


Summary
Dopamin-Substitutionstherapie Pharmakotherapie mit L-DOPA ist das am häufigsten verwendete symptomatischen Behandlung der Parkinson-Krankheit, sondern wird durch Nebenwirkungen einschließlich der zwangsweisen abnormen Bewegungen, bezeichnet Dyskinesie begleitet
Abstract
MALDI-Imaging-Massenspektrometrie (IMS) ist eine leistungsfähige Methode, die die räumliche Analyse von Molekülen in biologischen Gewebeproben 2 (Abb. 1) erleichtert. Ein 12 um dünne Gewebeschnitt mit einer MALDI-Matrix die Desorption und Ionisation von intakten Peptiden und Proteinen, die mit einem Massenanalysator detektiert werden, kann typischerweise unter Verwendung eines MALDI-TOF / TOF-Massenspektrometer ermöglicht bedeckt. Im Allgemeinen Hunderte von Peaks in einem einzigen Rattenhirngewebe Abschnitt bewertet werden. Im Gegensatz zu den häufig verwendeten bildgebenden Verfahren geht diese Vorgehensweise nicht erforderlich, eine vorherige Kenntnis der interessierenden Moleküle und erlaubt für den unüberwachten und umfassende Analyse von mehreren molekularen Spezies unter Beibehaltung der hohen molekularen Spezifität und Sensitivität 2. Hier beschreiben wir ein MALDI IMS basierenden Ansatz für die Aufklärung Region-spezifische Verteilung Profile von Neuropeptiden im Gehirn der Ratte von einem Tiermodell der Parkinson-Krankheit (PD).
PD ist eine häufige neurodegenerative Erkrankung mit einer Prävalenz von 1% für Personen über 65 Jahre alt 3,4. Die häufigste symptomatische Behandlung basiert auf Dopamin-Ersatz mit L-DOPA-5 basiert. Dies wird jedoch durch schwere Nebenwirkungen einschließlich der zwangsweisen abnorme Bewegungen, genannt L-DOPA-induzierten Dyskinesien (LID) 1,3,6 begleitet. Einer der prominentesten molekulare Veränderung in LID ist ein Hochregulation der Opioid-Vorläufer Prodynorphin mRNA-7. Die Dynorphin Peptide modulieren Neurotransmission im Gehirn Bereiche, die im Wesentlichen in Bewegungssteuerung 7,8 beteiligt sind. Allerdings sind die genauen Opioid-Peptide, die aus der Verarbeitung von dem Neuropeptid Vorläufer stammen bisher nicht charakterisiert worden. Daher verwendeten wir MALDI IMS in einem Tiermodell experimenteller Parkinson-Krankheit und L-DOPA induzierter Dyskinesie.
MALDI-Imaging-Massenspektrometrie erwiesen sich als besonders vorteilhaft im Hinblick auf Neuropeptid Charakterisierungtion, da häufig verwendete Antikörper Ansätze zielt bekannten Peptid-Sequenzen und zuvor beobachtet post-translationale Modifikationen. Im Gegensatz MALDI IMS kann entwirren neuartige Peptid-Processing-Produkten und damit zeigen neue molekulare Mechanismen der Neuropeptid Modulation der neuronalen Übertragung. Während die absolute Höhe der Neuropeptide können nicht durch MALDI IMS, bestimmt die relative Häufigkeit von Peptid-Ionen aus den Massenspektren abgegrenzt werden, geben Einblicke über das Ändern von Ebenen in Gesundheit und Krankheit. In den hier vorgestellten Beispielen wurden die Intensitäten von Dynorphin B, alpha-neoendorphin und Substanz P vor, signifikant in den dorsolateralen erhöht werden, aber nicht die dorsomedialen, Striatum von Tieren mit schwerer Dyskinesie mit Gesichts, des Rumpfes und orolingual Muskeln (Abb. 5). Darüber hinaus ergab MALDI IMS eine Korrelation zwischen Schweregrad und Dyskinesie Ebenen des-Tyrosin-alpha-neoendorphin, was einen bisher unbekannten Mechanismus der funktionellen Inaktivierungvation der Dynorphine im Striatum als die Entfernung von N-terminalen Tyrosin reduziert die Dynorphin der Opioid-Rezeptor-Bindung Kapazität 9. Dies ist die erste Studie über Neuropeptid Charakterisierung mittels MALDI LID in IMS und die Ergebnisse unterstreichen das Potenzial der Technik für die Anwendung in allen Bereichen der biomedizinischen Forschung.
