

Summary
Farmacoterapia de reemplazo de dopamina mediante L-DOPA es el tratamiento más comúnmente utilizado sintomático de la enfermedad de Parkinson, pero se acompaña de efectos secundarios como movimientos involuntarios anormales, discinesia denomina
Abstract
MALDI imágenes de espectrometría de masas (IMS) es un enfoque poderoso que facilita el análisis espacial de las especies moleculares en muestras de tejidos biológicos 2 (Fig. 1). Una sección 12 micras tejido delgado está cubierta con una matriz de MALDI, lo que facilita la desorción y la ionización de péptidos y proteínas intactas que pueden ser detectadas con un analizador de masa, típicamente usando una TOF MALDI / espectrómetro TOF. Por lo general cientos de picos se puede evaluar en una sección de tejido cerebral de rata sola. En contraste con las técnicas de imagen comúnmente utilizados, este enfoque no requiere conocimiento previo de las moléculas de interés y permite el análisis no supervisado y completa de múltiples especies moleculares mientras se mantiene una alta especificidad y sensibilidad molecular 2. Aquí se describe un método MALDI basada en IMS a fin de elucidar los perfiles específicos de la región de distribución de neuropéptidos en el cerebro de la rata de la enfermedad de un modelo animal de Parkinson (EP).
Enfermedad de Parkinson es una enfermedad común neurodegenerativa con una prevalencia del 1% para las personas mayores de 65 años de edad 3,4. El tratamiento sintomático más común se basa en la sustitución de la dopamina mediante L-DOPA 5. Sin embargo, esto se acompaña de efectos secundarios graves, incluyendo movimientos involuntarios anormales, llamados L-DOPA discinesias inducidas (LID) 1,3,6. Uno de los cambios moleculares más destacado en la tapa es una regulación del ARNm precursor de prodinorfina opioides 7. Los péptidos dinorfina modular la neurotransmisión en áreas del cerebro que están esencialmente involucrados en el control de movimiento de 7,8. Sin embargo, hasta la fecha los péptidos opioides exactas que se originan en el procesamiento del precursor neuropéptido no se han caracterizado. Por lo tanto, hemos utilizado MALDI IMS en un modelo animal experimental de la enfermedad de Parkinson y la L-DOPA discinesia inducida.
MALDI imágenes espectrometría de masas demostrado ser particularmente ventajoso con respecto a neuropéptido caracterizaciónción, ya que los enfoques basados en anticuerpos de uso común dirigido a las secuencias de péptidos conocidos y previamente observados modificaciones post-traduccionales. Por el contrario MALDI IMS puede desvelar nuevos productos de procesamiento de péptidos y así revelar nuevos mecanismos moleculares de la modulación de neuropéptido de la transmisión neuronal. Mientras que la cantidad absoluta de los neuropéptidos no se puede determinar por MALDI IMS, la abundancia relativa de los iones de péptidos se pueden delinear a partir de los espectros de masas, ofrecerá una visión de cambio en los niveles de salud y la enfermedad. En los ejemplos que aquí se presentan, las intensidades máximas de dinorfina B, alfa neoendorphin y la sustancia P se encontró que se incrementó significativamente en el dorso-lateral, pero no el cuerpo estriado dorsomedial, de los animales con discinesia grave en el tronco facial, y los músculos orolingual (Fig. 5). Por otra parte, MALDI IMS reveló una correlación entre la severidad de la discinesia y los niveles de des-tirosina alfa-neoendorphin, lo que representa un mecanismo previamente desconocido de la inactivación funcionalvación de dinorfinas en el cuerpo estriado como la eliminación de la N-terminal de la tirosina reduce la dinorfina de los receptores opiáceos capacidad de unión 9. Este es el primer estudio sobre la caracterización de neuropéptido Y en tapa con MALDI IMS y los resultados destacan el potencial de la técnica para su aplicación en todos los ámbitos de la investigación biomédica.
