Opisano wytyczne dotyczące komputerowej charakterystyki strukturalnej i funkcjonalnej białka za pomocą potoku I-TASSER. Począwszy od sekwencji białka zapytania, modele 3D są generowane przy użyciu wielu wyrównań gwintów i iteracyjnych symulacji montażu strukturalnego. Następnie wyciąga się wnioski funkcjonalne na podstawie dopasowań do białek o znanej strukturze i funkcjach.
Method Article
Protokół komputerowego przewidywania struktury i funkcji białek
Opens in a new tab
In This Article
Summary
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
Abstract
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
Projekty sekwencjonowania genomu zaszyfrowały miliony sekwencji białek, które wymagają wiedzy o ich strukturze i funkcji, aby lepiej zrozumieć ich biologiczną rolę. Chociaż metody eksperymentalne mogą dostarczyć szczegółowych informacji na temat niewielkiej części tych białek, modelowanie obliczeniowe jest potrzebne w przypadku większości cząsteczek białek, które nie zostały scharakteryzowane eksperymentalnie. Serwer I-TASSER to stół warsztatowy on-line do modelowania struktury i funkcji białek w wysokiej rozdzielczości. Biorąc pod uwagę sekwencję białka, typowy wynik z serwera I-TASSER obejmuje przewidywanie struktury drugorzędowej, przewidywaną dostępność rozpuszczalnika każdej reszty, homologiczne białka matrycowe wykrywane przez gwintowanie i dopasowanie struktury, do pięciu pełnowymiarowych trzeciorzędowych modeli strukturalnych oraz adnotacje funkcjonalne oparte na strukturze do klasyfikacji enzymów, terminów ontologii genów i miejsc wiązania białko-ligand. Wszystkie przewidywania są oznaczone współczynnikiem ufności, który informuje, jak dokładne są przewidywania bez znajomości danych eksperymentalnych. Aby ułatwić realizację specjalnych życzeń użytkowników końcowych, serwer udostępnia kanały do akceptowania określonych przez użytkownika map odległości między pozostałościami oraz map kontaktów w celu interaktywnej zmiany modelowania I-TASSER; Pozwala również użytkownikom określić dowolne białka jako matrycę lub wykluczyć dowolne białka matrycowe podczas symulacji składania struktury. Informacje strukturalne mogą być gromadzone przez użytkowników w oparciu o dowody eksperymentalne lub spostrzeżenia biologiczne w celu poprawy jakości prognoz I-TASSER. Serwer został oceniony jako najlepszy program do przewidywania struktury i funkcji białek w ostatnich eksperymentach CASP w całej społeczności. Obecnie z serwera on-line I-TASSER korzysta > 20 000 zarejestrowanych naukowców z ponad 100 krajów.
Protocol
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
Przegląd metody
Zgodnie z paradygmatem sekwencja do struktury-funkcji, procedura I-TASSER1-4 do modelowania struktury i funkcji obejmuje cztery następujące po sobie kroki: (a) identyfikacja szablonu przez LOMETS5; b) ponowne składanie struktury fragmentów poprzez wymianę replik symulacji Monte Carlo6; c) udoskonalanie struktury na poziomie atomowym przy użyciu REMO7 i FG-MD8; oraz (d) interpretacje funkcji opartych na strukturze przy użyciu COFACTOR9.
Identyfikacja szablonu: Dla sekwencji zapytania przesłanej przez użytkownika, sekwencja jest najpierw przepuszczana przez reprezentatywną bibliotekę struktur PDB przez lokalnie zainstalowany serwer meta-wątkowy LOMETS. Gwintowanie to procedura dopasowania sekwencja-struktura stosowana do identyfikacji białek matrycowych, które mogą mieć podobną strukturę lub zawierać podobny motyw strukturalny jak białko pytające. Aby zwiększyć zasięg wykrywania matryc homologicznych, LOMETS łączy wiele najnowocześniejszych algorytmów obejmujących różne metodologie gwintowania. Ponieważ różne programy gwintowania mają różne systemy punktacji i czułość wyrównania, jakość wygenerowanych osiowań gwintów z każdego programu gwintowania jest oceniana za pomocą znormalizowanego wskaźnika Z, który jest zdefiniowany jako:

gdzie Z-score jest wynikiem w jednostkach odchylenia standardowego w stosunku do średniej statystycznej wszystkich dopasowań wygenerowanych przez program; a Z0 to specyficzna dla programu wartość odcięcia Z-score określona na podstawie testów porównawczych gwintowania na dużą skalę5 w celu rozróżnienia "dobrych" i "złych" szablonów. Szablon z wysokim wskaźnikiem Z oznacza, że najlepsze szablony mają wynik wyrównania znacznie wyższy niż większość innych szablonów, co zwykle oznacza, że wyrównanie odpowiada dobremu modelowi. Jeśli większość najlepszych szablonów wątków ma wysokie znormalizowane wskaźniki Z, dokładność końcowego modelu I-TASSER jest zwykle wysoka. Jeśli jednak białko jest duże, a pokrycie dopasowań gwintów jest ograniczone do małego regionu białka zapytania, wysoki znormalizowany wskaźnik Z niekoniecznie oznacza wysoką dokładność modelowania dla modelu o pełnej długości. Dwa górne wyrównania gwintów z każdego programu gwintowania są zbierane i wykorzystywane do następnego kroku montażu konstrukcji.
Iteracyjna symulacja montażu struktury: Zgodnie z procedurą wątków, sekwencja zapytań jest dzielona na regiony wyrównane i niewyrównane wątkowo. Ciągłe fragmenty w wyrównaniu gwintów są wycinane z szablonów i wykorzystywane bezpośrednio do montażu konstrukcji, podczas gdy niewyrównane obszary pętli są budowane przez modelowanie ab initio. Procedura montażu konstrukcji odbywa się na układzie kratowym prowadzonym przez wymianę replik Symulacje Monte Carlo6. Pole siłowe I-TASSER obejmuje interakcje wiązań wodorowych10, oparte na wiedzy statystyczne terminy energetyczne pochodzące ze znanych struktur białkowych w PDB11, przewidywania kontaktowe oparte na sekwencjach z SVMSEQ12 oraz ograniczenia przestrzenne zebrane z szablonów gwintowych LOMETS5. Wabiki konformacyjne generowane w replikach niskotemperaturowych podczas symulacji są grupowane przez SPICKER13 w celu identyfikacji struktur o niskich stanach swobodnej energii. Centroidy klastrów górnych klastrów uzyskuje się poprzez uśrednienie współrzędnych 3D wszystkich zgrupowanych wabików strukturalnych i wykorzystuje się je do końcowego generowania modelu. Procedura symulacji i grupowania jest powtarzana dwukrotnie w celu usunięcia kolizji sterycznych i dalszego udoskonalenia topologii globalnej.
