



Overview
Source: Alexander S. Gold1, Tonya M. Colpitts1
1 Department of Microbiology, Boston University School of Medicine, National Emerging Infections Diseases Laboratories, Boston, MA
Transduction is a form of genetic exchange between bacteria that utilizes bacteriophages, or phages, a class of virus that infects exclusively prokaryotic organisms. This form of DNA transfer, from one bacterium to another by way of a phage, was discovered in 1951 by Norton Zinder and Joshua Ledererg, who termed the process "transduction" (1). Bacteriophages were first discovered in 1915 by British bacteriologist Frederick Twort, then independently discovered again in 1917 by French-Canadian microbiologist Felix d'Herelle (2). Since then, the structure and function of these phages have been widely characterized (3), dividing these phages into two classes. The first of these classes are the lytic phages which upon infection multiply within the host bacterium, disrupting the bacterial metabolism, lysing the cell, and releasing progeny phage (4). As a result of this anti-bacterial activity and the increasing prevalence of antibiotic-resistant bacteria, these lytic phages have recently proved useful as a substitute treatment for antibiotics. The second of these classes are the lysogenic phages which can either multiply within the host via the lytic cycle or enter a quiescent state in which their genome is integrated into that of the host (Figure 1), a process known as lysogeny, with the ability for phage production to be induced in multiple later generations (4).
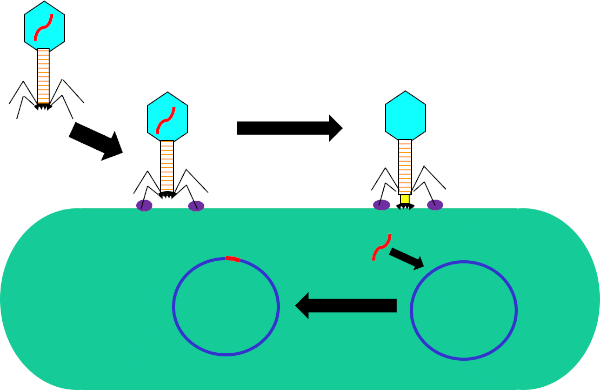
Figure 1: Infection of host cell by bacteriophage. Adsorption by the phage to the bacterial cell wall via interactions between the tail fibers and receptor (purple). Once on the cell surface, the phage is irreversibly attached to the bacterial cell using the base plate (black) which is moved to the cell wall by the contractile sheath (yellow). Phage genome (red) then enters the cell and integrates into the host cell genome.
While bacterial transduction is a naturally occurring process, using modern technology it has been manipulated for the transfer of genes into bacteria in the laboratory setting. By inserting genes of interest into the genome of a lysogenic phage, such as phage, one is able to transfer these genes into the genomes of bacteria and consequently express them within these cells. While other methods of gene transfer, such as transformation, use a plasmid for gene transfer and expression, the insertion of the phage genome into that of the recipient bacterium not only has the potential to confer new traits to this bacterium, but also allows for naturally occurring mutations and other factors of the cellular environment to alter the function of the transferred gene.
Compared to other methods of horizontal gene transfer, such as conjugation, transduction is fairly flexible in the criteria required for donor and recipient cells. Any genetic element that can fit inside the genome of the phage being used can be transferred from any strain of donor bacteria to any strain of recipient bacteria as long as both are permissive to the phage, requiring the expression of necessary phage receptors on the cell surfaces. Once this gene is moved out of the donor genome and packaged into the phage, it can be transferred to the recipient. Following transduction, it is necessary to select for recipient bacteria that contain the gene of interest have to be selected for. This could be done by use of a genetic marker, such as a FLAG-tag or polyhistidine-tag, to mark the gene of interest, or the intrinsic function of the gene, in the case of genes that encode for antibiotic resistance. Additionally, PCR could be used to further confirm successful transduction. By using primers for a region within the gene of interest and comparing signal to a positive control, bacteria that has the gene of interest, and a negative control, bacteria that underwent the same steps as the transduction reaction with no phage. While bacterial transduction is a useful tool in molecular biology, it has and continues to play an important role in the evolution of bacteria, particularly with regard to the recent rise of antibiotic resistance.
In this experiment, bacterial transduction was used to transfer the gene encoding for resistance to the antibiotic ampicillin from the W3110 strain of E.coli to the J53 strain via the P1 bacteriophage (5). This experiment consisted of two main steps. First, the preparation of P1 phage containing the ampicillin resistance gene from the donor strain. Second, the transfer of this gene to the recipient strain by transduction with the P1 phage (Figure 1). Once carried out, the successful transfer of the ampicillin resistance gene could be determined by qPCR (Figure 2). If transduction was successful, the J53 strain of E. coli would be resistant to ampicillin, and the gene conferring this resistance detectable by qPCR. If unsuccessful, there would be no detection of the ampicillin resistance gene and ampicillin would still function as an effective antibiotic against the J53 strain.
