między regionami mózgu pozwala na budowę złożonych systemów in vitro , które odbudowują wiele szczegółowych systemów projekcyjnych, np. obwodów kora-zwoje podstawy 27-30.
Po 4 – 6 tygodniach in vitro, pojedyncze wycinki kory 31 i wycinki kory współhodowane z prążkowiem 26 lub wzgórzem 32 wykazują spontaniczne stany górno-dolne, zwykle występujące in vivo u szczura znieczulonego uretanem 33. Delikatna organizacja czasowa tych stanów górnych nosi znamię zagnieżdżonych oscylacji θ i γ, co wskazuje na elektrofizjologicznie dojrzałą sieć neuronów piramidowych i szybko rosnących interneuronów GABA-ergicznych31. Co ważne, przy braku stymulacji receptora dopaminowego D2, dojrzewanie interneuronów korowych parwalbuminowo-dodatnich jest opóźnione o około 2 tygodnie w kulturach warstw kory mózgowej 34. Zgodnie z tymi odkryciami, przebieg rozwojowy zagnieżdżonych oscylacji θ-, β i γ jest dopasowany do tego in vivo, gdy wycinki kory są współhodowane z brzusznym obszarem nakrywki (VTA), który zawiera neurony dopaminergiczne rzutujące do kory 6.
Badania te wskazują, że badając lawiny neuronalne, które w decydującym stopniu zależą od szybkiego hamowania dojrzałego GABA-ergicznego i znajdują się w powierzchownych warstwach kory 2,4, należy zachować dużą ostrożność w celu zapewnienia prawidłowego dojrzewania tkanki korowej. Podczas gdy lawiny neuronalne powstają w pojedynczych kulturach kory mózgowej w ciągu 2 – 5 tygodni 4, gdy wymagają przebiegu czasu rozwojowego dopasowanego do rozwoju in vivo, wycinki kory potrzebują odpowiedniej stymulacji receptora dopaminy, np. poprzez współhodowlę wycinków kory mózgowej z VTA 6.
Disclosures
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
Nie stwierdzono konfliktu interesów.
Acknowledgements
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
To badanie zostało sfinansowane przez Division of Intramural Research Program (DIRP) Narodowego Instytutu Zdrowia Psychicznego, National Institutes of Health.
Materials
| Name | Company | Catalog Number | Comments |
|---|---|---|---|
| Zintegrowana planarna matryca | wieloelektrodowa Multi Channel System MCS GmbH Multi Channel System MCS GmbH | 200/30iR-ITO-w/o | Elektrody z azotanu tytanu (TiN) (średnica 30 mm) mają dużą powierzchnię, co skutkuje niską impedancją ( ~1,5 kΩ przy 1 kHz) i doskonałymi nagraniami szerokopasmowymi ( bez -– bez pierścienia) |
| Szkło komorowe | www.aceglass.com | 7620-32 | Gwintowany szklany |
| cylinder Pokrywa komory | www.aceglass.com | 7622-114 | Plastikowa nasadka z wkładką teflonową |
| Sylgard 184 | World Precision Instruments, Inc. | SYLG184 | Dwuskładnikowy elastomer silikonowy |
| Poli-D-lizyna | Sigma-Aldrich | P6407-5mg | γ-napromieniowany, liofilizowany proszek, testowany na hodowli komórkowych. Przed użyciem rozpuścić w 5 ml wody dejonizowanej. |
| Gnaj" s Zrównoważony roztwór soli | Sigma-Aldrich | G9779-500mL | sterylne, przefiltrowane i przebadane |
| osocze kurczaka | Sigma-Aldrich | P3266-5mL | Liofilizowane, przed użyciem rozpuść w 5 ml wody dejonizowanej. |
| trombina | Sigma-Aldrich | T6634-1KU | z osocza bydlęcego, liofilizowana w postaci proszku. |
| surowica konia | Sigma-Aldrich | H1138-100mL | stado dawców, inaktywowana termicznie, badana na hodowli komórkowej |
| Basal Medium Eagle | Invitrogen | 21010-046 | 1x, 500 ml - (+) Earle' s Sole, (-) L-glutamina), |
| Hank' s Bufor soli | Invitrogen | 24020-117 | 500 ml - (+) Magnez, (+) wapń, z czerwienią fenolową) |
| Szkiełka komorowe | Lab-Tek | 177429 | |
| Urydyna | Sigma-Aldrich | U3003 | |
| ARA-C cytozyna-β-D-arabinofuranozyd | Sigma-Aldrich | C6645 | |
| 5-fluoro-2'-deoksyurydyna | Sigma-Aldrich | F0503 |
References
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
- Spitzer, N. C. Electrical activity in early neuronal development. Nature. 444, 707-712 (2006).
- Beggs, J. M., Plenz, D. Neuronal avalanches in neocortical circuits. J. Neurosci. 23, 11167-11177 (2003).
- Beggs, J. M., Plenz, D. Neuronal avalanches are diverse and precise activity patterns that are stable for many hours in cortical slice cultures. J Neurosci. 24, 5216-5229 (2004).
- Stewart, C. V., Plenz, D. Inverted-U profile of dopamine-NMDA-mediated spontaneous avalanche recurrence in superficial layers of rat prefrontal cortex. J. Neurosci. 26, 8148-8159 (2006).
- Stewart, C. V., Plenz, D. Homeostasis of neuronal avalanches during postnatal cortex development in vitro. J. Neurosci. Meth. 169, 405-416 (2007).