Protocol
Das Protokoll wird für die Zwecke der statistischen Auswertung von MALDI IMS Daten aus mehreren Rattenhirnschnitten, typischerweise 20-30 Sektionen angepasst und besteht aus fünf verschiedenen Stufen umfasst Präparation von Gewebe-, Matrix-Anwendung, MALDI-TOF-MS-Analyse, Auswertung und Neuropeptid Identifikation. Die Verfahren werden skizziert und beschrieben in detaillierter unter:
1. Gewebepräparation
Dieses Verfahren umfasst die Sammlung der entsprechenden Gewebeproben sowie Gewebe-Schnitte für IMS-Analyse. Ein besonderes Ziel in Protein-und Peptid-Analyse ist es, proteolytischen Abbau zu vermeiden. Deshalb ist es wichtig, schnell und fleißig während Gewebedissektion arbeiten.
- Opfere Ratten (typischerweise 250-300 g) durch Enthauptung, zu entfernen Rattenhirn innerhalb von maximal Post-mortem-Zeit von <30 s und Einfrieren der pulverisierten Trockeneis vor der Übertragung auf -80 ° C Gefrierschrank. Schnelleres Einfrieren mit flüssigem Stickstofferhöhen das Risiko von Mikrorissen im Hirngewebe, welche sich in den Matrix-Kristallisation und somit die Verringerung MS Qualität (Abb. 2D). Ganze Gehirn kann mehrere Jahre vor dem Schneiden ohne Verlust der Signalqualität MS gespeichert werden.
- Schneiden Sie gefrorenem Gewebe auf einem Kryostat-Mikrotom bis 12 um Scheiben und Tauwetter-Mount Gewebeschnitte auf leitfähigen MALDI Glasträger (Indium-Zinnoxid beschichtete Objektträger, Bruker Daltonics) oder MALDI-Target (Abb. 2A-C).
- Trockene Abschnitte für 15 min unter Vakuum und Objektträger bei -80 ° C bis zur weiteren Verwendung. Gewebeschnitte sollten innerhalb der kürzest möglichen Zeit nach dem Schneiden analysiert werden, auch wenn bei -80 ° C Wir finden, dass MS-Signal Qualität spürbar werden nach einem Jahr bei der Lagerung reduziert werden. Um eine Oxidation von Proteinen und Peptiden zu verringern, kann die Luft im Vorratsbehälter mit einem inerten Gas (zB Argon oder Stickstoff) ersetzt werden.
2. Matrix-Anwendung
Die Matrix-AnwendungSchritt hat einen signifikanten Einfluss auf Spektrenqualität und erfordert Optimierung mehrerer Parameter in Abhängigkeit von der Art des Gewebes sowie den Analyten von Interesse. Zu diesen Faktoren zählen chemische Parameter wie die Art von Matrix, Matrix-Konzentration, pH-, Gewebe-Wasch-und organische Modifier sowie Instrumental-Einstellungen einschließlich der Einlagevolumen, laterale Auflösung und die Anzahl der Ablagerungen 10 (Abb. 2D). Für Großversuche, ist es von großer Bedeutung, um die Varianz zu reduzieren, beispielsweise durch Anwendung der Matrix auf allen Abschnitten innerhalb eines Tages und vom selben Betreiber. Obwohl es viele Strategien zur Matrixlösung, wie durch Sublimation oder durch Sprühtrocknung, das automatisierte Abscheidung von Anordnungen von kleinen Tröpfchen Matrix, etwa 100-150 Picoliter in der Größe, wurde erfolgreich für die Analyse von kleinen Proteinen und Neuropeptide in verschiedenen Geweben verwendet gelten , einschließlich Gehirn § § 9, 10,11, 12, 13.
- Abtauen die Abschnitte in einem Exsikkator für 1 hunsere.
- Stellen Sie sicher, dass das Experiment von einer anderen Person als dem Betreiber wird geblendet. Re-Label alle Proben.
- Waschen Segmente 1 in 70% Ethanol (EtOH, bei Raumtemperatur RT) für 10 Sekunden und zweimal in 95% EtOH (RT) für 10 sek. Für große Experimente, führen Sie die Wäsche für alle Glas-Folien zusammen mit einer Küvette, um Schwankungen zu minimieren.
- Trocknen der Abschnitte in einem Exsikkator für 10 min.