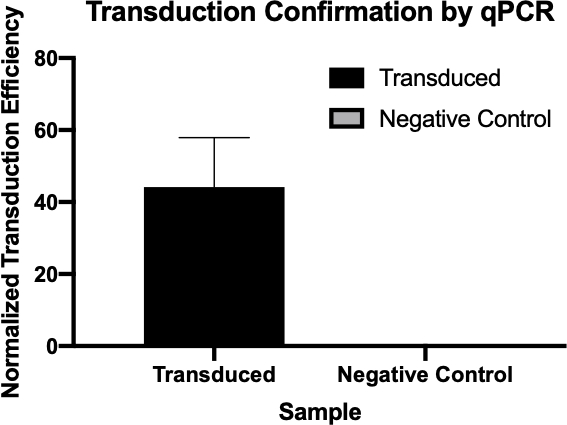
Figure 2: The confirmation of successful transduction by qPCR. By comparing the Cq values detected for the gene of interest from the transduction reaction and the negative control reaction, and normalizing these values against a housekeeping gene, one was able to confirm that bacterial transduction was successful.
Procedure
1. Set-up
- Before commencing any work involving microbes, sterilize the workspace using 70% ethanol. Always use the necessary PPE (lab coat and gloves).
- Ensure that LB agar plates with 1x ampicillin, commercially available P1 phage lysate solution, chloroform, 1 M sodium citrate, glycerol, and a box of pre-sterilized plastic pipette tips and cell spreaders are close at hand.
- Prepare sterile LB by autoclaving and use it to make three 1 mL aliquots of LB salt solution.
- mM MgCl2 (952.11-2.3803 μg), 5 mM CaCl2 (11.098 mg), 0.1-0.2% glucose (100-200 μg)
- mM MgSO4 (12.0366 mg), 5 mM CaCl2 (11.098 mg)
- mM sodium citrate (25.806 mg)
- Once finished, sterilize all surfaces as well as gloves with 70% ethanol and wash hands.
2. Protocol
- Donor Phage Lysate Preparation
- Prepare a 1 mL culture of donor W3110 strain E. coli in LB with 1x ampicillin grown overnight at 37 °C with aeration and shaking at 220 rpm.
- Dilute this overnight culture 1:100 in 1 mL of fresh LB supplemented with 10-25 mM MgCl2, 5 mM CaCl2, and 0.1-0.2% glucose.
- Grow this bacterial dilution at 37 °C for 2 hours with aeration and shaking at 220 rpm.
- Once these cells have reached early logarithmic growth phase (noticeable growth and slight turbidity), add 40 μL of commercially available P1 phage and leave at 37 °C with aeration and shaking at 220 rpm.
- Before phage addition, the measured optical density at 600 nm of these cells should be approximately 0.4 (6).
- Monitor cells for 1-3 hours until the culture has lysed.
- Lysis will result in increased cellular debris as well as a notable decrease in turbidity (i.e. cells will be considered lysed once one is able to see through the culture).
- Add several drops (50-100 μL) of chloroform to the lysate and mix by vortex.
- Chloroform sterilizes the phage lysate by killing any remaining donor cells, leaving only phage and increasing the titer of this lysate.
- Centrifuge lysate at 14,000 rpm for 2 minutes to remove debris and transfer supernatant to a fresh tube.
- Add a few drops of chloroform and store at 4 °C for no more than one day.
- Transduction
- Prepare a 1 mL culture of recipient J53 strain E. coli grown overnight in LB at 37 °C with aeration and shaking at 220 rpm.
- Transfer 100 μL of donor phage lysate (2.1) into a 1.5 mL microfuge tube and incubate with cap open at 37 °C for 30 minutes.
- This incubation allows for any remaining chloroform in the P1 lysate solution to evaporate before being added to the recipient cells.
- Gently pellet recipient strain cells by centrifugation at 6,000 rpm for 5 minutes.
- Resuspend these cells in 300 μL of fresh LB containing 100 mM MgSO4 and 5 mM CaCl2. (P1 phage requires calcium to be infectious).
- Set up two reactions using the recipient bacteria cells and prepared donor phage lysate: 1) transduction reaction combining 100 μL recipient J53 strain E. coli and 100 μL donor phage lysate, and 2) negative control combining 100 μL recipient J53 strain E. coli and 100 μL of LB containing 100 mM MgSO4 and 5 mM CaCl2.
