

Summary
Contamination during the genomic sequencing of microscopic organisms remains a large problem. Here, we show a method to sequence the genome of a tardigrade from a single specimen with as little as 50 pg of genomic DNA without whole genome amplification to minimize the risk of contamination.
Abstract
Tardigrades are microscopic animals that enter an ametabolic state called anhydrobiosis when facing desiccation and can return to their original state when water is supplied. The genomic sequencing of microscopic animals such as tardigrades risks bacterial contamination that sometimes leads to erroneous interpretations, for example, regarding the extent of horizontal gene transfer in these animals. Here, we provide an ultralow input method to sequence the genome of the tardigrade, Hypsibius dujardini, from a single specimen. By employing rigorous washing and contaminant exclusion along with an efficient extraction of the 50 ~ 200 pg genomic DNA from a single individual, we constructed a library sequenced with a DNA sequencing instrument. These libraries were highly reproducible and unbiased, and an informatics analysis of the sequenced reads with other H. dujardini genomes showed a minimal amount of contamination. This method can be applied to unculturable tardigrades that could not be sequenced using previous methods.
Introduction
Tardigrades are microscopic animals that can enter an ametabolic state called anhydrobiosis when facing desiccation. They recover by the absorption of water1,2. In the ametabolic state, tardigrades are capable of tolerating various extreme environments, which include extreme temperatures3 and pressures4,5, a high dosage of ultraviolet light6, X-rays, and gamma rays7,8, and cosmic space9. Genomic data is an indispensable foundation for the study of molecular mechanisms of anhydrobiosis.
Previous attempts to sequence the genome of tardigrades have shown signs of bacterial contamination10,11,12,13,14. Genomic sequencing from such small organisms requires a large number of animals and is prone to bacterial contamination; therefore, we have previously established a sequencing protocol using an ultralow input method starting from a single specimen of tardigrade, to minimize the risk of contaminations15. Using these data, we have further conducted a high-quality resequencing and reassembly of the genome of H. dujardini16,17. Here we describe in detail this method for genomic sequencing from a single tardigrade individual (Figure 1). The validation of this sequencing method is beyond the focus of this paper and has already been thoroughly discussed in our previous report16.
This method is comprised of two parts: the isolation of a single tardigrade with the lowest contamination possible, and the high-quality extraction of pictogram levels of DNA. The tardigrade is starved and rinsed thoroughly with water, as well as antibiotics, and observed under a microscope with 500X magnification to ensure the removal of any bacterial contamination. Previous estimates and measurements show that a single individual of tardigrade contains approximately 50 - 200 pg of genomic DNA16, which is extracted by cracking the chitin exoskeleton by freeze-thaw cycles or by manual homogenization. This genomic DNA is submitted to library construction and sequenced on a DNA sequencing instrument. An additional informatics analysis shows high-quality sequencing, as well as low levels of contamination in comparison to previous tardigrade sequencing projects.
Subscription Required. Please recommend JoVE to your librarian.
Protocol
1. Preparation
- Prepare 2% agarose gel using distilled water (DW) as the solvent in a 90-mm plastic culture dish, and 10 mL of a 1% penicillin/streptomycin cocktail with DW. The gel can be stored for 2 - 3 weeks in an incubator set at 18 °C.
NOTE: Avoid any storage of the gel below 10 °C, for the low temperature will shrink the agarose gel, resulting in a minuscule gap between the gel and the culture dish wall in which the tardigrades can be trapped.
2. Sample Preparation and Contaminant Exclusion
- Collect a single tardigrade, place it on the prepared agar plate, and wash it 2x - 3x with DW to remove any remaining particles.
- Incubate the tardigrade at 18 - 22 °C for 24 h to remove any excess food from the intestines.
- Place the starved tardigrade in penicillin/streptomycin antibiotics for 2 - 6 h to remove any bacterial contamination and place the decontaminated animal on a clean slide glass using a P10 pipette.
- Observe the tardigrade under a microscope at 500X magnification and confirm that there are no remaining bacteria.
NOTE: The high magnification with a stereomicroscope is optimal, but an optical microscope may be used alternatively without applying a coverslip. - Collect the individual using a P10 pipette with a maximum of 5 µL of liquid, place it into a low-binding polymerase chain reaction (PCR) tube, and remove as much excess liquid as possible.