Budowa i udoskonalanie modelu na poziomie atomowym: Centroidy klastrów uzyskane po grupowaniu SPICKER są zredukowanymi modelami białkowymi (każda reszta jest reprezentowana przezjej α C i środek masy łańcucha bocznego) i mają ograniczone zastosowanie biologiczne. Budowa modelu pełnoatomowego z modeli zredukowanych odbywa się w dwóch etapach. W pierwszym etapie REMO7 jest używany do konstruowania pełnoatomowych modeli ze śladów C-alfa poprzez optymalizację sieci wiązań H. W drugim etapie, pełne modele atomowe REMO są dalej udoskonalane przez FG-MD14, który poprawia kąty skręcania szkieletu, długości wiązań i orientacje rotamerów łańcucha bocznego, poprzez symulacje dynamiki molekularnej, zgodnie z wytycznymi fragmentów strukturalnych przeszukiwanych ze struktur PDB przez TM-align. Udoskonalone modele FG-MD są wykorzystywane jako modele końcowe do prognozowania struktur trzeciorzędowych przez I-TASSER.
Jakość generowanych modeli jest szacowana na podstawie wyniku ufności (C-score), który jest definiowany na podstawie wyniku Z dopasowań gwintów LOMETS i zbieżności symulacji I-TASSER, matematycznie sformułowanych jako:

gdzie M jest liczbą wabików strukturalnych w klastrach strukturalnych zidentyfikowanych przez SPICKER13; Mtot oznacza łączną liczbę wabików zgłoszonych do klastra; oznacza średnią wartość RMSD zgrupowanych wabików na centroidy klastrów; Norm.Z-Score(i) jest znormalizowanym wskaźnikiem Z-score (Równanie 1) górnego wyrównania gwintu uzyskanego z itego serwera gwintowania w LOMETS5; N to liczba serwerów używanych w LOMETS.
Wynik C jest silnie skorelowany z jakością modeli I-TASSER. Łącząc C-score i długość białka, dokładność pierwszych modeli I-TASSER można oszacować ze średnim błędem 0,08 dla TM-score i 2 A dla RMSD15. Ogólnie rzecz biorąc, oczekuje się, że modele z C-score > - 1,5 będą miały prawidłowe złożenie. W tym przypadku RMSD i TM-score są dobrze znanymi miarami podobieństwa topologicznego między modelem a strukturą natywną. Wartości TM-score mieszczą się w [0, 1], gdzie wyższy wynik oznacza lepsze dopasowanie struktury16,17. Jednak w przypadku modeli o niższej randze (tj. 2-5 tysięcy) korelacja C-score z TM-score i RMSD jest znacznie słabsza (~ 0,5) i nie może być wykorzystana do wiarygodnego oszacowania bezwzględnej jakości modelu.
Czy pierwszy model jest zawsze najlepszym modelem w symulacjach I-TASSER? Odpowiedź na to pytanie zależy od typu celu. W przypadku łatwych celów pierwszy model jest zwykle najlepszym modelem, a jego współczynnik C jest zwykle znacznie wyższy niż w pozostałych modelach. Jednak w przypadku twardych celów, gdzie gwintowanie nie ma znaczących trafień szablonu, pierwszy model niekoniecznie jest najlepszym modelem, a I-TASSER faktycznie ma trudności z wyborem najlepszego szablonu i modeli. Dlatego zaleca się przeanalizowanie wszystkich 5 modeli pod kątem twardych celów i wybranie ich na podstawie informacji eksperymentalnych i wiedzy biologicznej.
Przewidywania funkcji: W ostatnim kroku, końcowe modele 3D wygenerowane z FG-MD są używane do przewidywania trzech aspektów funkcji białka, a mianowicie: a) liczby18 Komisji Enzymatycznej (EC) i (b) terminów Gene Ontology (GO)19 oraz (c) miejsc wiązania dla ligandów małych cząsteczek. Dla wszystkich trzech aspektów interpretacje funkcjonalne są generowane przy użyciu COFACTOR, który jest nowym podejściem do przewidywania funkcji białka w oparciu o globalne i lokalne podobieństwo do białek matrycowych w PDB o znanej strukturze i funkcjach. Po pierwsze, globalna topologia przewidywanych modeli jest dopasowywana do funkcjonalnych bibliotek szablonów za pomocą programu do wyrównywania strukturalnego TM-align20. Następnie z biblioteki wybiera się zestaw białek najbardziej podobnych do modeli docelowych na podstawie ich globalnego podobieństwa struktury, a następnie przeprowadza się szeroko zakrojone wyszukiwanie lokalne w celu zidentyfikowania podobieństwa struktury i sekwencji w pobliżu regionu miejsca aktywnego/wiązania. Wynikowe wyniki podobieństwa globalnego i lokalnego są wykorzystywane do uszeregowania białek matrycowych (homologów funkcjonalnych) i przeniesienia adnotacji (numery EC i terminy Gene Ontology19) na podstawie najlepszych trafień punktowych. Podobnie, reszty miejsca wiązania liganda i tryb wiązania liganda są wnioskowane na podstawie lokalnego dopasowania zapytania do znanych reszt miejsca wiązania liganda w szablonach funkcji najwyższego punktowania9.
Jakość przewidywania funkcji (termin EC i GO) w I-TASSER jest oceniana na podstawie wyniku homologii funkcjonalnej (Fh-score), który jest miarą globalnego i lokalnego podobieństwa między zapytaniem a szablonem, i jest zdefiniowany jako:

gdzie C-score jest oszacowaniem jakości przewidywanego modelu zgodnie z definicją w równaniu (2); TM-score mierzy globalne podobieństwo strukturalne między białkiem modelowym a białkom matrycowym; RMSDali jest RMSD między modelem a strukturą szablonu w strukturalnie wyrównanym obszarze od TM-align20; Cov reprezentuje zakres dopasowania strukturalnego (tj. stosunek strukturalnie dopasowanych pozostałości podzielony przez długość zapytania); IDali jest tożsamością sekwencji w wyrównaniu TM. Szacowany współczynnik ufności dla przewidywań liczby EC obejmuje również termin oceny aktywnego dopasowania witryny (AcM) między zapytaniem a szablonem w zdefiniowanym regionie lokalnym, obliczanym jako:

gdzie Nt oznacza liczbę pozostałości matrycowych obecnych na danym obszarze, Nali oznacza liczbę dopasowanych par pozostałości zapytanie-szablon, dii oznacza odległośćmiędzy α między itą parą wyrównanych pozostałości, d0 = 3,0 A jest punktem odcięcia odległości, Mii to wyniki BLOSUM między i-tą parą wyrównanych reszt. Ogólnie rzecz biorąc, wynik Fh mieści się w zakresie [0, 5], a wynik AcM mieści się w zakresie [0, 2], gdzie wyższe wyniki wskazują na bardziej pewne przypisania funkcjonalne. Wynik AcM jest również używany do oceny lokalnej struktury i podobieństwa sekwencji w pobliżu miejsc wiązania ligandów, co jest określane jako wynik BS.