- Incubate at 37 °C for 30 minutes with shaking at 220 rpm.
- Add 1 mL LB and 200 μL 1M sodium citrate (pH=5.5) and incubate for 1 hour at 37 °C with shaking at 220 rpm.
- Citrate is used to reduce the infectivity of P1 by chelating with calcium, preventing the lysis of recipient bacteria.
- Incubation of this solution allows for the expression of the ampicillin resistance marker.
- Pellet cells by centrifugation at 6,000 rpm for 5 minutes.
- Resuspend cell pellets in 100 μL of LB with 100 mM sodium citrate (pH 5.5). Vortex and plate entire solution for both reactions on two LB agar plates.
- LB plate should have 1x Amp for the transduced sample and no Amp for the negative control.
- P1 phage contamination on this plate requires re-streaking before freezer stocks can be prepared.
- If phage is not removed, cultures grown from these colonies will not grow unless in the presence of a calcium chelator.
- Pick about 3-4 colonies from both plates and streak again on two LB agar plates spread with 100 μL of 1 M sodium citrate (pH=5.5).
- LB plate should have 1x Amp for the transduced sample and no Amp for the negative control.
- Incubate the plates at 37 °C overnight to allow colonies free of phage to grow.
- Pick colonies from both plates and use them to grow overnight cultures in 5 mL of LB at 37 °C with aeration and shaking at 220 rpm.
- Isolate DNA from these cultures by DNA miniprep using 4.5 mL of the total culture volume.
- Elute DNA using 35 μL of nuclease-free water.
- Measure the resulting concentration by Nanodrop. Pure DNA will generate an absorbance ratio (A260/280) of approximately 1.8.
- Use the remaining 0.5 mL of each culture to prepare 1 mL glycerol stocks by making a 1:1 mixture of 100% glycerol and bacterial culture.
- Store bacterial glycerol stocks at -80 °C.
3. Data Analysis and Results
- Confirmation of transduction by qPCR
- Prepare two qPCR master mixes for six qPCR reactions, three using qPCR primers for the ampicillin resistance gene, and the other three using qPCR primers for a housekeeping gene (14.5 μL per reaction): 12.5 μL qPCR buffer mix + 1 μL forward primer + 1 μL reverse primer.
- In this experiment, we used SYBR Green master mix.
- Housekeeping gene primers were designed to amplify a segment of DNA within the bacterial gene encoding for DNA gyrase B (7).
- For each qPCR reaction, combine 100 μg of DNA from each reaction (10.5 μL) with 14.5 μL of qPCR master mix.
- Using a qPCR machine and the thermocycling protocol listed in Table 1, amplification was measured for the ampicillin resistance and housekeeping genes for all six reactions.
- Cq values generated by qPCR were used to calculate the normalized transduction efficiency of the ampicillin resistance gene (Figure 3), confirming the successful transduction of the ampicillin resistance gene.
- The Cq value, or cycle quantification value, of a sample is the earliest PCR cycle number at which a signal exceeded the background threshold is detected. Low Cq values correspond to more target sequence, vice versa.
- Normalized transduction efficiency of a gene within a sample can be calculated using these Cq values by subtracting the value of the housekeeping gene from that of the target gene, generating a ΔCq value, which can be used to calculate normalized transduction efficiency by 2(-ΔCq).
- Prepare two qPCR master mixes for six qPCR reactions, three using qPCR primers for the ampicillin resistance gene, and the other three using qPCR primers for a housekeeping gene (14.5 μL per reaction): 12.5 μL qPCR buffer mix + 1 μL forward primer + 1 μL reverse primer.
| Temperature | Time | |
| Denaturation | 94 °C | 2 min |
| 40 cycles: | ||
| Denaturation | 94 °C | 15 sec |
| Annealing, extension, and fluorescence read | 60 °C or 5 °C below lowest primer Tm | 1 min |
Table 1: qPCR Thermocycling Protocol
Bacteria can adapt quickly to a fast-changing environment by exchanging genetic material and one way they can do this is via transduction, the exchange of genetic material mediated by bacterial viruses. A bacteriophage, often abbreviated to phage, is a type of virus that infects bacteria by first attaching to the surface of the host and then injecting its DNA into the bacterial cell. It then degrades the host cell's own DNA and replicates its viral genome, whilst hijacking the cell's machinery to synthesize many copies of its proteins. These phage proteins then self-assemble and package the phage genomes to form multiple progeny. However, due to the low fidelity of the DNA packaging mechanism, occasionally, the phage packages fragments of bacterial DNA into the phage capsid. After inducing the lysis of the host, the phage progeny are released and, once such a phage infects another host cell, it transfers the DNA fragment of its previous host. This can then recombine and become permanently incorporated into the new host's chromosome, thereby mediating gene transfer between the two bacteria.