NOTE: The microtubes, PCR tubes, and pipette tips should all be low binding to minimize the loss of DNA.
NOTE: Conduct a homogenization as soon as possible, for the rapid desiccation or death of the animal can significantly damage the genomic DNA.
3. Homogenization and DNA Extraction
- Homogenize the animal to obtain its genomic DNA (gDNA) with one of the following methods.
NOTE: It is critical to use the specified kit shown in the Table of Materials in the following DNA extraction steps, for other kits (either column and bead based) are not effective at this extremely low input. The genomic lysis buffer has been supplied with 0.5% beta-mercaptoethanol prior to use.- Homogenize the animal with freeze-and-thaw cycles18.
- Immediately following step 2.5, add 100 µL of lysis buffer to the PCR tube containing the tardigrade.
- Place the PCR tube in liquid nitrogen for 10 min and, for 10 min, move it to a heat block warmed to 37 °C. Repeat this step 2x.
NOTE: This step can be repeated when the homogenization is not sufficient or can be performed after the manual crushing.
- Manually crush the animal.
- Immediately following step 2.5, under a stereomicroscope, crush the individual with the P10 tip of the pipette by pressing the animal against the PCR tube wall, and immediately add 100 µL of lysis buffer.
NOTE: It is critical to observe this procedure under a stereomicroscope, because the tardigrade can easily slip away, and inefficient crushing will result in a failure to extract gDNA. Make sure that the tardigrade cuticle is broken so that the lysis buffer can infiltrate the organism.
- Immediately following step 2.5, under a stereomicroscope, crush the individual with the P10 tip of the pipette by pressing the animal against the PCR tube wall, and immediately add 100 µL of lysis buffer.
- Homogenize the animal with freeze-and-thaw cycles18.
- Incubate the tube for 30 min at room temperature for lysis to occur.
NOTE: A minimum of 30 min is required for an efficient lysis, but the incubation can be longer. - Transfer the complete volume (100 µL) of the lysis mixture to a clean 1.5-mL low-binding microtube.
- Add 100 µL of lysis buffer to the low-binding PCR tube that was used for the homogenization and is now empty, and after pipetting the mixture, transfer it to the 1.5-mL low-binding microtube used in step 3.3. Repeat this step 2x.
- As in step 3.4, add 300 µL of lysis buffer to the low-binding PCR tube, and, after pipetting, move the mixture to the 1.5-mL low-binding microtube. The 1.5-mL low-binding microtube should contain 600 µL of the mixture after this step.
NOTE: These steps are to minimize the loss of gDNA bound to the PCR tube wall, by washing the sample multiple times. - Add the total of 600 µL of lysis mixture to the spin column placed in a collection tube and centrifuge it at 10,000 x g for 1 min.
- Re-apply the flow through to the column and centrifuge it at 10,000 x g for 1 min.
NOTE: This step is critical to ensure that most of the gDNA is bound to the column. - Add 500 µL of wash buffer to the spin column and centrifuge it at 10,000 x g for 1 min. Transfer the spin column to a clean 1.5-mL microtube.
- Apply 20 µL of 10 mM Tris-HCl, pH 8.5, to the spin column, wait for 5 min at room temperature, and centrifuge it at 10,000 x g for 1 min.
NOTE: The elution buffer must not contain EDTA, for it interferes with the library preparation enzymes. - Re-apply the flow through the spin column, and after 5 min of incubation at room temperature, centrifuge it for 1 min at 10,000 x g.
NOTE: This step is critical to ensure the maximal elution of the gDNA bound to the column.
4. Library Construction Sequence
- DNA fragmentation
- Transfer 15 µL of the gDNA eluant to a 15-µL microtube for DNA fragmentation and centrifuge the tube for 1 min using a tabletop microcentrifuge.
NOTE: The specified microtube shown in the Table of Materials is optimal for a low input. The fragmentation with low-volume sonication is critical, and this cannot be substituted with enzymatic fragmentation because of the extremely low concentration of DNA. - Fragment the gDNA to 550 bp.
NOTE: The 550 bp settings we used are as follows: peak incident power = 30 W, duty factor = 20%, cycles per burst = 50, treatment time = 23 s. - After a thorough pipetting, transfer 10 µL of the fragmented DNA mixture to a clean low-binding PCR tube.