- Bewerten Gewebeschnitten unter dem Mikroskop zu überprüfen und für das Tissue Verzerrung, Mikrorisse und kleine Risse, die MALDI-MS-Qualität (Abb. 2D) beeinträchtigt wird.
- Ansetzen Matrix Lösung bestehend aus 50 mg / ml DHB in 50% Methanol, 10% 150 mM Ammoniumacetat (AMAC) und 0,3% Trifluoressigsäure (TFA) in Wasser.
- Matrix-Anwendung wird von diskreten Tröpfchen-Abscheidungs in einem rechteckigen Muster unter Verwendung eines chemischen Tintenstrahldrucker (CHIP, Shimadzu) durchgeführt. Der erste Schritt ist, um die experimentellen Parameter der Matrix-System für Neuropeptid-Analyse includi optimierenng die Anzahl der Tröpfchen pro Durchgang, Anzahl der Durchläufe. Dieses Experiment wird durch die Anwendung mehrerer Matrix-Arrays mit unterschiedlichen Parametern Anwendung auf dem gleichen Gewebeschnitt, gleichzeitig aber dafür sorgen, dass jedes Array, das ähnliche Hirnregionen wie Balken, Cortex und Striatum durchgeführt. Das gleiche Experiment muss jedes Mal durchgeführt Parameter geändert werden, einschließlich verschiedener Hirnstrukturen, verschiedene Matrices Targeting bestimmter Analyte und falls abweichend Matrix Lösungsmittel für die Extraktion von spezifischen Analyten erforderlich sein.
- Scannen Sie das Glas Diahalter mit dem Gewebeschnitt und richten Sie den Halter. Definieren Sie Ihre Array für Matrix-Anwendung auf dem Gewebeschnitt und geben Sie die räumliche Auflösung, dh Ort, um Abstand zu erkennen. Bewerben Matrix mit der optimierten Protokoll über die chemische Tintenstrahldrucker. Für dieses Experiment haben wir ein optimiertes Protokoll für die Peptid-Bildgebung mit den folgenden Druck-Parameter verwendet: 10 Tropfen (100 pL / Drop), übergibt 10-Anwendung und ein Ort, um d zu erkennenistance von 300 um.
- Scan endgültige Matrix gesichtet Abschnitte und speichern Sie das Bild für die Registrierung vor der MALDI Datenerfassung (Schritt 3.4).
- Lagern Sie die Teile bis zur weiteren Verwendung in einem Exsikkator unter Vakuum.
3. MALDI-MS-Datenerfassung und-verarbeitung
MS-Analyse von Neuropeptiden basiert auf einem MALDI-Flugzeit-Instrument (Ultraflex II, Bruker Daltonics, Deutschland), die in Reflektor-Modus durchgeführt, unter Verwendung von Software gestützte Datenerfassung von jedem einzelnen Matrix-Spot 14. Deshalb genaue räumliche Lehre ist zwingend erforderlich. Es ist wichtig, dass der MALDI-Optimierung, Erwerb und vor allem die Ziel-Registrierung Versuche vom selben Betreiber, die vorzugsweise mit den experimentellen Gruppen blenden lassen sollte durchgeführt werden. In einem großen Experiment mit mehreren Glasplatten können die MALDI Experimente von einem Bediener durchgeführt werden, während eine andere Person den Betrieb der chemischen Tintenstrahldrucker.
- Legen Glasträger in das Massenspektrometer.
- Überprüfen Sie die Kalibrierung des MALDI Erwerb Methode unter Verwendung eines niedermolekularen Standardkalibrierung Mix (Bruker Daltonics).
- Optimieren Sie Aufnahme-Parameter.
- Um MS-Signal zu optimieren und zu vermeiden, Abtragen Matrix-Matrix von benachbarten Einlagen, sollte die Größe des Lasers und der optimalen Fokus auf Gewebe bestimmt werden.
- Die Laserenergie wird gesetzt, um die maximale Qualität von MS so viele Matrix-Ablagerungen wie möglich zu gewährleisten, ohne Erhöhung der Grundlinie, die Verringerung Peak-Auflösung oder Sättigung des Detektors.
- Beurteilen Sie die maximale Anzahl der Aufnahmen pro Matrix-Spot, bis nur noch Rauschen erkannt wird, oft 1000-2000 Schüsse. Schätzen Sie die Anzahl der Aufnahmen, die angesammelt werden und die Anzahl der Schüsse vor der Laser-Position in einem Ort erworben sollte sich ändern sollte. Um jeden Punkt Matrix gleichmäßig zu probieren, reichern wir 600 Aufnahmen in 25 Schuss Schritte, für eine Gesamtzahl von 24 Schritten mit Hilfe eines RANdom Bewegungsmuster von jeder Matrix Abscheidung.