1. Przesyłanie sekwencji białka
- Odwiedź stronę internetową I-TASSER pod adresem http://zhanglab.ccmb.med.umich.edu/I-TASSER aby rozpocząć eksperyment z modelowaniem struktury i funkcji.
- Skopiuj i wklej sekwencję aminokwasów do dostarczonego formularza lub prześlij ją bezpośrednio ze swojego komputera, klikając przycisk "Przeglądaj". Serwer I-TASSER akceptuje obecnie sekwencje zawierające do 1500 reszt. Białka dłuższe niż 1500 reszt są zwykle białkami wielodomenowymi i zaleca się ich podzielenie na poszczególne domeny przed przesłaniem do I-TASSER.
- Podaj swój adres e-mail (obowiązkowo) i imię i nazwisko dotyczące stanowiska (opcjonalnie).
- Użytkownicy mogą opcjonalnie określić zewnętrzne ograniczenia kontaktu/odległości między pozostałościami, dodać dodatkowy szablon lub wykluczyć niektóre białka matrycowe podczas procesu modelowania struktury. Dowiedz się więcej o korzystaniu z tych opcji w sekcji "Dyskusja".
- Aby przesłać sekwencję, kliknij przycisk "Uruchom I-TASSER". Przeglądarka zostanie przekierowana na stronę potwierdzenia, na której wyświetlane są informacje określone przez użytkownika, numer identyfikacyjny zadania (Job ID) oraz link do strony internetowej, na której wyniki zostaną zdeponowane po zakończeniu zadania. Użytkownicy mogą dodać ten link do zakładek lub zanotować numer identyfikacyjny zadania do wykorzystania w przyszłości.
2. Dostępność wyników
- Sprawdź status przesłanego zadania, odwiedzając stronę kolejki I-TASSER pod adresem http://zhanglab.ccmb.med.umich.edu/I-TASSER/queue.php. Kliknij kartę wyszukiwania i użyj numeru identyfikacyjnego zadania lub sekwencji zapytania, aby wyszukać przesłane zadanie.
- Po zakończeniu modelowania struktury i funkcji zostanie wysłana do Ciebie wiadomość e-mail z powiadomieniem zawierającą obraz przewidywanych konstrukcji i link internetowy. Kliknij ten link lub otwórz link dodany do zakładek w kroku 1.5, aby wyświetlić i pobrać wyniki.
3. Przewidywania dotyczące struktury drugorzędowej i dostępności rozpuszczalnika
- Sprawdź sekwencję zapytań w formacie FASTA wyświetlaną w górnej części strony wyników. Jeśli podczas przesyłania sekwencji określono jakiekolwiek dodatkowe ograniczenie/szablon, można również zobaczyć link do strony internetowej wyświetlającej informacje określone przez użytkownika (Rysunek 1A).
- Zbadaj przewidywanie struktury drugorzędowej wyświetlane jako: alfa helisa (H), nić beta (S) lub cewka (C) oraz wynik ufności przewidywania (0 = niski, 9 = wysoki) dla każdej pozostałości. Poszukaj regionu z długimi odcinkami regularnych przewidywań struktury drugorzędowej (H lub S), aby oszacować region rdzenia w białku. Klasa strukturalna białka może być również analizowana na podstawie rozkładu elementów struktur drugorzędowych. Ponadto długie obszary elementów cewki w białku zwykle wskazują na regiony nieustrukturyzowane/nieuporządkowane.
- Wyświetl przewidywaną dostępność rozpuszczalnika (rysunek 1C), aby ustalić obszary zakopane i odsłonięte przez rozpuszczalnik w zapytaniu. Wartości przewidywanej dostępności rozpuszczalnika mieszczą się w zakresie od 0 (pozostałość zakopana) do 9 (pozostałość odsłonięta). Region zawierający w większości zakopane pozostałości można wykorzystać do wyznaczenia regionu rdzenia w białku, podczas gdy regiony z odsłoniętymi rozpuszczalnikami i pozostałościami hydrofilowymi są potencjalnymi miejscami hydratacji/funkcjonowania.
4. Przewidywania dotyczące struktury trzeciorzędowej
- Przewiń w dół, aby wyświetlić przewidywane trzeciorzędowe struktury białka zapytania, wyświetlane w interaktywnym aplecie Jmol (rysunek 2). Kliknij lewym przyciskiem myszy na aplet, aby zmienić wygląd wyświetlanej struktury, przybliżyć konkretny region, wybrać określone typy pozostałości w przewidywanym modelu lub obliczyć odległości między pozostałościami.
- Przeanalizuj modele pod kątem obecności długich regionów bez struktury. Regiony te zwykle odpowiadają nieuporządkowanym regionom białka lub wskazują na brak wyrównania matrycy. Obszary te mają na ogół niską dokładność modelowania, a usunięcie tych obszarów podczas modelowania z obszaru N i C-końca poprawi dokładność modelowania.
- Pobierz pliki struktury modelu w formacie PDB, klikając łącze "Pobierz model". Pliki te można otworzyć w dowolnym oprogramowaniu do wizualizacji molekularnej (np. Pymol, Rasmol itp.) w celu dalszej analizy cech strukturalnych.
- Analizuj współczynnik ufności (C-score) modelowania struktur, aby oszacować jakość przewidywanych konstrukcji. Wartości C-score (Równanie 2) mieszczą się zazwyczaj w zakresie [-5, 2], przy czym wyższy wynik odzwierciedla model o lepszej jakości. Szacowany wynik TM i RMSD pierwszego modelu jest pokazany jako "Szacowana dokładność Modelu 1". W przypadku długich białek zaleca się ocenę jakości modelu na podstawie TM-score, ponieważ TM-score jest bardziej wrażliwy na zmiany topologiczne niż RMSD.
- Kliknij link "więcej o C-score", aby przeanalizować C-score, rozmiar klastra i gęstość klastra wszystkich modeli. Szacowany wynik TM i RMSD są prezentowane tylko dla pierwszego modelu I-TASSER, ponieważ wynik C modeli o niższej pozycji w rankingu nie jest silnie skorelowany z wynikiem TM lub RMSD. Jakość modeli o niższej pozycji w rankingu może być częściowo oceniona na podstawie ich gęstości skupień i wielkości klastrów w stosunku do pierwszego modelu, przy czym modele z większych klastrów i większej gęstości są średnio bliższe strukturze natywnej.