NOTE: The experiment can be stopped here. Preserve the DNA at 4 °C or -20 °C.
- Transfer 15 µL of the gDNA eluant to a 15-µL microtube for DNA fragmentation and centrifuge the tube for 1 min using a tabletop microcentrifuge.
- Library construction sequence
NOTE: It is absolutely critical to use the specified kit in the Table of Materials in the following procedures, due to the low amount of input DNA.- Add 2 µL of the template preparation buffer and 1 µL of the template preparation enzyme and mix them thoroughly with a pipette.
- Perform the template preparation reaction on a thermal cycler with the following conditions: 22 °C for 25 min, 55 °C for 20 min, 4 °C hold, and a heated lid at 101 - 105 °C. Once the reaction is completed, proceed to the next step.
- Add 1 µL of the library synthesis buffer and 1 µL of the library synthesis enzyme to the template preparation reaction product and incubate the mixture at 22 °C for 40 min (with a 4 °C hold). Once the reaction is completed, proceed to the next step.
- Add 30 µL of the library amplification master mix (25 µL of the library amplification buffer, 1 µL of the library amplification enzyme, and 4 µL of nuclease-free water) and 5 µL of the indexing reagent.
- Mix everything thoroughly with a pipette and centrifuge the mixture briefly with a tabletop centrifuge.
- Perform a PCR with the conditions presented in Table 1.
| Temperature | Time | Cycles |
| 72 ˚C | 3 minutes | |
| 85 ˚C | 2 minutes | |
| 98 ˚C | 2 minutes | |
| 98 ˚C | 20 seconds | 4 Cycles |
| 67 ˚C | 20 seconds | |
| 72 ˚C | 40 seconds | |
| 98 ˚C | 20 seconds | 16 Cycles |
| 72 ˚C | 50 seconds | |
| 4 ˚C | Hold |
Table 1: PCR conditions.
- Purification of the PCR products
NOTE: This step can be substituted with other purification methods. If an alternative method is used, make sure to check for the resulting DNA purity using a spectrophotometer. Absorbance ratios of 260/280 and 260/230 must both be above 1.8.- Add 50 µL of magnetic beads, pipette the solution 10x, and centrifuge it briefly with a tabletop microcentrifuge.
- Incubate the solution for 2 min at room temperature.
- Incubate it on a magnetic stand for 5 min or until the solution becomes completely clear, and remove the supernatant.
- Add 200 µL of freshly prepared 80% ethanol to the low-binding PCR tube on a magnet stand, wait for 30 s, and remove the supernatant. Repeat this step 2x. Do not disturb the beads.
- Briefly centrifuge the low-binding PCR tube with a tabletop centrifuge and remove any excess ethanol on the magnetic stand. Air-dry the beads but avoid overdrying.
- Resuspend the beads with 15 µL of 10 mM Tris-HCl, pH 8.5, thoroughly pipette the solution so that the magnetic beads are homogeneously distributed, incubate the tube for 2 min at room temperature, and after a brief centrifugation, incubate it on a magnetic stand for 2 min until the solution is clear.
NOTE: The elution buffer should not contain EDTA, for it interferes with the sequencing chemistry. - Transfer the supernatant, without disturbing the pellet, to a new low-binding PCR tube.
NOTE: The experiment can be stopped here. Preserve the DNA at 4 °C or at -20 °C for long-time storage.
5. Quality Check, Quantification, and Sequence of the DNA
NOTE: A quality check is not conducted prior to this step due to the low amount of DNA.
- Validation of the DNA library size distribution
NOTE: Other high-sensitive electrophoresis systems with a digital reporting of size distribution can be used.- Return the electrophoresis buffer reagent and gel cartridge (25 - 1,000 bp in range) to room temperature.
- Add 3 µL of electrophoresis buffer reagent with 1 µL of the sequencing library and mix them thoroughly for 1 min with a vortex, and briefly centrifuge them with a tabletop centrifuge.
- Conduct electrophoresis and validate the library size distribution with the associated software. The main fragment peak should be broadly ranging from about 300 to 1,000 bp.