- Registrieren Sie den Scan aller Sektionen gesichtet, um Motor-Koordinaten des MALDI-Bühne mit dem FlexImaging (v.2.0) 10 und führen die Datenerfassung im Batch-Modus durch AutoXexuteBatchRunner.exe Software.
- Verarbeiten jeden einzelnen Spektren durch Subtraktion Baseline (Konvexe Hülle V3), Glättung und externe Kalibrierung (optional), durch den Export als ASCII-Datei (*. Dat, *. Txt oder *. Csv-Format) gefolgt. 15
4. Die Auswertung der Daten
Abschließende Auswertung der Daten umfasst Daten-Nachbearbeitung und Datenreduktion von Focus nur an Spitzentagen Informationen, die durch statistische Analyse.
- In einem ersten Schritt wurden die MALDI IMS Abschnitte für overnormalization Wirkungen ausgewertet. Dies kann leicht durch den Einsatz von Daten Visualisierungs-Tools wie FlexImaging (Bruker Daltonics) oder BioMap (Novartis) erreicht werden. In einem ersten Schritt die Summe der Ionen-Bilder sind evaluated vor Totalionenstrom (TIC) Normalisierung, die durch manuelle Inspektion der einzelnen Ionenverteilung Bilder von verschiedenen prominenten Peptidpeaks gefolgt. Suchen Sie nach charakteristischen Peak Intensitätsverteilungen und wenn sie an Gewebemerkmale (Schadenersatz) verwandt, Schmierblutungen Qualität oder Normalisierung Effekte (Abb. 3).
- Abgrenzung von Regionen Interessen (z. B. das Striatum) nach histologischen Merkmale und exportieren Sie die entsprechenden Spektren im ASCII-Dateiformat. Vorzugsweise können die Normalisierung der Spektren auf das Gesamtgewicht Ionenstrom (TIC) zu diesem Zeitpunkt durchgeführt werden.
- ASCII-Dateien in einem Daten-Handling-Software wie Origin (V.8.1, Originlab), MATLAB (MathWorks, Natick, MA, USA) oder R 16. Peak-Erkennung kann mit Spitzensuche Tools in der Software enthalten sein, zum Beispiel "Peak-Analyse" in Origin oder "mspeaks" in Matlab. Exportieren Sie die peaklists aus allen Spektren als eine einzige Registerkarte Textdatei.
- Um bin Grenzen für erkannte Peptid bestimmenGipfeln wird Binning Analyse unter Verwendung geeigneter Software-Werkzeuge (z. B. pBIN 17) oder in-house geschriebene Skripte für MATLAB oder R. Hier wird die Textdatei mit allen Spitzen gepflückt Daten werden in die Software geladen und die Parameter für die Peak-Grenze Bestimmung angegeben wie, wie oft ein Höhepunkt sollte in Spektren, um relevant zu sein für das Experiment. Zum Beispiel enthält das Experiment 2-Gruppen von Tieren, 5 Tiere in jeder Gruppe, und 100-Spektren von jedem Tier und Region von Interesse gesammelt. Angenommen, ein Spitzenwert ist potentiell interessant, wenn sie in mindestens der Mehrheit der Tiere in einer Gruppe (3/5) und in mindestens der Hälfte der Spektren dieser Tiere (3x50 = 150 Spektren) ist, ergibt dies einen Gesamtanteil von 15% für die 150 positiven Spektren von insgesamt 1000 (2x5x100) Spektren. Mit dem Werkzeug pBIN, ergibt dieser Schritt eine einzelne Datei mit allen binrange bin Breiten von den erfassten Daten bestimmt. Um zu überprüfen, dass Bin Grenzenangemessen sind, ist es einfach, die Behälter in Origin gemeinsam visualisieren mit den Original-Spektren Spuren.
- Peak-Bereich Integration zu reduzieren Varianz, die wichtig ist für die statistische Analyse. Wir verwenden ein eigenes geschriebenes Skript für R, um die Fläche unter der Kurve zwischen Spitzen-Grenzen in Schritt 4 ermittelt berechnen. Integrierten Peakflächen werden in MS Excel (v.2007) und statistische Analysen mit Hilfe von nicht-parametrischen Tests unter Verwendung des ungepaarten SAM-Tool durchgeführt wird, 18 importiert.