To carry out phage transduction in the laboratory requires a donor strain that contains a gene of interest, a recipient strain that lacks it, a phage that can infect both the strains, and a method to select the transduced bacteria. In most cases, this will be a selective solid growth media that supports the growth of transduced bacteria but inhibits the growth of non-transduced ones. To begin, the donor strain that contains the gene of interest is cultured in a liquid growth medium. When all the bacteria are actively dividing in the log phase of their growth, the culture is inoculated with the target phage. After three to four hours of incubation, when nearly all the bacteria have lysed and released the phage particles, the donor phage lysate is inoculated into a freshly grown culture of the recipient bacterial strain. After a brief incubation of one hour, the culture should now contain a mixture of transduced and non-transduced bacterial cells and this is screened for the transduced cells by spreading a fraction of the suspension onto an appropriate selective solid growth media. Upon further incubation, the transduced cells should grow and multiply to yield visible colonies. These colonies can then be selected for further analysis using a variety of methods to further confirm successful transduction, such as colony PCR, DNA sequencing, or quantitative PCR.
Before starting the procedure, put on any appropriate personal protective equipment, including a lab coat and gloves. Next, sterilize the workspace with 70% ethanol and wipe down the surface.
After this, prepare three one-milliliter aliquots of LB salt solution. Now, prepare a donor strain culture by adding 100 microliters of E. coli to a 15 milliliter conical vial containing five milliliters of LB growth medium with 500 micrograms of ampicillin. Then, grow the culture overnight at 37 degrees Celsius with aeration and shaking at 220 rpm. The next day, wipe down the bench top with 70% ethanol before removing the culture from the shaking incubator. Next, dilute the overnight culture one to 100 by adding 10 microliters of donor strain to 990 microliters of fresh LB supplemented with salt solution.
Allow the bacterial dilution to grow at 37 degrees Celsius for two hours with aeration and shaking at 220 rpm. Once the cells have reached early log phase, remove the culture from the incubator, add 40 microliters of P1 phage to the culture and incubate again. Continue to monitor the cells for one to three hours until the culture has lysed. Next, add 50 to 100 microliters of chloroform to the lysate and mix by vortexing. Then, centrifuge the lysate to remove debris and transfer the supernatant to a fresh tube. Add a few drops of chloroform to the supernatant and store it at four degrees Celsius for no more than one day.
To begin the transduction procedure, obtain a one milliliter culture of recipient strain. Next, transfer 100 microliters of donor phage lysate into a 1.5 milliliter microcentrifuge tube and incubate it at 37 degrees Celsius with the cap open for 30 minutes to allow any remaining chloroform to evaporate. While the donor phage lysate incubates, pellet the recipient strain cells via gentle centrifugation. Discard the supernatant and resuspend the cell pellet in 300 microliters of fresh LB containing 100 millimolar magnesium sulfate and five millimolar calcium chloride.
Next, set up the transduction reaction by combining 100 microliters of the recipient strain and 100 microliters of the donor phage lysate in a microcentrifuge tube. Then, set up the negative control by combining 100 microliters of the recipient strain and 100 microliters of the LB with magnesium sulfate and calcium chloride. After incubation, add 200 microliters of one molar sodium citrate and one milliliter of LB to both tubes, and mix by gently pipetting up and down. Then, after the tubes have been incubated for an hour, gently pellet the cells via centrifugation.
After centrifuging, discard the supernatant and resuspend the pelleted cells in 100 microliters of LB with 100 millimolar sodium citrate. Vortex the solutions and pipette the entire transduced sample onto an LB agar plate with 1X ampicillin. Finally, pipette the entire volume of the negative control cell mixture onto an LB agar plate without ampicillin. After incubating the plates overnight at 37 degrees Celsius, use a sterile pipette tip to pick three to four colonies from the transduction plate and streak them onto a new LB agar plate containing 1X ampicillin and 100 microliters of one molar sodium citrate. Repeat this plating method for the negative control on another LB agar plate containing only 100 microliters of one molar sodium citrate. Then, incubate the plates at 37 degrees Celsius overnight to allow colonies free of phage to grow.