- DNA quantification
NOTE: Other fluorescence-based methods or quantitative PCR can also be used, but spectrophotometry should be avoided, as it is not accurate enough for quantifying sequencing libraries.- Add 796 µL of solution buffer and 4 µL of fluorescent reagent and mix them thoroughly. Dispense 190 µL of the working solution to two assay tubes and 197 µL to one assay tube.
- Add 10 µL of standards with known concentrations of DNA to each assay tube containing 190 µL of the working solution, and 3 µL of the prepared library to the assay tube containing 197 µL of the working solution.
- Vortex the tubes briefly and centrifuge them on a tabletop centrifuge. Quantify the DNA using a fluorometer with 3-µL settings.
- DNA library sequence
Note: The sequencing platform must be compatible with the specified library construction kit.- Prepare the sequencing library based on the manufacturer's protocol.
- Set the reagent cassette and flow cell into the sequencing instrument and enter the sequencing run information following the manufacturer's protocol.
- Run the sequencing.
NOTE: We have conducted two sequencing runs: one sample/run, as well as four samples multiplexed in one run.
6. Computational Analysis
- Base-call and, if required, demultiplex the reads.
- Validate the quality of the sequence data with FastQC19.
NOTE: For a more in-depth validation of the obtained data, see our previous report16.
Subscription Required. Please recommend JoVE to your librarian.
Representative Results
Contaminant Exclusion:
This protocol involves a thorough washing of the tardigrade and a sterilization with antibiotics treatment to minimize contamination. It also involves a visual checking process to ensure the completeness of these processes. A microscope image made during the validation (step 2.4 of the protocol) is shown in Figure 2. When observed at a 500X magnification, bacterial cells can be seen as small particles that move around the tardigrade individual.
Validation of the DNA Library Quality:
The total amount of the constructed DNA-Seq library is approximately 109.5 ng (7.3 ng/µL x 15 µL)16. To validate the length distribution of the fragmentation, an electrophoresis pattern should be similar to Figure 3. As we set the fragmentation size to 550 bp with a DNA shearing system, the library should be 550 - 600 bp, including the sequencing adaptors. It can be observed that the majority of the sequence library is contained between 200 - 1000 bp and is consistent between replicates (N1 - N4).
Sequence Data Analysis:
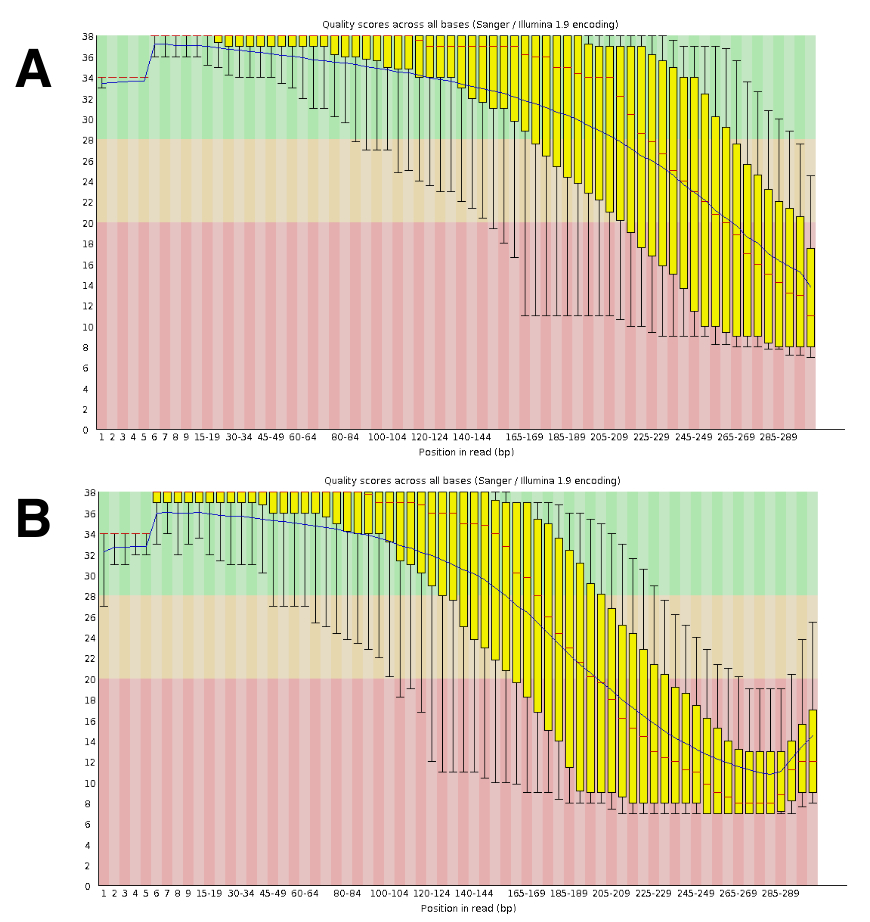
The DNA sequencing generated roughly 20- to 25-M paired reads per run. The validation of the quality was conducted using FastQC (Figure 4). The distribution of the quality along the sequenced read is typical of a 300-bp paired run.