- Przewidywania niskiego wskaźnika C-score zwykle wskazują na niską dokładność przewidywania. W większości takich przypadków białko zapytania nie ma dobrego szablonu w bibliotece i ma rozmiar wykraczający poza zakres modelowania ab initio (tj. >120 reszt). W takich przypadkach użytkownicy mogą szukać dodatkowych ograniczeń przestrzennych i używać ich do ulepszania modelowania I-TASSER (patrz sekcja Dyskusja). Zachęca się również do przesyłania sekwencji do naszego serwera QUARK (http://zhanglab.ccmb.med.umich.edu/QUARK/) w celu czystego modelowania ab initio, jeśli rozmiar białka jest mniejszy niż 200 reszt.
5. Wyrównanie szablonu docelowego LOMETS
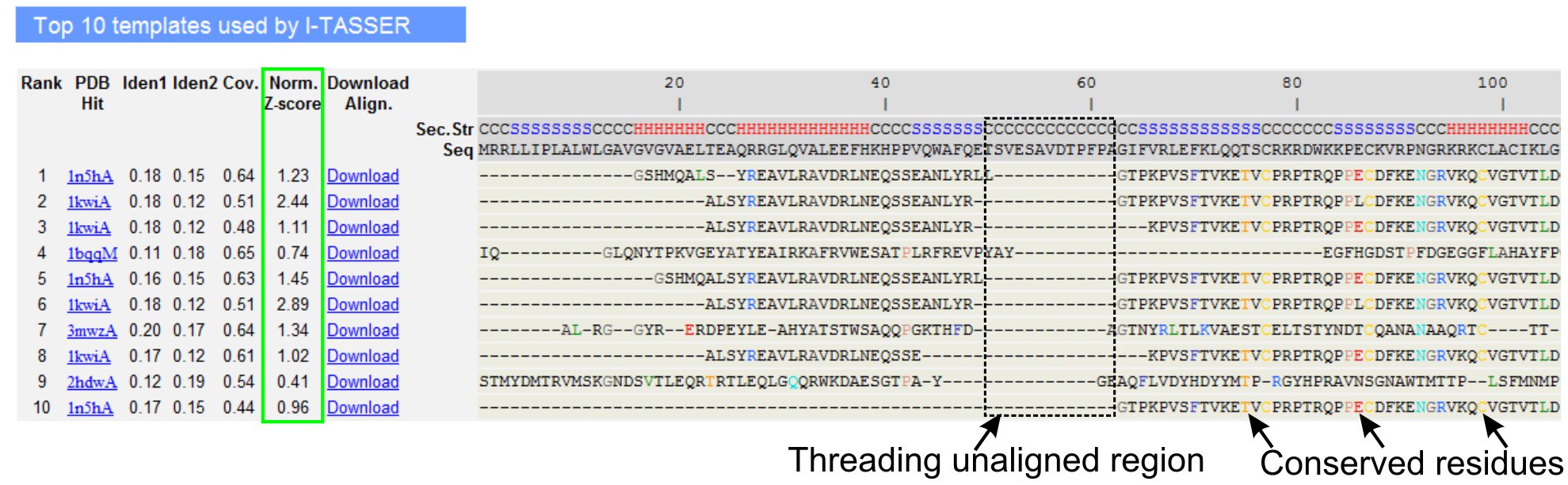
- Przewiń w dół, aby przeanalizować dziesięć pierwszych szablonów wątków białka zapytania, zidentyfikowanych przez programy do nawlekania LOMETS (Rysunek 3). Wyświetl znormalizowany wskaźnik Z (równanie 1), pokazany w kolumnie "Norm. Z-score", aby przeanalizować jakość wyrównania gwintów. Dopasowania ze znormalizowanym wskaźnikiem Z > 1 odzwierciedlają pewne wyrównanie i najprawdopodobniej mają takie samo fałdowanie jak białko zapytania.
- Przeanalizuj tożsamość sekwencji w regionie wyrównanym do wątków (kolumna "Iden. 1") i dla całego łańcucha (kolumna "Iden. 2"), aby ocenić homologię między zapytaniem a białkami matrycowymi. Tożsamość o wysokiej sekwencji jest wskaźnikiem ewolucyjnego pokrewieństwa między białkami zapytania i matrycy.
- Wyświetl wyrównane resztki gwintowania pokazane w kolorze, aby wizualnie zidentyfikować konserwatywne pozostałości/motywy w zapytaniu i białka matrycowe. Wyższa identyczność sekwencji w regionie wyrównanym do wątków w porównaniu z wyrównaniem całego łańcucha również wskazuje na obecność zachowanych motywów/domen strukturalnych w zapytaniu.
- Oceń pokrycie wyrównania gwintu, przeglądając kolumnę "Cov." i sprawdzając wyrównanie. Jeśli pokrycie górnych wyrównań jest niskie i ograniczone tylko do małego regionu białka zapytania lub nieobecne w długim segmencie sekwencji zapytania, wówczas białko zapytania zwykle zawiera więcej niż jedną domenę i zaleca się podzielenie sekwencji i modelowanie domen indywidualnie (Rysunek 3).
- Pobierz pliki wyrównania struktury sekwencyjnej w formacie PDB, klikając łącza "Pobierz wyrównanie". Te pliki wyrównania można otworzyć w dowolnym programie do wizualizacji molekularnej wymienionym w sekcji Materiały, a także można go użyć do dodania dodatkowych umocowań podczas modelowania konstrukcji (krok 1.4).
6. Analogi strukturalne w PDB
- Zapoznaj się z następną tabelą (Rysunek 4) na stronie wyników, aby określić dziesięć pierwszych analogów strukturalnych pierwszego przewidywanego modelu, zidentyfikowanych przez program do wyrównania strukturalnego TM-align20. Wynik TM >0,5 wskazuje, że wykryty analog i model mają podobną topologię i mogą być wykorzystane do określenia klasy strukturalnej/rodziny białek białka zapytania16, podczas gdy te z wynikiem TM <0,3 oznacza losowe podobieństwo struktury.
- Przeanalizuj tożsamość sekwencji i RMSD w strukturalnie wyrównanym regionie pokazanym w kolumnach "IDENa" i "RMSDa", aby ocenić zachowanie motywów przestrzennych w modelu i analogu strukturalnym. Sprawdź wzrokowo kolorowe i wyrównane pary pozostałości w wyrównaniu, aby zidentyfikować te strukturalnie zachowane pozostałości i motywy.
- Kliknij kod PDB pokazany w kolumnie "Trafienie PDB", aby odwiedzić stronę internetową RCSB i dowiedzieć się więcej o ich klasyfikacji strukturalnej (SCOP, CATH i PFAM) oraz informacjach funkcjonalnych (numer EC, powiązane terminy GO i związany ligand).
7. Przewidywanie funkcji za pomocą COFACTOR
- Przewiń w dół na stronie wyników, aby przeanalizować interpretacje funkcjonalne dla białka zapytania. Funkcje białek są wyliczone w trzech tabelach kontekstowych, wyświetlających: numery Komisji ds. Enzymów (EC), terminy ontologii genów (GO) i miejsca wiązania ligandów.