Protocol
El protocolo se ajusta a los efectos de un análisis estadístico de los datos de IMS MALDI de múltiples secciones de cerebro de rata, por lo general 20-30 secciones, y consta de cinco pasos diferentes que comprenden la preparación del tejido, la matriz de la aplicación, MALDI-TOF MS análisis, la evaluación de datos, y el neuropéptido identificación. Los procedimientos se describen y se describe con más detalle a continuación:
1. Preparación de tejidos
Este procedimiento incluye la recolección de las muestras de tejido respectivos así como el tejido de seccionamiento para el análisis de IMS. Un objetivo particular de la proteína y el análisis de péptidos es para evitar la degradación proteolítica. Por lo tanto, es esencial trabajar rápido y diligente durante la disección de los tejidos.
- Sacrificio ratas (normalmente 250-300 g) por decapitación, retire el cerebro de ratas en un plazo máximo post-mortem tiempo de 30 menores y la congelación en hielo seco en polvo antes de pasar a -80 ° C congelador. Más rápido de congelación con nitrógeno líquidoaumentar el riesgo de microdesgarros en el tejido cerebral, que afectará negativamente a la cristalización de la matriz y con ello reducir la calidad de la EM (Fig. 2D). Cerebros enteros se pueden almacenar durante varios años antes de seccionar sin pérdida de calidad de la señal EM.
- Cortar tejido congelado en un micrótomo criostato a 12 micras rodajas y montaje de deshielo en cortes de tejido conductores portaobjetos de vidrio MALDI (óxido de estaño indio diapositivas recubiertos, Bruker Daltonics) o MALDI objetivo (Fig. 2A-C).
- Secciones en seco durante 15 minutos bajo vacío y se desliza tienda a -80 ° C hasta su uso posterior. Las secciones de tejido debe ser analizado en el menor tiempo posible después de la sección, incluso si se almacenan a -80 ° C. Encontramos que la calidad de la señal EM se reduce notablemente después de un año en el almacenamiento. Con el fin de reducir la oxidación de las proteínas y péptidos, el aire en el recipiente de almacenamiento puede ser reemplazado con un gas inerte (por ejemplo argón o nitrógeno).
2. Matriz de la aplicación
La aplicación de matrizpaso tiene un impacto significativo sobre la calidad del espectro y requiere la optimización de múltiples parámetros en función del tipo de tejido, así como el analito de interés. Estos factores incluyen parámetros químicos tales como el tipo de matriz, la concentración de la matriz, el pH, el lavado de tejidos y modificadores orgánicos, así como configuraciones instrumentales incluyendo el volumen de depósitos, resolución lateral y el número de deposiciones 10 (Fig. 2D). Para los experimentos a gran escala, es de gran importancia para reducir las variaciones, por ejemplo mediante la aplicación de la matriz para todas las secciones dentro de un día y por el mismo operador. Aunque hay muchas estrategias para aplicar la solución de matriz tal como por sublimación o por pulverización, la deposición automatizado de las matrices de las gotitas de matriz pequeños, sobre 100-150 picolitros de tamaño, se ha utilizado con éxito para el análisis de proteínas pequeñas y neuropéptidos en diversos tejidos , incluyendo las 9 secciones del cerebro, 10,11, 12, 13.
- Descongele las secciones en un desecador durante 1 hnuestra.
- Asegúrese de que el experimento está cegado por una persona que no sea el operador. Volver a etiquetar todas las muestras.
- Lavar secciones 1x en 70% de etanol (EtOH, a temperatura ambiente, la RT) durante 10 segundos y dos veces en EtOH al 95% (RT) durante 10 segundos. Para los experimentos de gran tamaño, realizar el lavado de todo el vidrio se desliza junto con una cubeta con el fin de minimizar la variación.
- Secar las secciones en un desecador durante 10 min.
- Evaluar las secciones de tejido bajo un microscopio y comprobar si hay deformación de los tejidos, microdesgarros y pequeñas grietas que perjudicará la calidad MALDI MS (Fig. 2D).