- Gireesh, E. D., Plenz, D. Neuronal avalanches organize as nested theta- and beta/gamma-oscillations during development of cortical layer 2/3. Proc. Natl. Acad. Sci. U. S. A. 105, 7576-7581 (2008).
- Petermann, T. Spontaneous cortical activity in awake monkeys composed of neuronal avalanches. Proc. Natl. Acad. Sci. U. S. A. 106, 15921-15926 (2009).
- Kinouchi, O., Copelli, M. Optimal dynamical range of excitable networks at criticality. Nature Physics. 2, 348-351 (2006).
- Shew, W. L., Yang, H., Petermann, T., Roy, R., Plenz, D. Neuronal avalanches imply maximum dynamic range in cortical networks at criticality. J. Neurosci. 29, 15595-15600 (2009).
- Shew, W. L., Yang, H., Yu, S., Roy, R., Plenz, D. Information capacity is maximized in balanced cortical networks with neuronal avalanches. J Neurosci. 5, 55-63 (2011).
- Karpiak, V., Plenz, D. Preparation and maintenance of organotypic cultures for multi-electrode array recordings. Current Protocols in Neuroscience. 1, 6-6 (2002).
- Hammerle, H., Egert, U., Mohr, A., Nisch, W. Extracellular recording in neuronal networks with substrate integrated microelectrode arrays. Biosens. Bioelectron. 9, 691-696 (1994).
- Nisch, W., Bock, J., Egert, U., Hammerle, H., Mohr, A. A thin film microelectrode array for monitoring extracellular neuronal activity in vitro. Biosens. Bioelectron. 9, 737-741 (1994).
- Egert, U. A novel organotypic long-term culture of the rat hippocampus on substrate-integrated multielectrode arrays. Brain Res. Protoc. 2, 229-242 (1998).
- Altman, J., Bayer, S. A. Atlas of Prenatal Rat Brain Development. , CRC Press. Boca Raton. (1995).
- Clauset, A., Shalizi, C. R., Newman, M. E. J. Power-law distributions in empirical data. arXiv. , (2009).
- Plenz, D., Thiagarajan, T. C. The organizing principles of neuronal avalanches: cell assemblies in the cortex? Trends Neurosci. 30, 101-110 (2007).
- Cäser, M., Bonhoeffer, T., Bolz, J. Cellular organization and development of slice cultures from rat visual cortex. Exp. Brain Res. 477, 234-244 (1989).
- Cäser, M., Schüz, A. Maturation of neurons in neocortical slice cultures. A light and electron microscopic study on in situ and in vitro material. J. Hirnforsch. 33, 429-443 (1992).
- Götz, M., Bolz, J. Development of vasoactive intestinal polypeptide (VIP)-containing neurons in organotypic slice cultures from rat visual cortex. Neurosci. Lett. 107, 6-11 (1989).
- Götz, M., Bolz, J. Formation and Preservation of cortical layers in slice cultures. J. Neurobiol. 23, 783-802 (1992).
- Bolz, J., Novak, N., Götz, M., Bonhoeffer, T. Formation of target-specific neuronal projections in organotypic slice cultures from rat visual cortex. Nature. 346, 359-362 (1990).
- Bolz, J., Novak, N., Staiger, V. Formation of specific afferent connections in organotypic slice cultures from rat visual cortex cocultured with lateral geniculate nucleus. J. Neurosci. 12, 3054-3070 (1992).
- Novak, N., Bolz, J. Formation of specific efferent connections in organotypic slice cultures from rat visual cortex cocultured with lateral geniculate nucleus and superior colliculus. Eur. J. Neurosci. 5, 15-24 (1993).
- Plenz, D., Aertsen, A. Neural dynamics in cortex-striatum co-cultures. I. Anatomy and electrophysiology of neuronal cell types. Neurosci. 70, 861-891 (1996).
- Plenz, D., Aertsen, A. Neuronal dynamics in cortex-striatum co-cultures. II. Spatio-temporal characteristics of neuronal activity. Neurosci. 70, 893-924 (1996).
- Plenz, D., Kitai, S. T. Organotypic cortex-striatum-mesencephalon cultures: the nigro-striatal pathway. Neurosci. Lett. 209, 177-180 (1996).
- Plenz, D., Kitai, S. T. Up' and 'down' states in striatal medium spiny neurons simultaneously recorded with spontaneous activity in fast-spiking interneurons studied in cortex-striatum-substantia nigra organotypic cultures. J. Neurosci. 18, 266-283 (1998).
- Plenz, D., Herrera-Marschitz, M., Kitai, S. T. Morphological organization of the globus pallidus-subthalamic nucleus system studied in organotypic cultures. J. Comp. Neurol. 397, 437-457 (1998).
- Plenz, D., Kitai, S. T. A basal ganglia pacemaker formed by the subthalamic nucleus and external globus pallidus [see comments]. Nature. 400, 677-682 (1999).
- Plenz, D., Kitai, S. T. Generation of high-frequency oscillations in local circuits of rat somatosensory cortex cultures. J Neurophysiol. 76, 4180-4184 (1996).
- Klostermann, O., Wahle, P. Patterns of spontaneous activity and morphology of interneuron types in organotypic cortex and thalamus-cortex cultures. Neurosci. 92, 1243-1259 (1999).
- Cowan, R. L., Wilson, C. J. Spontaneous firing patterns and axonal projections of single cortico-striatal neurons in the rat medial agranular cortex. J. Neurophysiol. 71, 17-32 (1994).
- Porter, L. L., Rizzo, E., Hornung, J. P. Dopamine affects parvalbumin expression during cortical development in vitro. J Neurosci. 19, 8990-9003 (1999).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request Permission