Figure 1: Workflow of this protocol. This figure shows a summary of this protocol. Please click here to view a larger version of this figure.

Figure 2: Representative photo of a bacteria-free tardigrade. This figure shows images of a contaminated (left) tardigrade and cleaned (right) tardigrade (Hypsibius dujardini), along with further magnified images (bottom). Rod-shaped cells around the tardigrade are contaminants and are indicated with an arrow. The scale bar indicates 100 µm. Please click here to view a larger version of this figure.

Figure 3: Validation of the fragment length distribution of the constructed DNA library. This panel shows the distribution of the sequencing library size. The purple and green lines indicate the upper and lower markers at 1,500 and 25 bp, respectively. L = Ladder, S = 1 sample/run, N1 - N4 = 4 replicates/run. Please click here to view a larger version of this figure.

Figure 4: Example of the validation of the DNA-Seq quality with FastQC. DNA-Seq data were submitted to FastQC to validate the sequence performance. A representative result for DRR055040 per base sequence quality is shown (DDBJ Sequence Read Archive DRA00445516). (A) This panel shows the forward reads (R1). (B) This panel shows the reverse reads (R2). Please click here to view a larger version of this figure.
Subscription Required. Please recommend JoVE to your librarian.
Discussion
Bacterial contamination poses a threat to the genomic sequencing of microscopic organisms. While previous studies on tardigrade genome sequencing have filtered out contamination using extensive informatics methods12,20, we sequenced the genome from a single individual to minimize the risk of contaminations. Since an individual tardigrade contains approximately 50 - 200 pg of genomic DNA16 and is encased in a thick layer of chitin exoskeleton, the exclusion of contaminants and high-quality DNA extraction are the critical points in this protocol. Existing tardigrades cultures are not aseptic, and those collected from the wild carry a lot of contaminants on the surface, as well as the remains of food in their intestines. Previous genome sequencing projects of tardigrades have sequenced 10,000 - 100,000 individuals collectively as one sample12,14, which means the results are very likely to be influenced by bacterial contaminants. In their report, Boothby et al. collected H. dujardini individuals by using their negative phototaxis behavior14, and the group did not employ any anti-bacterial methods.
To visually examine if there are contaminants, we incubated the tardigrade in antibiotics (penicillin/streptomycin) and examined the individual under a 500X microscope. By isolating a single individual and carefully inspecting it for any contaminants, we minimized the risk of possible contaminations. Low levels of contamination were confirmed from the sequencing data as well16. As for DNA extraction, we employed manual homogenization, as well as thermal homogenization18. By submitting the tardigrade individual to liquid nitrogen and 37 °C, cracks were induced in the chitin exoskeleton, and the lysis buffer was able to penetrate the body and lyse the cells. When the DNA yield remains lower than anticipated, both thermal and manual homogenization may be conducted to maximize the yield.
The method stated in this article has several limitations. First, homogenization by freeze-and-thaw cycles was applied from a study on nematodes; thus, the method may only be effective against ecdysozoa. Secondly, due to the amplification of DNA fragments during the DNA sequence library phase, the possibility of PCR errors cannot be ignored. Thus, the sequence data is not recommended for analysis that requires high-accuracy reads (i.e., SNP analysis). Furthermore, as we have stated in the protocol, the usage of the specified SNA-Seq kit shown in the Table of Materials is absolutely critical, due to the low amount of input DNA. This DNA library construction kit ligates Illumina adaptor sequences prior to the amplification; therefore, this library cannot be applied for long-read sequencing using PacBio or Nanopore technology. Finally, a quality check of the constructed DNA library during this protocol occurs only once, after the sequencing library construction. This is due to the low input of DNA since most DNA quantification and electrophoresis methods cannot detect 50 - 200 pg of DNA. Therefore, we have conducted quality checks, such as the electrophoresis (Figure 1) and fluorescence-based quantifications, only after the PCR amplification.
