



Overview
Source: Laboratory of Dr. Paul Bower - Purdue University
The method of standard additions is a quantitative analysis method, which is often used when the sample of interest has multiple components that result in matrix effects, where the additional components may either reduce or enhance the analyte absorbance signal. That results in significant errors in the analysis results.
Standard additions are commonly used to eliminate matrix effects from a measurement, since it is assumed that the matrix affects all of the solutions equally. Additionally, it is used to correct for the chemical phase separations performed in the extraction process.
The method is performed by reading the experimental (in this case fluorescent) intensity of the unknown solution and then by measuring the intensity of the unknown with varying amounts of known standard added. The data are plotted as fluorescence intensity vs. the amount of the standard added (the unknown itself, with no standard added, is plotted ON the y-axis). The least squares line intersects the x-axis at the negative of the concentration of the unknown, as shown in Figure 1.
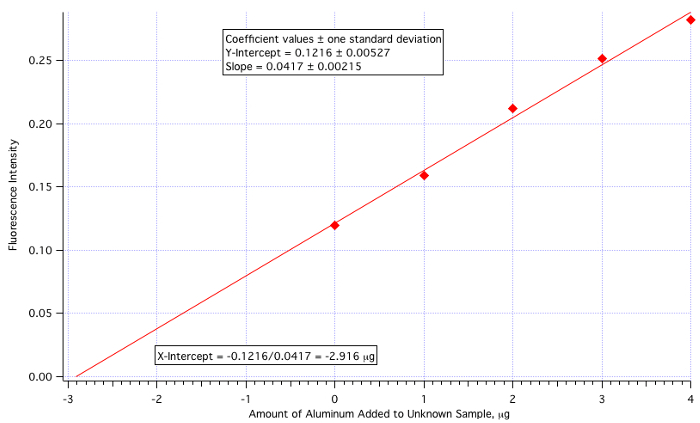
Figure 1. Graphic representation of method of standard addition.
Principles
In this experiment, the method of standard additions is demonstrated as an analytical tool. The method is a procedure for the quantitative analysis of a species without the generation of a typical calibration curve. Standard addition analysis is accomplished by measuring spectroscopic intensity before and after the addition of precise aliquots of a known standard solution of the analyte.
This experiment studies non-fluorescent species by reacting them in such a way as to form a fluorescent complex. This approach is commonly used in the investigation of metal ions. Aluminum ions (Al3+) will be determined by forming a complex with 8-hydroxyquinoline (8HQ). The Al3+ is precipitated by 8HQ from aqueous solution and then is extracted into chloroform; the fluorescence of the chloroform solution is measured and related to the concentration of the original Al3+ solution. Sensitivity in the part-per-million (ppm or μg/mL) range is expected for this experiment.
The reaction is

The amount of aluminum in each sample during this experiment is calculated as follows:
| Blank | 0 | ||
| Unknown + 0 mL Standard | VUnknown(CUnknown) = 25 mL(CUnknown) | ||
| Unknown + 1 mL Standard | VUnknown(CUnknown) + VStandard(CStandard) = 25 mL(CUnknown) + 1 mL(1 μg/mL) | ||
| Unknown + 2 mL Standard | VUnknown(CUnknown) + VStandard(CStandard) = 25 mL(CUnknown) + 2 mL(1 μg/mL) | ||
| Unknown + 3 mL Standard | VUnknown(CUnknown) + VStandard(CStandard) = 25 mL(CUnknown) + 3 mL(1 μg/mL) | ||
| Unknown + 4 mL Standard | VUnknown(CUnknown) + VStandard(CStandard) = 25 mL(CUnknown) + 4 mL(1 μg/mL) | ||
Subscription Required. Please recommend JoVE to your librarian.
Procedure
1. Preparing the Reagents
- 100 ppm standard Al3+ solution: Dissolve 0.9151 g aluminum nitrate (Al(NO3)3•9H2O) into a 1-L volumetric flask with DI water.