- Preparar una solución fresca matriz que consta de 50 mg / ml DHB en metanol al 50%, 10% de acetato de amonio 150 mM (AMAC) y 0,3% de ácido trifluoroacético (TFA) en agua.
- Matriz de aplicación se realiza mediante deposición de gotículas discretas en un patrón rectangular utilizando una impresora de chorro de tinta química (CHIP, Shimadzu). El primer paso es optimizar los parámetros experimentales de aplicación matriz para neuropéptido includi análisisng el número de gotas por pasada, el número de pasadas. Este experimento se lleva a cabo mediante la aplicación de múltiples arrays de la matriz con los parámetros de aplicación diferentes en la sección de tejido de la misma, mientras se asegura de que cada serie está cubriendo las mismas regiones cerebrales, como cuerpo calloso, la corteza, y el cuerpo estriado. El mismo experimento se tiene que realizar cada parámetros de tiempo se cambian, incluidas las estructuras cerebrales diferentes, matrices diferentes dirigidos analitos específicos, y si disolventes diferentes de la matriz son necesarios para la extracción de analitos específicos.
- Analizar el soporte de diapositivas de cristal con la sección de tejido y alinear el titular. Definir la matriz para la aplicación de la matriz en la sección de tejido y especificar el lugar, es decir la resolución espacial para detectar la distancia. Aplicar la matriz mediante el protocolo optimizado de la impresora de inyección de tinta química. Para este experimento se utilizó un protocolo optimizado para obtener imágenes de péptido con los parámetros de impresión: 10 gotas (100 PL / gota), 10 de aplicación pasa y un punto al punto dIstance de 300 micras.
- Scan final de la matriz visto secciones y guardar la imagen para su registro antes de la MALDI de adquisición de datos (paso 3.4).
- Almacenar las secciones hasta su uso posterior en un desecador a vacío.
3. MALDI MS de adquisición de datos y el procesamiento
MS análisis de los neuropéptidos se realiza en un tiempo de vuelo por instrumentos MALDI (Ultraflex II, Bruker Daltonics, Alemania) que funcionan en modo de espejo, con la adquisición de datos de software asistida desde todos los rincones sola matriz 14. Por lo tanto la enseñanza espacial precisa es imprescindible. Es esencial que la optimización MALDI, la adquisición y especialmente los experimentos de registro objetivo se llevan a cabo por el mismo operador que preferiblemente debe ser cegado a los grupos experimentales. En un experimento a gran escala con múltiples portaobjetos de vidrio, los experimentos MALDI puede ser realizado por un operador, mientras que otra persona está utilizando la impresora de chorro de tinta química.
- Cargar portaobjetos de vidrio en el espectrómetro de masas.
- Compruebe la calibración del método de adquisición MALDI usando una mezcla de bajo peso molecular estándar de calibración (Bruker Daltonics).
- Optimizar los parámetros de adquisición.
- Con el fin de optimizar la señal MS y para evitar la ablación de la matriz de los depósitos de la matriz de vecinos, el tamaño del láser y el enfoque óptimo en el tejido debe ser determinada.
- La energía del láser se establece para garantizar la máxima calidad de la MS como los depósitos de la matriz como sea posible sin elevar la línea de base, reduciendo la resolución de pico o la saturación del detector.
- Evaluar el número máximo de disparos por punto de la matriz hasta que el ruido sólo se detecta, a menudo 1000-2000 disparos. Estimar el número de disparos que deben ser acumulados y el número de disparos adquiridos antes de la posición del láser dentro de un terreno debe cambiar. Con el fin de tomar muestras de cada punto de la matriz en forma pareja, se acumulan 600 disparos en incrementos de 25 tiros, para un total de 24 pasos con un random patrón de movimiento, de cada deposición de la matriz.