- Wyświetl kolumny 'TM-score', 'RMSDa', 'IDENa' i 'Cov.' w każdej tabeli, aby przeanalizować parametry struktury globalnej, podobieństwa i zachowania wzorców przestrzennych między modelem a zidentyfikowanymi homologami funkcjonalnymi (szablonami).
8. Przewidywanie liczby Komisji ds. Enzymów
- Zapoznaj się z pięcioma najważniejszymi potencjalnymi homologami enzymów białka kwerendy przedstawionymi w tabeli "Przewidywane liczby EC" (Figura 5). Poziom ufności przewidywania liczby EC przy użyciu tych szablonów jest pokazany w kolumnie "EC-Score". Na podstawie analizy porównawczej23 podobieństwo funkcjonalne (pierwsze 3 cyfry numeru EC) między zapytaniem a białkiem matrycowym można wiarygodnie zinterpretować za pomocą wskaźnika EC-score >1,1.
- Poszukaj konsensusu funkcji (liczby EC) wśród szablonów, które mają podobny krotność (tj. TM-score >0,5) jak białko zapytania. Jeśli wiele szablonów ma ten sam numer EC, a wynik WE > 1,1, poziom ufności przewidywania jest bardzo wysoki. Jeśli jednak EC-Score jest wysoki, ale nie ma konsensusu wśród zidentyfikowanych trafień, prognoza staje się mniej wiarygodna i zaleca się użytkownikom zapoznanie się z przewidywaniami dotyczącymi terminu GO.
- Kliknij na link podany pod numerami EC, aby odwiedzić bazę danych enzymów ExPASy i szczegółowo przeanalizować funkcję, w tym katalizowaną reakcję, wymagania dotyczące kofaktora i szlak metaboliczny białka matrycowego.
9. Przewidywania terminów Gene Ontology (GO)
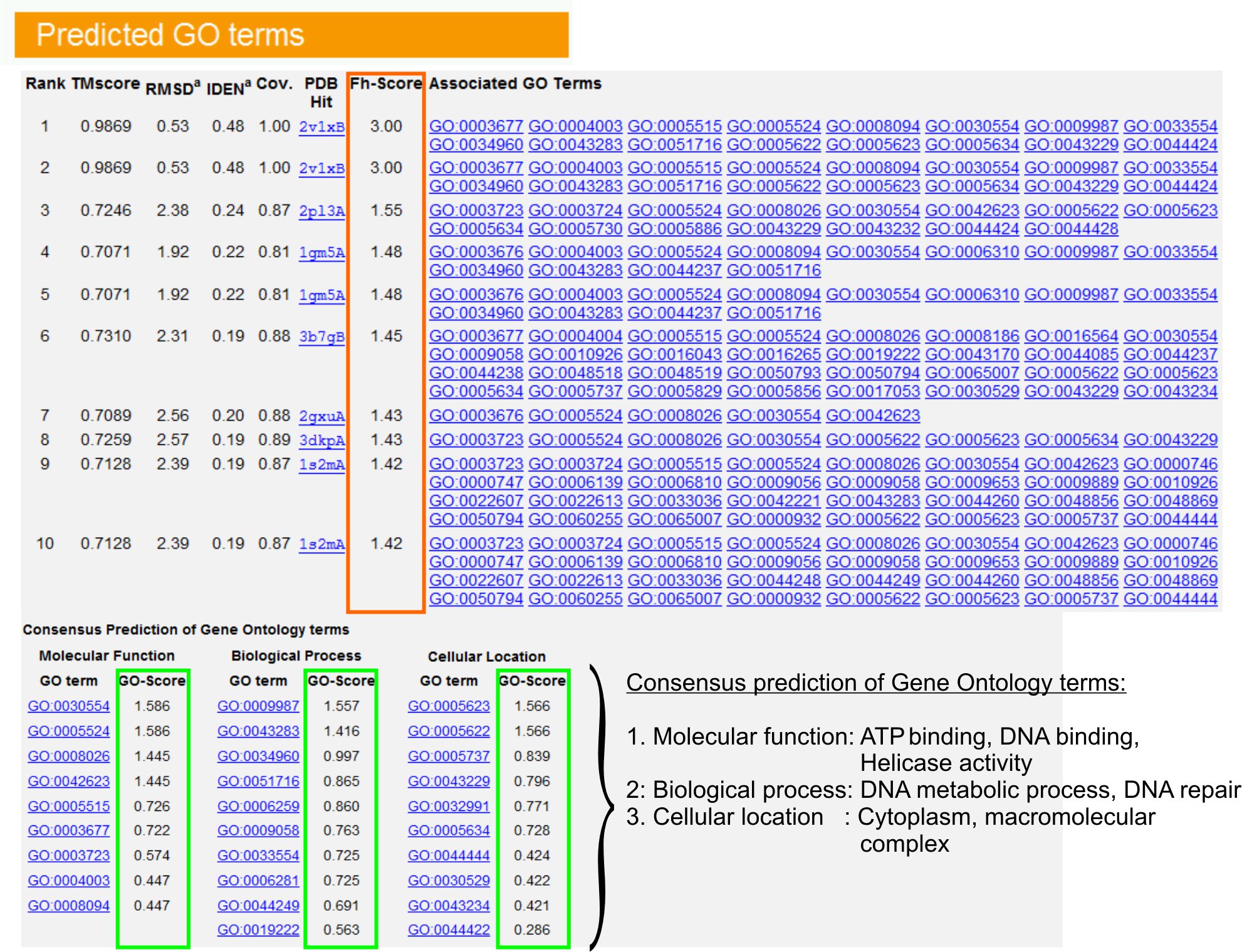
- Zapoznaj się z tabelą "Przewidywane warunki GO" (Rysunek 6), aby zidentyfikować dziesięć pierwszych homologów białka zapytania w bibliotece PDB, opatrzonych adnotacjami za pomocą terminów Gene Ontology (GO). Każde białko jest zwykle kojarzone z wieloma terminami GO, opisującymi jego funkcje molekularne (MF), procesy biologiczne (BP) i składnik komórkowy (CC). Kliknij na każdy termin, aby odwiedzić stronę internetową Amigo i przeanalizować jego definicję oraz rodowód.
- Przeanalizuj kolumnę Fh-score (Functional homology score), aby uzyskać dostęp do podobieństwa funkcjonalnego między białkami zapytania i matrycy oraz oszacować poziom ufności transferu adnotacji funkcjonalnej z tych białek. W naszym badaniu porównawczym23 50% natywnych terminów GO można było poprawnie zidentyfikować na podstawie pierwszego zidentyfikowanego szablonu przy użyciu wartości granicznej Fh-score wynoszącej 0,8, z ogólną dokładnością 56%.