- 8HQ solution in 1 M acetic acid (2% wt/vol): Add 2.0 g of 8-hydroxyquinoline to a 100-mL volumetric flask.
- Carefully add 5.74 mL glacial acetic acid to the 100-mL flask, then dilute to the mark with DI water. This allows the 8-hydroxyquinoline to dissolve in aqueous phase.
- 1 M NH4+/NH3 buffer (pH~8): Add 20 g of ammonium acetate (NH4OAc) to a 100-mL bottle.
- Add 7 mL of 30% ammonium hydroxide to this 100-mL bottle, and dilute to the mark with DI water. This helps neutralize the acid in the 8HQ solution when combined.
- Other reagents include anhydrous sodium sulfate (Na2SO4) and chloroform (Spec grade).
2. Preparing the Samples
- Prepare a 1.00 ppm standard Al3+ solution by adding 1.0 mL of the 100-ppm stock Al3+ solution with a pipette to a 100-mL volumetric flask.
- Place six 125-mL separatory funnels onto the rings that are on a large ring stand located in the hood. They should be labeled as follows: BL, 0, 1, 2, 3, 4. Make sure all glassware is scrupulously clean, as it is difficult to obtain quantitative results if little beads of chloroform stick to the walls of the glassware.
- Add 25.00 mL of the unknown Al3+ solution to the five separatory funnels labeled 0, 1, 2, 3, & 4. In this example, the unknown concentration is 0.110 ppm.
- Add 0, 1.00, 2.00, 3.00, and 4.00 mL of the 1.00-ppm standard Al3+ solution, respectively, to the 5 funnels with a 1-mL pipette.
- Prepare a BLANK by adding 25.00 mL of distilled water to the separatory funnel labeled BL.
- Add 1.0 mL of the 8-hydroxyquinoline solution with a pipette to each of the six solutions.
- Add 3.0 mL of buffer solution with a pipette to each of the six solutions.
- Extract each solution twice with 10 mL of chloroform, shaking vigorously for 1 min each time. Remember to occasionally vent the separatory funnel to release pressure buildup. (NOTE: A good extraction only takes place when there is a lot of liquid-liquid contact between the phases).
- Collect the chloroform in a clean, dry 100-mL labeled beaker. Chloroform has a density of close to 1.5 g/cm3, so it is the lower layer. There should be no trace of yellow color left in the aqueous phase after a complete extraction.
- Transfer the combined chloroform extract from each beaker into their respective 25-mL volumetric flask and dilute to the mark with chloroform. Be sure to place stoppers in each volumetric flask to keep any chloroform from evaporating.
- Add ~1 g of anhydrous sodium sulfate (Na2SO4) to each of the six 100-mL beakers from step 2.9. The sodium sulfate helps remove any trace of water that may be present in the chloroform extract.
- Transfer the solutions back to their respected beakers. Swirl carefully to facilitate dehydration of any water in the sample.
- Decant the chloroform extracts into a quartz fluorimeter cell (Note: Chloroform will dissolve a plastic polystyrene cell).
3. Selecting the Excitation Wavelength
Determine the excitation and emission wavelengths by running scans, then simply read and record the fluorescence intensity of all samples at those values. The excitation and emission bandwidths are preset at 5 nm. The complex absorbs in the near UV, so the excitation wavelength should be about 385 nm. Initially, monitor the fluorescence at 500 nm in the emission branch.
- On the Fluorimeter, verify that both the internal and outer cooling fans in the fluorimeter are turned on prior to powering up the xenon lamp. The xenon lamp gets very hot, and requires continual cooling.
- Turn on the high voltage (HV) to the PMT detector to 400 V.
- Open both of the shutters.
- Open the data acquisition program on the computer, here it's "100nmFluorScan".
- Place the "Sample + 2 mL added" (2) solution into the quartz cell to use for determining the best excitation and emission wavelengths.
- With the emission wavelength initially set at 500 nm, run an excitation scan from 335-435 nm with a scan speed of 2 nm/s.