- Registro de la exploración de todos los sectores manchados a las coordenadas de motor de la etapa de MALDI utilizando el FlexImaging (v.2.0) 10 y llevar a cabo la adquisición de datos en modo batch por un software AutoXexuteBatchRunner.exe.
- Procesar cada espectro de una sola línea de base por medio de la resta (Convex casco V3), suavizado y calibración externa (opcional), seguida de la exportación como un archivo ASCII (*. Dat, *. Txt o *. Csv) 15.
4. Evaluación de los datos
Evaluación de datos final comprende el procesamiento de datos y posterior reducción de datos por el foco sólo en la información de pico, seguido por el análisis estadístico.
- Como primer paso, las secciones de MALDI IMS se evaluaron los efectos overnormalization. Esto se puede conseguir fácilmente mediante el empleo de herramientas de visualización de datos, tales como FlexImaging (Bruker Daltonics) o BioMap (Novartis). Como un paso inicial de las imágenes de iones totales son evaluated antes de corriente total de iones (TIC) la normalización, seguido por la inspección manual de las imágenes individuales de iones de distribución de los distintos picos de péptidos prominentes. Busque características distribuciones pico de intensidad y si están relacionados con las características del tejido (daños), al ver los efectos de calidad o normalización (Fig. 3).
- Delimitar las regiones de interés (por ejemplo, el cuerpo estriado), de acuerdo a las características histológicas y exportación de los espectros correspondientes en formato ASCII. Preferiblemente, la normalización de los espectros de la corriente iónica total (TIC) se puede realizar en esta etapa.
- Importar archivos ASCII en un software de manejo de datos tales como el origen (v.8.1, OriginLab), MATLAB (The MathWorks, Natick, MA, EE.UU.) o R 16. La detección de picos se puede realizar utilizando herramientas de punta para encontrar incluidos en el software, por ejemplo, "el análisis de pico" en el origen o "mspeaks" en Matlab. Exportar los peaklists de todos los espectros como un archivo de texto delimitado por una sola ficha.
- Con el fin de determinar las fronteras bin para el péptido detectadopicos, el análisis de hurgar en la basura se lleva a cabo usando las herramientas apropiadas de software (por ejemplo, pBIN 17) o en la casa escrito guiones para MATLAB o R. A continuación, el archivo de texto único que contenga todos los picos recogidos los datos se cargan en el software y los parámetros para la determinación de las fronteras de pico se especifican tales como la frecuencia con un pico deben estar presentes en los espectros con el fin de ser relevante para el experimento. Por ejemplo, el experimento contiene 2 grupos de animales, 5 animales en cada grupo, y los espectros 100 están recogidos de cada animal y la región de interés. Supongamos un pico es potencialmente interesante si está presente en al menos la mayoría de los animales en un grupo (3.5) y en al menos la mitad de los espectros de estos animales (3x50 = 150 espectros), esto dará un porcentaje total del 15% para el espectro de 150 positivos de un total 1000 (2x5x100) espectros. Con la herramienta pBIN, este paso se obtiene un archivo binrange única que contiene todos los anchos de caja determinados a partir de los datos adquiridos. Con el fin de verificar que las fronteras de basurason apropiados, es fácil de visualizar los contenedores en origen, junto con las huellas espectros originales.
- Pico del área de la integración puede reducir la varianza que es importante para el análisis estadístico. Nosotros usamos una secuencia de comandos en la casa preparada para el R para calcular el área bajo la curva entre las fronteras de pico determinado en el paso 4. Integradas las áreas de pico se importan a MS Excel (v.2007) y el análisis estadístico por medio de la no-paramétrica de la prueba no apareada con la herramienta SAM se lleva a cabo 18.