- Zapoznaj się z tabelą "Przewidywanie konsensusu warunków GO", aby przeanalizować zbieżność funkcji między szablonami. Te typowe funkcje są używane do przewidywania warunków GO (MF, BP i CC) białka zapytania i oceny poziomu ufności (GO-score) przewidywań terminów GO. Na podstawie testu porównawczego23 najlepsze wskaźniki wyników fałszywie dodatnich i fałszywie ujemnych uzyskuje się dla prognoz z odcięciem GO-score = 0,5, przy malejącym pokryciu prognoz na głębszych poziomach ontologii.
10. Przewidywania dotyczące miejsca wiązania białko-ligand
- Przewiń w dół strony, aby wyświetlić dziesięć pierwszych prognoz miejsca wiązania ligandów dla białka zapytania. Przewidywane miejsca wiązania są klasyfikowane na podstawie liczby przewidywanych konformacji ligandów, które mają wspólną kieszeń wiążącą. Najlepiej zidentyfikowane miejsce wiązania jest już wyświetlane w aplecie Jmol. Kliknij przyciski opcji, aby przeanalizować inne przewidywania i zwizualizować pozostałości oddziałujące z ligandem.
- Przeanalizuj kolumnę BS-score, aby ocenić lokalne podobieństwo między modelem a witryną powiązania szablonu. Na podstawie testu porównawczego9, wynik BS > 1,1 wskazuje na wysokie podobieństwo sekwencji i struktury w pobliżu przewidywanego miejsca wiązania w modelu i znanego miejsca wiązania w szablonie.
- Pobierz plik struktury kompleksu w formacie PDB, klikając link "Pobierz". Użytkownicy mogą otwierać te pliki w dowolnym programie do wizualizacji molekularnej i interaktywnie przeglądać przewidywane miejsce wiązania i interakcje ligand-białko na swoim komputerze lokalnym.
11. Reprezentatywne wyniki

Rysunek 1. Fragment strony wyników I-TASSER przedstawiający (A) sekwencję zapytań w formacie FASTA; (B) przewidywana struktura drugorzędowa i związane z nią oceny ufności; oraz (C) przewidywaną dostępność pozostałości w postaci rozpuszczalnika. Analizowany region rdzenia i potencjalne miejsce nawodnienia w zapytaniu są wyróżnione odpowiednio w kolorze niebieskozielonym i czerwonym prostokątach.

Rysunek 2. Przykład strony wyników I-TASSER przedstawiającej przewidywania struktury trzeciorzędowej dla białek zapytania. Przewidywane modele są wyświetlane w interaktywnym aplecie Jmol, co pozwala użytkownikowi na zmianę sposobu wyświetlania cząsteczki. Modele można również pobrać, klikając linki "Pobierz". Współczynnik ufności służący do oszacowania jakości modelu jest podawany jako wskaźnik C.

Rysunek 3. Przykładowa strona wyników I-TASSER pokazująca dziesięć najczęściej zidentyfikowanych szablonów gwintowania i wyrównań przez programy gwintowe LOMETS5. Jakość wyrównania gwintów jest oceniana na podstawie znormalizowanego wskaźnika Z (zaznaczonego na zielono), gdzie wartość >1 odzwierciedla pewne wyrównanie. Wyrównane reszty w szablonie, które są identyczne z odpowiadającymi im pozostałościami zapytania, są wyróżnione kolorem, aby wskazać obecność konserwatywnej pozostałości/motywu, podczas gdy brak wyrównania w większości górnych szablonów wskazuje na obecność wielu domen w białku zapytania, a niedopasowane reszty odpowiadają regionom łącznika domeny. Kliknij tutaj aby zobaczyć pełnowymiarową wersję rysunku 3.
{kind=link}

Rysunek 4. Przykładowa strona wyników przedstawiająca dziesięć najważniejszych zidentyfikowanych analogów strukturalnych i wyrównań strukturalnych, zidentyfikowanych przez program wyrównania strukturalnego TM-align20. Ranking analogów pokazanych na rysunku opiera się na wyniku TM (zaznaczonym na niebiesko) wyrównania strukturalnego. Wynik TM >0,5 wskazuje, że dwie porównywane struktury mają podobną topologię, podczas gdy wynik TM <0,3 oznacza podobieństwo między dwiema losowymi strukturami. Strukturalnie wyrównane pary reszt są podświetlone kolorem w oparciu o ich właściwości aminokwasowe, podczas gdy niewyrównane obszary są oznaczone znakiem "-". Kliknij tutaj, aby zobaczyć pełnowymiarową wersję rysunku 4.
{kind=link}

Rysunek 5. Przykładowa strona wyników I-TASSER przedstawiająca zidentyfikowane homologi enzymatyczne białka pytającego w bibliotece PDB. Poziom ufności przewidywania liczby EC jest analizowany na podstawie wyniku EC (zaznaczonego na zielono), gdzie wynik EC > 1,1 wskazuje na podobieństwo funkcjonalne (te same pierwsze 3 cyfry numeru EC) między białkiem zapytania a białkiem matrycowym.

Rysunek 6. Przykład strony wyników I-TASSER przedstawiającej przewidywania terminów GO dla białka zapytania. Funkcjonalne homologi dla białka zapytania w bibliotece szablonów Gene Ontology są uszeregowane na podstawie ich wyniku Fh (w pomarańczowym prostokącie). Typowe cechy funkcjonalne z tych najwyżej punktowanych trafień są wyprowadzane w celu wygenerowania końcowych prognoz terminu GO dla białka zapytania. Jakość przewidywanych warunków GO jest szacowana na podstawie GO-score (pokazanego na zielono), gdzie GO-score >0,5 oznacza wiarygodną prognozę. Kliknij tutaj, aby zobaczyć pełnowymiarową wersję rysunku 6.
{kind=link}

Rysunek 7. Przykładowa strona wyników I-TASSER przedstawiająca przewidywania dotyczące miejsca wiązania ligandów białkowych dziesięciu najlepszych przy użyciu algorytmu COFACTOR9. Klasyfikacja przewidywanych miejsc wiązania jest oparta na liczbie przewidywanych konformacji ligandów, które mają wspólną kieszeń wiązania w zapytaniu. Wynik BS (zaznaczony na czerwono) jest miarą lokalnej sekwencji i podobieństwa struktury między przewidywanym a miejscem wiązania szablonu; i jest przydatny do analizy zachowania kieszeni w miejscu wiązania.

Rysunek 8. Przykład plików utwierdzeń zewnętrznych stosowanych do określania umocnień kontaktowych/odległościowych dla pozostałości/pozostałości.

Rysunek 9. Przykład plików unieruchamiania używanych do określania białka matrycowego dla serwera I-TASSER. Użytkownik może określić wyrównanie szablonu zapytania w formacie (A) FASTA; lub (B) w formacie 3D.