A full discussion of bioinformatics analyses of this data is beyond the scope of this article; however, we have briefly stated several analyses we have conducted. A quality check of the sequencing data with FastQC19 calculates the per-base qualities, sequence duplication, etc. Sequence data that have been validated can be submitted to the genome assembly. We have assembled a 132 Mb genome with MaSuRCA v3.1.321 and have compared the mapping statistics calculated with BWA22 and QualiMap23 of this DNA sequencing library with other H. dujardini genome assemblies16. Furthermore, we also have used this DNA sequencing data for the exclusion of contaminants in our study17, and have observed that the sequenced reads are distributed evenly throughout the genome.
Most projects on non-model organisms start from culturing enough sample material, as was the case with tardigrades24. Technical advances in culture techniques have enabled high quantities of tardigrade culture, but current culture methods are not yet aseptic, and since most tardigrades are still unculturable in labs, it has been nearly impossible to conduct genome or transcriptome sequencing. This DNA sequencing method from a single individual makes it possible to analyze rare tardigrade species, including marine species that have been studied less. By conducting comparative genomics at a wider phyletic area, a further understanding of anhydrobiosis mechanisms in tardigrades may be achieved.
Subscription Required. Please recommend JoVE to your librarian.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors thank Nozomi Abe, Yuki Takai, and Nahoko Ishii for their technical support in genomic sequencing. This work was supported by Grant-in-Aid for the Japan Society for the Promotion of Science (JSPS) Research Fellow, KAKENHI Grant-in-Aid for Young Scientists (No.22681029), and KAKENHI Grant-in-Aid for Scientific Research (B), No. 17H03620 from the JSPS, by a Grant for Basic Science Research Projects from The Sumitomo Foundation (No.140340), and partly by research funds from the Yamagata Prefectural Government and Tsuruoka City, Japan. Chlorella vulgaris used to feed the tardigrades was provided courtesy of Chlorella Industry Co. LTD.
Materials
| Name | Company | Catalog Number | Comments |
| SZ61 microscope | OLYMPUS | ||
| BactoAgar | Difco Laboratories | 214010 | |
| Penicillin Streptomycin (10,000 U/mL) | Gibco by life technologies | 15140-148 | |
| VHX-5000 System | Keyence | ||
| 0.2mL Silicone coating tube | Bio Medical Science | BC-bmb20200 | |
| Quick-DNA Microprep Kit | ZYMO Research | D3021 | Use of this kit is absolutey critical; see step 3.1 |
| 1.5 mL microtube | greiner bio-one | 616-201 | See 4.1.1 |
| HIgh speed refrigerated micro centrifuge | TOMY | MX-307 | |
| Covaris M220 | Covaris Inc. | 4482277 | |
| ThruPLEX DNA-Seq kit | Rubicon Genomics | CAT. NO. R400406 | Use of this kit is absolutey critical; see step 4.2 |
| Thermal Cycler | Bioer Technology | TC-96GHbC | |
| AMPure XP reagent | BECKMAN COULTER Life Science | A63881 | |
| Ethanol | Wako | 054-027335 | |
| EB buffer | QIAGEN | 19086 | |
| 2200 TapeStation | Agilent | G2965AA | |
| D1000 Reagents | Agilent | 5067-5583 | |
| D1000 ScreenTape | Agilent | 5067-5582 | |
| Qubit dsDNA BR Buffer/Reagent | ThermoFisher Scientific | Q32850 | |
| Cubee Mini-Centrifuge | RecenttecGenereach | R5-AQBD01aqbd | |
| MiSeq 600 cycle v3 | Illumina Inc. | MS-102-3003 | |
| MiSeq Sequencer | Illumina Inc. | SY-410-1003 |
References
- Crowe, J. H., Hoekstra, F. A., Crowe, L. M. Anhydrobiosis. Annual Review of Physiology. 54 (1), 579-599 (1992).