- From the resulting fluorescence plot, determine the maximum fluorescence excitation wavelength (EXλmax), and set the instrument to that value.
4. Selecting the Emission Wavelength
- Set the fluorimeter emission wavelength to 450 nm.
- Set the emission wavelength range to run a 100 nm scan from 450-550 nm.
- To start the scan, click the "start trial" button on the program at the same time of pressing the "START" button on the front panel of the fluorimeter.
- From the resulting fluorescence plot, determine the maximum fluorescence emission wavelength (EMλmax), and set the instrument to that value (Figure 2).

Figure 2. Determining optimum EXλmax and EMλmax Wavelengths.
5. Measuring the Fluorescence of the Samples
- All samples are run at EMλmax and EXλmax. Scans are not needed for each sample, but just the fluorescence value at these conditions. Starting with the most dilute sample (blank), place in a quartz cell and then in instrument. Record the fluorescent intensity in lab notebook.
- Repeat for all other samples.
- Remember that the Blank Relative Intensity must be subtracted from the Relative Intensities of each solution prior to creating the calibration chart.
6. Creating the Standard Addition Plot
- Plot the fluorescence intensity vs. µg of Al3+ added.
- Determine the least-squares value of the resulting plot, and record both the slope and intercept.
- Determine the µg of Al3+ in the unknown sample from the equation µg of Al3+ = -b/m
- Knowing that the unknown aluminum had a volume of 25.0 mL added to each sample, determine the concentration of the aluminum in the unknown.
The method of standard addition is a quantitative analysis technique used to minimize matrix effects that interfere with analyte measurement signals.
Unknown component concentrations are often elucidated through a range of analytical techniques, such as light spectroscopy, mass spectrometry, and electrochemistry. However, the measurement can be affected by other components in the sample, called the matrix, and cause the inadvertent reduction or enhancement of the signal, called matrix effects. These effects can skew results and cause significant errors in analysis.
The method of standard addition can be used to minimize matrix effects on measurement signals. This is performed by adding precise volumes of a known analyte solution to the sample.
This video will introduce the basics of the standards addition method, and demonstrate how to perform the technique in the laboratory using a fluorescence measurement.
Matrix effects can arise in complex samples where a number of other molecules interact with the analyte. For example, this can occur when molecules bind or agglomerate with the analyte, thereby changing its ability to fluoresce. Or the matrix may change the ionic strength of the overall solution, changing specific properties of the analyte.
To mitigate these effects using the method of standard addition, a range of volumes of an analyte standard solution is added to equal volumes of the sample. The solution volumes are then made equal using solvent.
Then, the signal is measured for the samples with and without the standard addition. The data are plotted as intensity versus the amount of the standard added to the sample, rather than a classic calibration curve. The actual concentration of the analyte in any given flask is defined by the following equation. The instrumental response will equal some constant times the concentration of analyte. The resulting equation takes the linear form y=mx+b. Thus, when the plot is extrapolated to zero absorbance, the intercept is equal to the unknown concentration of the sample.
The signal plot must be linear over the concentration range of concern. Also, the interference should not vary as the ratio of analyte to sample matrix changes. Finally, the matrix itself should not generate any measurement signals on its own.
The following experiment studies aluminum, a non-fluorescent species, by reacting it with 8-hydroxyquinoline, or 8HQ, to form the fluorescent ALQ3 complex.
The fluorescence of the aluminum complex in an organic solvent is measured, and then related to the concentration of the original aluminum solution. This approach is common in the analysis of metal ions.
Now that the basics of the method of standard addition have been outlined, and the basics of the experiment explained, let's perform the technique in the laboratory.
First, prepare the 100-ppm aluminum stock solution in water, and then use it to prepare a 1-ppm standard solution.
Next, add 2 g of 8-hydroxyquinoline, or 8HQ, to a 100-mL volumetric flask.