The next day, wipe down the bench top with 70% ethanol before removing your plates from the incubator. Using a sterile pipette tip, pick three colonies from the transduction plate and add them each to a separate tube containing five milliliters of LB media. Then, select three colonies from the negative control plate and add them to another tube containing five milliliters of LB media. Grow the cultures overnight at 37 degrees Celsius with aeration and shaking at 220 rpm. After sterilizing the bench top as previously demonstrated, use a DNA miniprep kit to isolate DNA from 4.5 milliliters of each culture according to the manufacturer's instructions. Then, elute the DNA with 35 microliters of nuclease-free water and measure the resulting concentration by lab spectrophotometer. Finally, prepare glycerol stocks by adding the remaining 0.5 milliliters of both bacterial solutions to 0.5 milliliters of 100% glycerol.
To confirm transduction, first prepare two qPCR master mixes for 24 qPCR reactions. For the first master mix, add 150 microliters of qPCR buffer mix to a microcentrifuge tube and 12 microliters each of a forward and reverse primer designed to amplify the ampicillin resistance gene. Next, prepare a second qPCR master mix by adding 150 microliters of qPCR master mix to a microcentrifuge tube and then adding 12 microliters each of a forward primer and reverse primer designed to amplify a housekeeping gene.
For each qPCR reaction, combine 100 micrograms of experimental DNA from each reaction with 14.5 microliters of qPCR master mix. Now, prepare the remaining reactions as previously demonstrated. Transfer the reactions to a thermocycler preheated to 94 degrees Celsius and then initiate the program. Finally, use the cycle quantification, or Cq, values generated by qPCR to calculate the normalized transduction efficiency of the ampicillin resistance gene.
The cycle quantitation, or Cq, values for the genes of interest were tabulated for each of the negative controls and transduced samples. Low Cq values, typically below 29 cycles, like the transduced samples in this example indicate high amounts of the target sequence.
A housekeeping gene, also tabulated here, is used as a loading control to normalize the amount of DNA in each reaction and as a positive control to ensure the qPCR is working. Provided the same amounts of the housekeeping gene are loaded, it is found at relatively the same rate in each sample.
Next, to calculate the delta Cq value for each sample, subtract the Cq value of the housekeeping gene for each sample from the Cq value of its corresponding target gene. For example, the delta Cq of the first negative control is 13.54. Then, use this value to calculate the normalized transduction efficiency of each sample using the formula shown here. Finally, the average normalized transduction efficiency for each sample group can be calculated.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
The transfer of genes to and from bacteria by bacteriophage, while a natural process, has proved extremely useful for a multitude of research purposes. While other methods of gene transfer such as transformation and conjugation are possible, transduction uniquely uses bacteriophages; not only allowing for gene integration into the host genome, but also for gene delivery to multiple bacteria that are not susceptible to other methods. This process, while especially useful in the laboratory, has also been used in the recently emerging field of gene therapy, more specifically in alternative gene therapy, a therapeutic strategy that utilizes bacteria to deliver therapeutics to target tissues, many of which are not susceptible to other delivery methods and have much clinical relevance (8,9).
Subscription Required. Please recommend JoVE to your librarian.
References
- Lederberg J, Lederberg E.M., Zinder, N.D., et al. Recombination analysis of bacterial heredity. Cold Spring Harbor symposia Quantitative Biol. 1951;16:413-43.
- Duckworth DH. "Who Discovered Bacteriophage?". Bacteriology Reviews. 1976;40:793-802.
- Yap ML, Rossman, M.G. Structure and Function of Bacteriophage T4. Future Microbiol. 2014;9:1319-27.
- Sulakvelidze A, Alavidze, Z., Morris, J. G. Bacteriophage Therapy Antimicrobial Agents and Chemotherapy 2001;45(3):649-59.
- Moore S. Sauer:P1vir phage transduction 2010 [Available from: https://openwetware.org/wiki/Sauer:P1vir_phage_transduction].
- Kobayashi A, et al. Growth Phase-Dependent Expression of Drug Exporters in
- Escherichia coli and Its Contribution to Drug Tolerance. Journal of Bacteriology. 2006;188(16):5693-703.
- Rocha D, Santos, CS, Pacheco LG. Bacterial reference genes for gene expression studies by RT-qPCR: survey and analysis. Antonie Van Leeuwenhoek. 2015;108:685-93.
- Pálffy R. et al. Bacteria in gene therapy: bactofection versus alternative gene therapy. Gene Ther. 2006 13:101-5.
- O'Neill JM, et al. Intestinal delivery of non-viral gene therapeutics: physiological barriers and preclinical models. Drug Discovery Today. 2011;16:203-2018.