- Mobjerg, N., et al. Survival in extreme environments - on the current knowledge of adaptations in tardigrades. Acta Physiologica. 202 (3), 409-420 (2011).
- Becquerel, P. La suspension de la vieau dessous de 1/20 K absolu par demagnetization adiabatique de L'alun de fer dans le vide les plus eléve. Comptes Rendus de l'Académie des Sciences. 231, 261-264 (1950).
- Ono, F., et al. Effect of ultra-high pressure on small animals, tardigrades and Artemia. Cogent Physics. 3 (1), 1167575 (2016).
- Horikawa, D. D., et al. Tolerance of anhydrobiotic eggs of the Tardigrade Ramazzottius varieornatus to extreme environments. Astrobiology. 12 (4), 283-289 (2012).
- Horikawa, D. D., et al. Analysis of DNA repair and protection in the Tardigrade Ramazzottius varieornatus and Hypsibius dujardini after exposure to UVC radiation. PLoS One. 8 (6), e64793 (2013).
- Horikawa, D. D., et al. Radiation tolerance in the tardigrade Milnesium tardigradum. International Journal of Radiation Biology. 82 (12), 843-848 (2006).
- May, R. M., Maria, M., Gumard, J. Action différentielle des rayons x et ultraviolets sur le tardigrade Macrobiotus areolatus, a L'état actif et desséché. Bulletin Biologique de la France et de la Belgique. 98, 349-367 (1964).
- Jonsson, K. I., Harms-Ringdahl, M., Torudd, J. Radiation tolerance in the eutardigrade Richtersius coronifer. International Journal of Radiation Biology. 81 (9), 649-656 (2005).
- Bemm, F., Weiss, C. L., Schultz, J., Forster, F. Genome of a tardigrade: Horizontal gene transfer or bacterial contamination? Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3054-E3056 (2016).
- Delmont, T. O., Eren, A. M. Identifying contamination with advanced visualization and analysis practices: metagenomic approaches for eukaryotic genome assemblies. PeerJ. 4, e1839 (2016).
- Koutsovoulos, G., et al. No evidence for extensive horizontal gene transfer in the genome of the tardigrade Hypsibius dujardini. Proceedings of the National Academy of Sciences of the United States of America. 113 (18), 5053-5058 (2016).
- Boothby, T. C., Goldstein, B., et al. Reply to Bemm et al. and Arakawa: Identifying foreign genes in independent Hypsibius dujardini genome assemblies. Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3058-E3061 (2016).
- Boothby, T. C., et al. Evidence for extensive horizontal gene transfer from the draft genome of a tardigrade. Proceedings of the National Academy of Sciences of the United States of America. 112 (52), 15976-15981 (2015).
- Arakawa, K. No evidence for extensive horizontal gene transfer from the draft genome of a tardigrade. Proceedings of the National Academy of Sciences of the United States of America. 113 (22), E3057 (2016).
- Arakawa, K., Yoshida, Y., Tomita, M. Genome sequencing of a single tardigrade Hypsibius dujardini individual. Scientific Data. 3, 160063 (2016).
- Yoshida, Y., et al. Comparative genomics of the tardigrades Hypsibius dujardini and Ramazzottius varieornatus. PLoS Biology. 15 (7), e2002266 (2017).
- He, F. Total RNA Extraction from C. elegans. Bio-protocol. Bio101, e47 (2011).
- Andrews, S. FastQC a quality-control tool for high-throughput sequence data. , http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2015).
- Hashimoto, T., et al. Extremotolerant tardigrade genome and improved radiotolerance of human cultured cells by tardigrade-unique protein. Nature Communications. 7, 12808 (2016).
- Zimin, A. V., et al. The MaSuRCA genome assembler. Bioinformatics. 29 (21), 2669-2677 (2013).
- Li, H., Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 25 (14), 1754-1760 (2009).
- Okonechnikov, K., Conesa, A., Garcia-Alcalde, F. Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics. 32 (2), 292-294 (2016).
- Horikawa, D. D., et al. Establishment of a rearing system of the extremotolerant tardigrade Ramazzottius varieornatus: a new model animal for astrobiology. Astrobiology. 8 (3), 549-556 (2008).

{kind=link}