Carefully add 5.74 mL glacial acetic acid, then dilute to the 100-mL mark with deionized water. This step enables the 8HQ to dissolve in the aqueous phase.
Next, prepare the buffer by adding 20 g of ammonium acetate and 7 mL of 30% ammonium hydroxide to a 100-mL marked bottle and dilute. Verify the pH with a pH indicator stick. This buffer helps neutralize the acid in the 8HQ solution when combined.
Other reagents needed include anhydrous sodium sulfate and spectrophotometric grade chloroform.
Now prepare the samples, in this case by extracting the aqueous sample into the organic phase using liquid-liquid extraction. Place six 125-mL separatory funnels onto ring-stand rings inside the hood. Make sure all glassware is scrupulously clean, as dirty glassware will skew results. Sequentially label the funnels "blank", "0", "1", "2", "3", and "4".
Using a pipette, add 25 mL of the unknown aluminum solution to each of the five separatory funnels labeled "0" through "4". Prepare the blank by adding 25 mL of deionized water to the funnel labeled "blank".
Next, add 1, 2, 3, and 4 mL of the 1-ppm standard solution to the corresponding numbered funnels. Add no standard solution to the blank or 0 funnels.
Add 1 mL of the 8HQ solution and 3 mL of buffer solution to each of the 6 funnels.
Perform a liquid-liquid extraction by adding 10 mL of chloroform to each flask. Shake the funnel vigorously, and occasionally vent the funnel to release pressure buildup. Place the funnel back into the ring, and allow the liquid layers to separate.
Next, collect the chloroform phase in a clean, dry, and labeled 100-mL beaker. Since chloroform has a higher density than water, it is the lower layer in the funnel.
Transfer the chloroform extract into a 25-mL volumetric flask, and cap each flask to prevent evaporation.
Perform a second liquid-liquid extraction on the remaining aqueous solution, by adding 10 mL of chloroform to each funnel. Shake the funnel, as before, to transfer any remaining analyte to the chloroform phase. There should be no yellow color left in the top aqueous phase.
Repeat the second extraction for each funnel, then collect the chloroform phases in corresponding labeled beakers. Pour the collected chloroform into their respective volumetric flasks, and dilute to the mark with fresh chloroform.
To remove trace water, add about 1 g of anhydrous sodium sulfate to each of the six 100-mL beakers. Transfer the solutions back into their respective beakers, and swirl to facilitate dehydration of the sample.
Decant the chloroform extract into a quartz fluorimeter cell.
Set up the fluorimeter according to the manufacturers instructions and set the voltage to 400 V. Next, open the data acquisition program on the computer.
Use sample 2 to determine the best excitation and emission wavelengths. Set the emission wavelength to 500 nm, and run an excitation scan from 335–435 nm, with a scan speed of 2 nm/s.
From the fluorescence plot, determine the maximum wavelength for excitation. Set the instrument to that excitation wavelength value, in this case 399 nm.
Next, determine the emission wavelength by performing a scan from 450–550 nm. From the resulting fluorescence plot, determine the maximum wavelength and set the emission wavelength, in this case 520 nm.
Measure each sample, including the blank at the selected excitation and emission wavelength. Record each fluorescence intensity reading.
Subtract the measured fluorescence of the blank sample from each of the other 5 samples.
Plot the fluorescence intensity of each of the five samples versus the amount of aluminum added to the sample. Determine the least squares value of the resulting plot, and record the slope and intercept.
The plot of fluorescence intensity vs. amount of aluminum added yielded a least-square line as shown. The amount of aluminum in the sample can then be calculated using this line. Since the amount of unknown added was 25 mL, the determined value, 2.916 μg is divided by 25 mL. This gives a final result of 0.117 μg/mL, or 0.117 ppm. This is quite close to the known value of 0.110 ppm.
Now, let's look at some other analytical techniques that can have skewed results due to matrix effects.
Atomic absorption spectroscopy is an analytical method that measures the absorbance of light by a target analyte in the gaseous phase. For most samples, a simple calibration curve relating absorption to sample concentration, can serve as a reliable method to quantify an unknown concentration.