5. Péptido de identificación
Secuencia de verificación de las identidades de péptidos observados es esencial para la conclusión de relevancia biológica. El enfoque más preciso incluir cierto de arriba a abajo la determinación directa de tejido utilizando la fragmentación de péptidos mediante espectrometría de masas en tándem (MS / MS), a pesar de las altas concentraciones de péptidos son necesarios para este tipo de análisis 12,13. Para péptidos de bajo abundantes o péptidos múltiples con Closem / valores de z (± 0,5%), en el análisis de los tejidos se altera y el análisis de los tejidos fuera de uso de una estrategia peptidomic se utiliza, que incluye la extracción, separación e identificación basada en MS de neuropéptidos endógenos. Para el experimento se presenta aquí, el foco central estaba en la detección de péptido opioide, que es un desafío especial, ya que estos péptidos son abundantes bastante baja en comparación con otros neuropéptidos en los espectros. Además, estos péptidos son más bien polar que los hace relativamente hidrófila y difícil de retener con la extracción de péptido común y técnicas de separación .. Por lo tanto se aplicó un protocolo previamente reportado para la extracción de los tejidos y fraccionamiento previo de péptidos opioides en combinación con LC-MS/MS estándar según la identificación de péptidos 9,19.
- Recoger las secciones coronales de estructuras diana de interés (núcleo accumbens, NAc, caudado putamen, CPU). Montar el cerebro de rata congelada en un microtomo criostato y eliminar el material que rodea el cerebro (cortex, tabique, cuerpo calloso) con un bisturí. Recoger las secciones (30 m, n = 50) de la disección NAc y la CPU y la descongelación montar el NAc y partes de la CPU de las secciones sobre portaobjetos de vidrio diferentes.
- Extracto de péptidos frente a los tejidos mediante la adición de TFA 100 l 5% ACN/0.1%, se incuba durante dos minutos y se recoge en tubos eppendorf de unión a proteínas de baja. Repita este paso dos veces.
- Realizar fraccionamiento previo péptido por medio de cromatografía de intercambio catiónico fuerte utilizando paso a paso (n = 4) de elución en aumento de la fuerza iónica 19. Secar abajo muestras bajo vacío usando un concentrador SpeedVac.
- Analizar las fracciones peptídicas mediante cromatografía de nanoflujo C18 invertido en fase líquida (1100, Agilent Technologies, Santa Clara, CA) interconectados con electrospray espectrometría de masas (LC-MS/MS). Los experimentos de MS se realizaron en un híbrido lineal iontrap / transformada de Fourier resonancia ciclotrón de iones (FTICR) instrumento (LTQ FT 7T, Thermo Scientific, Waltham, MA), péptido espectros fulscan (m / z 150-2000) Fueron adquiridas con el analizador de FTICR en masas de alta resolución (100.000), seguida por la fragmentación posterior de los 5 picos de péptidos más intensos en el iontrap por medio de la colisión inducida por la disociación (CID) 9.
- Identificación de secuencia de péptidos se realiza por coincidencia de base de datos y puede ser complementado por novo análisis de secuenciación. Para la búsqueda de bases de datos, motores de búsqueda disponibles en el mercado (la mascota, XTandem o buscador de proteína) se emplean 20. Las búsquedas se realizan normalmente en bases de datos que contienen secuencias de neuropéptidos conocidos o previstos y las secuencias de proteínas precursoras neuropéptido 21.
6. Los resultados representativos
MALDI imágenes espectrometría de masas de secciones de tejido estriado como preparado de acuerdo con el protocolo descrito aquí como resultado la detección de más de 1000 picos correspondientes a aproximadamente 300 especies moleculares (monoisotopic espectros promedio muestraen la figura. 1). Visualización de los datos de prominentes picos de iones moleculares se logró utilizando el software de Flex y mostró imágenes de las distribuciones de intensidad de los picos característicos que están muy en consonancia con las características anatómicas (Fig. 3). Una característica adicional de MALDI IMS es su buena reproducibilidad relativa. En este experimento, el coeficiente global de varianza para las intensidades de los picos de todas las especies moleculares detectados fue del 30%, pero muchos picos muestran variación muy baja y alta reproducibilidad dentro de los grupos de tratamiento (Fig. 4). Los datos relativos máximos de intensidad de cuatro diferentes regiones de interés, incluyendo la parte dorsolateral y dorsomedial del striata tanto lesionado e intactos se sometieron a análisis estadístico. Con el fin de ajustar para comparaciones múltiples al mismo tiempo, el análisis estadístico se realizó mediante la prueba no paramétrica de que utilizan la herramienta SAM 18. Los cambios más notables se encuentran en la parte dorsolateral de la dopamina denervado, el cuerpo estriado de Parkinson. Aquí si cambios en gnificant entre los diferentes grupos de tratamiento se observaron dos péptidos dinorfina, dinorfina B y alfa-neoendorphin (Fig. 5). En concreto, un aumento relativo de las dos intensidades de los picos dinorfina en un 50-60% se observó en los animales de alta discinéticos en comparación con los animales bajo discinéticos y controles lesión (p <0,05, F (2, 15) = 12,8 DynB, F = 5,7 ANEO; fig. 5).