Rysunek 10. Przykładowy plik używany do wykluczania szablonu podczas procedury modelowania konstrukcji I-TASSER. Pierwsza kolumna zawiera identyfikator PDB białek matrycowych, które mają zostać wykluczone. Druga kolumna służy do określania wartości granicznej tożsamości sekwencji, która będzie używana dla innych podobnych szablonów w bibliotece szablonów.
Discussion
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
Przedstawiony powyżej protokół jest ogólną wskazówką dla modelowania struktury i funkcji za pomocą serwera I-TASSER. Chociaż ta zautomatyzowana procedura działa bardzo dobrze w przypadku większości białek, interwencje człowieka często pomagają znacznie poprawić dokładność modelowania, szczególnie w przypadku białek, które nie mają bliskich matryc w bibliotece PDB. Użytkownicy mogą interweniować podczas modelowania I-TASSER w następujący sposób: (a) rozszczepianie białek wielodomenowych; b) zapewnienie zewnętrznych ograniczeń w celu usprawnienia montażu konstrukcji; oraz (c) usuwanie szablonów podczas modelowania.
Podział białka wielodomenowego:
Wiele długich sekwencji białkowych często zawiera wiele domen połączonych elastycznymi regionami łącznikowymi, co utrudnia wyjaśnienie ich struktury przy użyciu zarówno technik eksperymentalnych, jak i obliczeniowych. Niemniej jednak, ponieważ domeny są niezależnie składającymi się jednostkami i mogą pełnić odrębną funkcję molekularną; Pożądane jest rozszczepienie długich białek wielodomenowych i modelowanie każdej domeny osobno. Indywidualne modelowanie domen nie tylko przyspieszy proces przewidywania, ale także zwiększy jakość dopasowania zapytania do szablonu, co skutkuje bardziej niezawodnymi przewidywaniami struktury i funkcji.
Granice domen w sekwencjach białek można przewidzieć za pomocą ogólnodostępnych zewnętrznych programów online, takich jak NCBI CDD24, PFAM25 lub InterProScan26. Ponadto, jeśli dla białka zapytania dostępne są wyrównania gwintów LOMETS, granice domeny można zlokalizować, wizualnie identyfikując długie odcinki niewyrównanych reszt w górnych szablonach gwintowania (patrz krok 5.4). Te niewyrównane regiony w większości odpowiadają regionom konsolidatora domen. Jeśli szablony wielodomenowe są już dostępne w bibliotece PDB szablonów z wyrównanymi wszystkimi domenami zapytań, białko zapytania można modelować jako pełną długość.
Zapewnienie zewnętrznych utwierdzeń
Symulacje montażu konstrukcji w I-TASSER opierają się głównie na ograniczeniach przestrzennych zebranych z szablonów gwintowania LOMETS. W przypadku białek zapytań, które mają dobre trafienie wątków (Norm. Z-score > 1) w bibliotece szablonów, pochodne ograniczenia przestrzenne są w większości bardzo dokładne, a I-TASSER wygeneruje modele strukturalne o wysokiej rozdzielczości dla tych białek. Z drugiej strony, w przypadku białek zapytań, które mają słabe trafienie gwintu lub nie mają go wcale (norma. Z-score < 1), zebrane ograniczenia przestrzenne często zawierają błędy z powodu niepewności szablonu i dopasowania. W przypadku tych docelowych białek informacje przestrzenne określone przez użytkownika mogą być bardzo pomocne w poprawie jakości przewidywanego modelu. Użytkownicy mogą wprowadzać zewnętrzne ograniczenia do serwera I-TASSER na dwa sposoby:
A. Określ umocowania kontaktu/odległości
Eksperymentalnie scharakteryzowane kontakty/odległości między pozostałościami, na przykład z eksperymentów NMR lub sieciowania, można określić poprzez przesłanie pliku utwierdzenia. Przykładowy plik pokazano na rysunku 8, gdzie kolumna 1 określa typ utwierdzenia, tj. "ROZPOR" lub "KONTAKT". W przypadku utwierdzenia odległości (DIST) kolumny 2 i 4 zawierają pozycje reszt (i, j), kolumny 3 i 5 zawierają typy atomów w reszcie, a kolumna 6 określa odległość między dwoma określonymi atomami. W przypadku urządzeń umoczających dla kontaktu (CONTACT) kolumny 2 i 3 zawierają pozycje (i, j) pozostałości, które powinny się stykać. Odległość między środkami łańcuchów bocznych tych stykających się par reszt jest ustalana na podstawie obserwowanych odległości w znanych strukturach w PDB. I-TASSER spróbuje narysować te pary atomów w pobliżu określonej odległości podczas symulacji udoskonalania struktury.
B. Określ szablon struktury białka
Programy do nawlekania LOMETS wykorzystują reprezentatywną bibliotekę PDB do znajdowania prawdopodobnych fałd dla białka zapytania. Chociaż użycie reprezentatywnej biblioteki struktur pomaga skrócić czas potrzebny do obliczenia dopasowań sekwencja-struktura, możliwe jest, że w bibliotece brakuje dobrego białka matrycowego lub matryca mogła nie zostać zidentyfikowana przez programy do wątkowania LOMETS, mimo że jest obecna w bibliotece. W takich przypadkach użytkownik powinien określić pożądaną strukturę białka jako matrycę.
Aby określić strukturę białka jako dodatkowy szablon, użytkownicy mogą przesłać plik struktury w formacie PDB lub określić identyfikator PDB zdeponowanej struktury białkowej w bibliotece PDB. I-TASSER wygeneruje wyrównanie szablonu zapytania za pomocą programu MUSTER23 i zbierze ograniczenia przestrzenne zarówno z szablonu określonego przez użytkownika, jak i szablonów LOMETS, aby poprowadzić symulację montażu konstrukcji. Ze względu na to, że dokładność zabezpieczeń LOMETS jest różna dla różnych celów, waga urządzeń ograniczających LOMETS jest większa w przypadku łatwych (homologicznych) celów niż w przypadku twardych (niehomologicznych) celów, które były systematycznie dostrajane w naszym treningu porównawczym.
Użytkownicy mogą również określać własne wyrównania szablonów zapytań. Serwer akceptuje wyrównanie w dwóch formatach: formacie FASTA (Rysunek 9A) i formacie 3D (Rysunek 9B). Format FASTA jest standardowy i opisany na http://zhanglab. ccmb.med.umich.edu/FASTA/. Format 3D jest podobny do standardowego formatu PDB (http://www.wwpdb.org/documentation/format32/sect9.html), ale do rekordów ATOM dodawane są dwie dodatkowe kolumny pochodzące z szablonów (patrz rysunek 9B):
Kolumny 1–30: Atom (tylko C-alfa) i nazwy reszt dla sekwencji zapytania.