5. Peptid-Identifikation
Sequenz Überprüfung der Identität beobachteten Peptid ist, um biologische Relevanz schließen wesentlich. Die genaueste Methode zählen wahre Top-down-Bestimmung direkt aus Gewebe mittels Peptid-Fragmentierung mittels Tandem-Massenspektrometrie (MS / MS), obwohl hohen Peptidkonzentrationen für diese Art der Analyse 12,13 erforderlich. Für niedrige reichlich Peptide oder mehrere Peptide mit closem / z-Werte (± 0,5%), basiert auf Analyse-Gewebe beeinträchtigt und aus Gewebe Analyse unter Verwendung einer peptidomic Strategie wird ausgenutzt, dass zählt die Gewinnung, Trennung und MS-basierte Identifikation endogener Neuropeptide. Für das Experiment hier präsentierten, war der zentrale Fokus auf Opioid-Peptid-Erkennung, die eine besondere Herausforderung ist, da diese Peptide ziemlich niedrig sind reichlich vorhanden im Vergleich zu anderen Neuropeptiden in den Spektren. Darüber hinaus sind diese Peptide nicht polaren, welche auch relativ hydrophil und schwer mit gemeinsamen Peptidextraktion und Trenntechniken erhalten .. Deshalb haben wir mit einer zuvor berichtet Protokoll für die Gewebeentnahme und Opioidpeptid Vorfraktionierung in Kombination mit Standard-basiertes Peptid LC-MS/MS Identifikation 9,19.
- Sammle koronalen Abschnitte der Zielstrukturen von Interesse (Nucleus accumbens, NAC; caudatus Putamen, CPU). Montieren Sie gefrorene Rattenhirn in einem Kryostat-Mikrotom und entfernen umliegenden Gehirn Material (AdRtex, Septum, Corpus callosum) mit einem Skalpell. Sammle Abschnitte (30 &mgr; m; n = 50) der sezierten NAC und CPU-und Tauwetter montieren die NAC-und CPU-Teile der Abschnitte auf verschiedene Glasplatten.
- Auszug aus Peptiden Gewebe durch Zugabe von 100 ul 5% ACN/0.1% TFA, inkubieren für zwei Minuten und sammeln in Eppendorf geringe Proteinbindung Röhren. Wiederholen Sie diesen Schritt zweimal.
- Führen Peptid Vorfraktionierung durch starke Kationenaustauschchromatographie unter Verwendung schrittweise (n = 4) Elution bei erhöhte Ionenstärke 19. Trocknen Sie sich Proben unter Vakuum mit Hilfe eines Speedvac Konzentrator.
- Analysieren Sie die Peptid-Fraktionen mittels Nanoflow C18 Umkehrphasen-Flüssigchromatographie (1100, Agilent Technologies, Santa Clara, CA) Schnittstelle mit Elektrospray-Tandem-Massenspektrometrie (LC-MS/MS). Die MS-Experimente wurden an einem Hybrid-lineare Ionenfallen / Fourier-Transform-Ionen-Zyklotron-Resonanz (FTICR) Instrument (LTQ FT 7T, Thermo Scientific, Waltham, MA,) Peptid fulscan Spektren (m / z 150 durchgeführt-2000) Wurden mit dem Analysator FTICR bei hoher Massenauflösung (100000) durch anschließende Fragmentierung der 5 intensivsten Peaks Peptid in der Ionenfallen durch Kollision herbeigeführte Dissoziation (CID) 9 an erworben.
- Peptidsequenz Identifizierung wird durch Datenbank-Abgleich durchgeführt und können von de-novo-Sequenzierung Analyse ergänzt werden. Für Datenbank-Suche, im Handel erhältlich sind Suchmaschinen (Mascot, XTandem oder Protein Prospector) 20 beschäftigt. Die Recherchen werden in der Regel gegen Datenbanken mit Sequenzen von bekannten oder vorhergesagten Neuropeptide und Sequenzen von Neuropeptid Vorläufer-Proteine 21 durchgeführt.