. Figura 1 medio MS traza obtenida a partir de dos regiones estrechamente relacionadas del cuerpo estriado, caudado putamen (CPU) y el núcleo accumbens (NAC). Las dos regiones muestran diferentes perfiles de MS con algunas especies moleculares únicamente expresadas en una región, o en diferentes niveles máximos de intensidad (insertar, m / z 2028). El patrón de distribución espacial de cada pico se puede visualizar utilizando el software de imágenes especializado (panel inferior).
2.jpg "/>
Figura 2 (A) El cerebro está montado sobre un mandril criostato utilizando un medio de empotrar OTC; flecha)., Se tiene cuidado de que la OTC no contamine el área de cerebro para ser seccionada desde la supresión de iones OTC causa de péptidos. (B, C) secciones delgadas (≈ 12 micras de espesor) son deshielo montado en portaobjetos de vidrio MALDI compatibles y se secó durante unos pocos segundos para evitar daños por congelación como se ve en C. (d) microdesgarros puede ser difícil de detectar a simple vista , pero poner en peligro la cristalización matriz de MALDI y borrar la señal de MALDI MS. En la misma sección se tiñeron con violeta de cresilo revela microdesgarros y grietas (fotomicrografía parte inferior derecha).

3 Figura. El primer paso en la evaluación de datos es visualizar varios picos diferentes en toda la gama de masas analizados (IA). En este caso, las secciones del cuerpo estriado de 9 ratones fueron imágenes con MALDI MS. La visualización de la media total ien la actual revelan áreas de notables fuerzas iónicas altas o bajas (flechas). Estas áreas pueden verse afectadas por exceso o por defecto-la normalización de los efectos y distorsionar el análisis de datos comprometer los resultados. Mala definición anatómica de la distribución de los picos revelan las secciones con el pico generalmente baja relación señal-ruido, por ejemplo, las secciones 3 y 9, F picos de hasta I.

Figura 4. Reproducibilidad de MS entre los grupos de tratamiento pueden ser evaluados mediante el cálculo de la media de seguimiento de MS y el error estándar para cada valor de m / z (inserciones, m / z 722 y 1749). Buena reproducibilidad asegura el análisis estadístico válido.
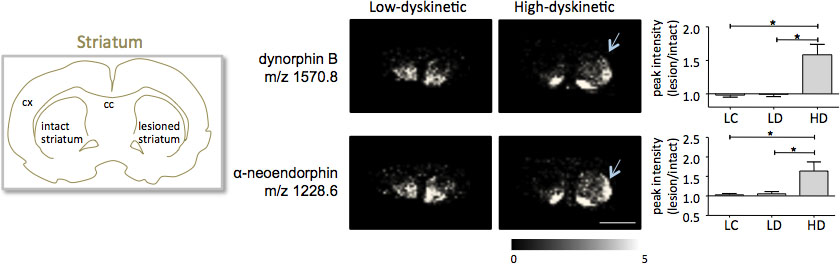
Figura 5. Dinorfina B y alfa-neoendorphin pico de intensidad se incrementó significativamente en la 6-OHDA-Lesionado, Parkinson, el cuerpo estriado de alta discinéticos animales (HD, flechas) en comparación con el grupo de control de bajo discinética (LD) y lesión (LC). Intensidades máximas de péptidos expresados como promedio de veces de cambio de lado intacto ± SEM (lesión / intacta lado). * P <0,05; cx corteza; cc cuerpo calloso. Barra de escala de 5 mm.