Kolumny 31–54: współrzędne atomów C-alfa zapytania skopiowane z odpowiednich atomów w szablonie.
Kolumny 55–59: Odpowiedni numer pozostałości we wzorze na podstawie dopasowania
Kolumny 60–64: Odpowiednia nazwa pozostałości we wzorze
Wykluczanie białek matrycowych
Białka są elastycznymi cząsteczkami i mogą przyjmować wiele stanów konformacyjnych, aby zmienić swoją aktywność biologiczną. Na przykład struktury wielu kinaz białkowych i białek błonowych zostały rozwiązane zarówno w konformacji aktywnej , jak i nieaktywnej . Również obecność lub brak związanego liganda może powodować duże ruchy strukturalne. Chociaż wszystkie stany konformacyjne szablonu są podobne dla programów wątkowych, pożądane jest modelowanie zapytania przy użyciu szablonów tylko w jednym określonym stanie. Nowa opcja na serwerze pozwala użytkownikowi wykluczyć białka matrycowe podczas modelowania struktury. Ta funkcja pozwoliłaby również użytkownikowi wybrać poziom homologii szablonów, które mają być używane do modelowania. Użytkownicy mogą wykluczyć białka matrycowe z biblioteki I-TASSER poprzez:
A. Określanie punktu odcięcia tożsamości sekwencji
Użytkownicy mogą użyć tej opcji, aby wykluczyć białka homologiczne z biblioteki szablonów I-TASSER. Poziom homologii jest ustalany na podstawie odcięcia tożsamości sekwencji, tj. liczby identycznych reszt między zapytaniem a białkiem matrycowym podzielonej przez długość sekwencji zapytania. Na przykład, jeśli użytkownik wpisze "70%" w podanym formularzu, wszystkie białka szablonowe, które mają tożsamość sekwencji >70% do białka zapytania I-, zostaną wykluczone z biblioteki szablonów I-TASSER.
B. Wykluczenie określonych białek matrycowych
Określone białka matrycowe można wykluczyć z biblioteki szablonów I-TASSER, przesyłając listę zawierającą identyfikatory PDB struktur, które mają zostać wykluczone. Przykładowy plik pokazano na rysunku 10. Ponieważ to samo białko może istnieć jako wiele wpisów w bibliotece PDB, serwer I-TASSER domyślnie wykluczy określone szablony (w kolumnie 1), a także wszystkie inne szablony z biblioteki, które mają tożsamość >90% z określonymi szablonami. Użytkownicy mogą również określić inną granicę tożsamości, np. 70%, gdzie wszystkie matryce o tożsamości >70% do określonych białek matrycowych zostaną wykluczone.
Disclosures
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
Nie stwierdzono konfliktu interesów.
Acknowledgements
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
Projekt jest częściowo wspierany przez Fundację Alfreda P. Sloana, Nagrodę Kariery NSF (DBI 1027394) oraz Narodowy Instytut Ogólnych Nauk Medycznych (GM083107, GM084222).
Materials
| Name | Company | Catalog Number | Comments |
|---|---|---|---|
| Nazwa materiału Typ | firmy | Numer katalogowy | |
| FASTA sformatowana sekwencja aminokwasowa białka, które ma być modelowane (patrz, http://www.ncbi.nlm.nih.gov/BLAST/fasta.shtml). | |||
| Komputer osobisty z dostępem do internetu i przeglądarką internetową. | |||
| Oprogramowanie do wizualizacji molekularnej, np. RASMOL lub PYMOL, do analizy przewidywanej struktury trzeciorzędowej i miejsc funkcjonalnych. |
References
Loading...
$$\rightleftharpoonup{xx}$$
$$\longleftharp{xx}$$,
$$\longrightharp{xx}$$,
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 69, 108-117 (2007).
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins. 77, 100-113 (2009).
- Wu, S., Skolnick, J., Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 5, 17-17 (2007).
- Roy, A., Kucukural, A., Zhang, Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 5, 725-738 (2010).
- Wu, S., Zhang, Y. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35, 3375-3382 (2007).
- Zhang, Y., Kihara, D., Skolnick, J. Local energy landscape flattening: parallel hyperbolic Monte Carlo sampling of protein folding. Proteins. 48, 192-201 (2002).
- Li, Y., Zhang, Y. REMO: A new protocol to refine full atomic protein models from C-alpha traces by optimizing hydrogen-bonding networks. Proteins. 76, 665-676 (2009).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , (2011).
- Roy, A., Zhang, Y. COFACTOR: protein-ligand binding site predictions by global structure similarity match and local geometry refinement. , Forthcoming (2011).
- Zhang, Y., Hubner, I. A., Arakaki, A. K., Shakhnovich, E., Skolnick, J. On the origin and highly likely completeness of single-domain protein structures. Proc. Natl. Acad. Sci. U. S. A. 103, 2605-2610 (2006).
- Zhang, Y., Kolinski, A., Skolnick, J. TOUCHSTONE II: a new approach to ab initio protein structure prediction. Biophys. J. 85, 1145-1164 (2003).
- Wu, S., Zhang, Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 24, 924-931 (2008).
- Zhang, Y., Skolnick, J. SPICKER: a clustering approach to identify near-native protein folds. J. Comput. Chem. 25, 865-871 (2004).
- Zhang, J., Zhang, Y. High-resolution protein structure refinement using fragment guided molecular dynamics simulations. , Forthcoming (2010).
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 9, 40-40 (2008).
- Xu, J., Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics. 26, 889-895 (2010).
- Zhang, Y., Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins. 57, 702-710 (2004).
- Barrett, A. J. Nomenclature Committee of the International Union of Biochemistry and Molecular Biology (NC-IUBMB). Enzyme Nomenclature. Recommendations 1992. Supplement 4: corrections and additions (1997). Eur J Biochem. 250, 1-6 (1997).
- Ashburner, M. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25-29 (2000).
- Zhang, Y., Skolnick, J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic. Acids. Res. 33, 2302-2309 (2005).
- Xu, D., Zhang, Y. RK Ab Intio Protein Structure Prediction. , Forthcoming (2011).
- Kim, D. E., Chivian, D., Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic. Acids. Res. 32, W526-W531 (2004).
- Roy, A., Mukherjee, S., Hefty, P. S., Zhang, Y. Inferring protein function by global and local similarity of structural analogs. , Forthcoming (2011).
- Marchler-Bauer, A., Bryant, S. H. CD-Search: protein domain annotations on the fly. Nucleic. Acids. Res. 32, W327-W331 (2004).
- Finn, R. D. The Pfam protein families database. Nucleic. Acids. Res. 38, D211-D222 (2010).
- Zdobnov, E. M., Apweiler, R. InterProScan--an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847-848 (2001).
Reprints and Permissions
Request permission to reuse the text or figures of this JoVE article
Request Permission