However, this technique can lose accuracy if other components of the mixture interact with the target analyte and suppress or enhance absorption. The standard addition method can be used in this case to account for the effects of these interactions, especially in samples where the matrix cannot be removed prior to analysis.
Instrument calibration plays a crucial role in the accuracy of a measurement. The method of standard addition is often used to aid in calibration of instruments such as ICP-MS. ICP-MS is a comparative method, meaning that the measurement of an unknown sample is based on the measurement of a chemical standard.
Thus, the uncertainty of a measurement of an unknown can't be better than the uncertainty of the calibration. The method of standard addition can therefore be used to create a calibration curve that is more accurate than the standard method, and accounts for matrix interactions in the sample.
Many biological molecules are analyzed using high-performance liquid chromatography, or HPLC. HPLC is a technique that separates and analyzes complex mixtures based on molecule properties such as polarity, charge, and size. The time at which the analyte leaves the column enables the user to identify each component in the mixture.
Biological molecules can often interact in a mixture, and are greatly affected by the matrix they are suspended in. Often, the method of standard addition is used to create a calibration curve that accounts for these affects.
You've just watched JoVE's introduction to the method of standard addition. You should now understand how to perform the technique to account for matrix effects in sample analysis.
Thanks for watching!
Subscription Required. Please recommend JoVE to your librarian.
Results
A scan of the excitation wavelength from 335–435 showed the highest absorption at 399 nm, so the excitation monochromator was set for that value. Then the emission scan was performed from 450–550 nm, and the strongest signal was found to be at 520 nm. These are the wavelengths that are used for all of the samples.
| Sample | Fluorescence Intensity | Corrected Fluorescence Intensity |
| Blank | 0.008 | 0.000 |
| Sample | 0.128 | 0.120 |
| Sample + 1 mL | 0.167 | 0.159 |
| Sample + 2 mL | 0.220 | 0.212 |
| Sample + 3 mL | 0.260 | 0.252 |
| Sample + 4 mL | 0.290 | 0.282 |
A plot of fluorescence (Figure 3) vs. µg of Al3+ added (Figure 4) yielded a least-squares line of:
Fluorescence Intensity = 0.0417 x (µg of Al3+ added) + 0.1216
Amount of Al3+ = -(Y-Int)/Slope = -0.1216/0.0417 = -2.916 µg/mL
Since the amount of unknown added was 25 mL, then the 2.916 µg/mL value needs to be divided by 25.
Unknown Aluminum Concentration = 2.916 µg/mL / 25.0 mL = 0.117 µg/mL = 0.117 ppm
which is quite close to the actual value of 0.110 ppm (6.4% error).

Figure 3. Fluorescence of the samples.

Figure 4. The standard addition calibration plot.
Subscription Required. Please recommend JoVE to your librarian.
Applications and Summary
The method of standard additions is often the technique utilized when accurate quantitative results are desired, used in analytical analysis such as atomic absorption, fluorescence spectroscopy, ICP-OES, and gas chromatography. This is often used when there are other components in the sample of interest that causes either a reduction or enhancement of the absorbance desired for quantitative results. When this is the case, one cannot simply compare the analytes signal to standards using the traditional calibration curve approach. In fact, matrix effect evaluation should be a mandatory part of the validation procedure.
Another example where standard additions can be used is when extracting silver from old photographic waste. The waste contains silver halides, and can be extracted so the silver can be reclaimed. By spiking the unknown "waste" with known amounts of silver, this method can predict the amount of silver obtained from the photographic film.
Workers who are exposed to benzene manufacturing plants are often tested to verify they are safely below the accepted levels of benzene. Their urine is tested for the chemical, and that is the biological matrix. Also, the amount of analyte suppression varies for different people, so a single calibration kit will not work. With the method of standard addition, every employee can be tested and evaluated accurately.
Subscription Required. Please recommend JoVE to your librarian.