Discussion
Hay varias ventajas del empleo de la espectrometría de masas MALDI imágenes en el estudio de los neuropéptidos. Un análisis imparcial de los datos de MS puede revelar que los núcleos del cerebro sólo específicos, o como en los resultados presentados aquí, donde sólo la parte dorsolateral del cuerpo estriado se asocia con un estado fisiopatológico determinado. Al retener la información espacial es entonces posible volver a definir regiones de interés para el análisis estadístico con mayor sensibilidad y una menor variabilidad en comparación con el análisis de las secciones de todo el cerebro o utilizando estudios Peptidomics tradicionales en extractos peptídicos. Además, es importante darse cuenta de MALDI IMS fácilmente puede detectar previamente desconocidos modificaciones post-traduccionales, pero los análisis estructurales deben seguir para determinar las posiciones exactas de aminoácidos que están modificados.
Errores comunes en la visualización de los datos de MALDI IMS incluyen el mapeo de la intensidad del pico máximo de una escala lineal óptica de blACK (0%) a color (100%) para todos y cada sección de la serie experimental (Figura 3), en lugar de mapear todas las secciones a una escala absoluta común donde 100% es la intensidad del pico máximo de todas las secciones (Figura 5) . Este último método permite la comparación de los datos del grupo y la visualización de las diferencias entre los grupos de tratamiento.
Un obstáculo importante en el análisis de MALDI IMS es la asignación de los péptidos a los picos de comunicación específicos. En los tejidos en tándem de espectrometría de masas es a veces posible, pero a menudo resulta muy difícil 13,14. Nos parece que un enfoque más tradicional, incluyendo un fraccionamiento de preparación en la cromatografía de intercambio catiónico fuerte, seguido por LC-MS/MS de fase reversa se puede utilizar para la secuencia con éxito muchos neuropéptidos y péptidos opioides en particular. Todavía no es raro obtener una buena calidad de MS / MS espectros que no coinciden con las entradas de base de datos usando los motores de búsqueda comunes, tales como mascota. En estos casos de novo de secuenciación a mano es la ONLy opción. La última prueba de la identidad de pico se puede obtener por MALDI IMS de las secciones de tejido del ratón knock-out adecuada, pero esto no siempre está disponible o posible. Una alternativa consiste en validar los resultados mediante un método diametralmente distinto, por ejemplo, inmunotransferencia occidental o inmunohistoquímica. A menudo, esto puede incluir la producción de anticuerpos y una cantidad significativa de trabajo la validación de los nuevos anticuerpos.
La estrategia general se presenta en este protocolo está optimizado para los experimentos a gran escala neuropéptido MALDI IMS, incluyendo varias secciones y las condiciones experimentales. El protocolo se ha optimizado especialmente para los péptidos opioides y tendrá un gran impacto en futuros estudios, tal como se emplea en diversos campos de investigación, incluyendo el dolor y los mecanismos subyacentes a la respuesta endógena a las drogas de adicción.
Disclosures
Los autores no tienen nada que revelar.
Acknowledgments
Damos las gracias a Hanna Warner por su contribución a los datos de la Figura 3 y el Prof. Jonas Bergquist para sus valiosos comentarios. El Consejo Sueco de Investigación (Grant 522-2006-6416 (MA), 521-2007-5407 (MA), la Fundación La Åke Wiberg (MA, JH), La Real Academia Sueca de las Ciencias (MA, JH), y el químico sueco Sociedad (JH) se agradece por el apoyo financiero.
References
- Obeso, J. A., Olanow, C. W., Nutt, J. G. Levodopa motor complications in Parkinson's disease. Trends Neurosci. 23, S2-S7 (2000).
- Caprioli, R. M., Farmer, T. B., Gile, J. Molecular imaging of biological samples: localization of peptides and proteins using MALDI-TOF MS. MALDI-TOF MS. Anal. Chem. 69, 4751-4760 (1997).
- Obeso, J. A. The evolution and origin of motor complications in Parkinson's disease. Neurology. 55, S13-S20 (2000).
- O, W. H. Noncommunicable Diseases and Mental Health Cluster, Noncommunicable Disease Prevention and Health Promotion Department, Ageing and Life Course. Active Ageing: A Policy framework. , (2002).
- Schapira, A. H. Movement disorders: advances in cause and treatment. Lancet Neurology. , 6-7 (2010).
- Obeso, J. A., Rodriguez-Oroz, M. C., Rodriguez, M., DeLong, M. R., Olanow, C. W. Pathophysiology of levodopa-induced dyskinesias in Parkinson's disease: problems with the current model. Ann. Neurol. 47, S22-S32 (2000).
- Cenci, M. A., Lee, C. S., Bjorklund, A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur. J. Neurosci. 10, 2694-2706 (1998).
- Andersson, M., Hilbertson, A., Cenci, M. A. Striatal fosB expression is causally linked with l-DOPA-induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson's disease. Neurobiol Dis. 6, 461-474 (1999).
- Hanrieder, J. Alterations of striatal neuropeptides revealed by imaging mass spectrometry. Molecular & Cellular Proteomics. , (2011).
- Cornett, D. S., Reyzer, M. L., Chaurand, P., Caprioli, R. M. MALDI imaging mass spectrometry: molecular snapshots of biochemical systems. Nat. Methods. 4, 828-833 (2007).
- Ljungdahl, Imaging Mass Spectrometry Reveals Elevated Nigral Levels of Dynorphin Neuropeptides in L-DOPA-Induced Dyskinesia in Rat Model of Parkinson's Disease. PLoS ONE. 6, e25653 (2011).
- Groseclose, M. R., Andersson, M., Hardesty, W. M., Caprioli, R. M. Identification of proteins directly from tissue: in situ tryptic digestions coupled with imaging mass spectrometry. J. Mass. Spectrom. 42, 254-262 (2007).
- Andersson, M., Groseclose, M. R., Deutch, A. Y., Caprioli, R. M. Imaging mass spectrometry of proteins and peptides: 3D volume reconstruction. Nat. Methods. 5, 101-108 (2008).
- Deininger, S. -O. Imaging Mass Spectrometry. Setou, M. , Springer. Japan. 199-208 (2010).
- Norris, J. L. Processing MALDI Mass Spectra to Improve Mass Spectral Direct Tissue Analysis. Int. J. Mass. Spectrom. 260, 212-221 (2007).
- Ihaka, R., Gentleman, R. R. A Language for Data Analysis and Graphics. Journal of Computational and Graphical Statistics. 5, 299-314 (1996).
- Mass Spectrometry Binning Software GAB. , Vanderbilt Center for Quantitative Sciences. Nashville, TN. Available from: http://www.vicc.org/biostatistics/software.php (2012).
- Tusher, V. G., Tibshirani, R., Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 98, 5116-5121 (2001).
- Bergstrom, L., Christensson, I., Folkesson, R., Stenstrom, B., Terenius, L. An ion exchange chromatography and radioimmunoassay procedure for measuring opioid peptides and substance P. Life. Sci. 33, 1613-1619 (1983).
- Falth, M. Neuropeptidomics strategies for specific and sensitive identification of endogenous peptides. Mol. Cell. Proteomics. 6, 1188-1197 (2007).
- Falth, M. SwePep, a database designed for endogenous peptides and mass spectrometry. Mol. Cell. Proteomics. 5, 998-1005 (2006).