6. Repräsentative Ergebnisse
MALDI-Imaging-Massenspektrometrie der striatalen Gewebeschnitten, die gemäß der hier beschriebene Protokoll für den Nachweis von mehr als 1000 Peaks, die etwa 300 monoisotopischen molekulare Spezies (durchschnittliche gezeigten Spektren zur Folge hattein Fig.. 1). Datenvisualisierung für prominente Molekülion Gipfel wurde erreicht mit dem Flex Imaging Software und zeigte charakteristische Peakintensität Distributionen, die gut sind im Einklang mit anatomischen Merkmalen (Abb. 3). Ein weiteres Merkmal der MALDI IMS ist seine relative gute Reproduzierbarkeit. In diesem Experiment war die Gesamt-Variationskoeffizient für die Peak-Intensitäten aller entdeckten molekularen Spezies 30%, aber viele Spitzen angezeigt mit geringen Schwankungen und hohe Reproduzierbarkeit innerhalb Behandlungsgruppen (Abb. 4). Die relativen Peak-Intensität Daten aus vier verschiedenen Regionen von Interesse, einschließlich der dorsolateralen und dorsomedialen Teil der beide verletzten und intakten striata wurden einer statistischen Analyse unterzogen. Um für mehrere Vergleiche gleichzeitig anzupassen, wurde die statistische Analyse mittels parametrischer Tests mit dem SAM-Werkzeug 18 durchgeführt. Die wichtigsten Änderungen wurden im dorsolateralen Teil des Dopamin-denervierten, Parkinson Striatum gefunden. Hier si Dynorphin B und Alpha-neoendorphin (Abb. 5); gnificant Änderungen zwischen den verschiedenen Behandlungsgruppen wurden für zwei Dynorphin Peptide beobachtet. Im Einzelnen eine relative Erhöhung beider Dynorphin Peakintensitäten um 50-60% in hoher dyskinetische Tieren im Vergleich zu niedrigen dyskinetische Tieren und Kontrollen Läsion (beobachtet p <0,05, F (2, 15) = 12,8 DynB, F = 5,7 Aneo; Abb.. 5).

. Abbildung 1 Durchschnittliche MS Spuren erhalten aus zwei eng verwandte Bereiche des Striatum, Putamen caudatus (CPU) und Nucleus accumbens (NAC). Die beiden Bereiche jeweils unterschiedliche MS-Profile mit einigen molekularen Spezies einzigartig in einer Region exprimiert, oder in verschiedenen Ebenen Peakintensität (Einsatz, m / z 2028). Das räumliche Verteilungsmuster der einzelnen Peaks können visualisiert werden mithilfe spezieller Bildbearbeitungssoftware (unten).
2.jpg "/>
Abbildung 2 (A) Das Gehirn ist auf einem Kryostat mit Chuck eine Einbettung Medien (OTC; Pfeil) montiert. Wird darauf geachtet, dass der OTC nicht verunreinigt den Bereich des Gehirns zu sein, da die Ursache OTC-Ionen-Unterdrückung von Peptiden geschnitten. (B, C) Dünnschnitte (≈ 12 um Dicke) für die MALDI kompatibel Glasobjektträger Tau-montiert und getrocknet für ein paar Sekunden, um Frostschäden zu vermeiden, wie in C (D) gesehen Mikrorisse kann schwierig sein, mit dem bloßen Auge erkannt , sondern beeinträchtigen MALDI-Matrix Kristallisation und auslöschen MALDI-MS-Signal. Der gleiche Abschnitt mit Kresylviolett gefärbt zeigt Mikrorisse und Risse (rechts unten Mikrophotographie).

3. Der erste Schritt bei der Datenauswertung ist, mehrere verschiedene Peaks über den Massebereich analysiert (AI) zu visualisieren. Hier wurden striatalen Abschnitten von 9 Mäusen, die mit MALDI-MS abgebildet. Visualisierung des durchschnittlichen Gesamtkosten idie auf den gegenwärtigen enthüllt Bereichen auffällig hohen oder niedrigen Ionenintensitäten (Pfeile). Diese Bereiche können durch Über-oder Unter-Normalisierung und Effekte verzerren Datenanalyse gefährden die Ergebnisse beeinflusst werden. Schlechte anatomische Definition der Peak-Distributionen zeigen Abschnitte mit allgemein niedrigen Peak-Signal-Rausch-, z. B. § § 3 und 9, Spitzen F bis I

Abbildung 4. MS Reproduzierbarkeit zwischen den Behandlungsgruppen kann durch Berechnung des durchschnittlichen MS Spur und die Standardfehler für die einzelnen m / z-Wert (Einsätze, m / z 722 und 1749) beurteilt werden. Gute Reproduzierbarkeit gewährleistet gültige statistische Analyse.
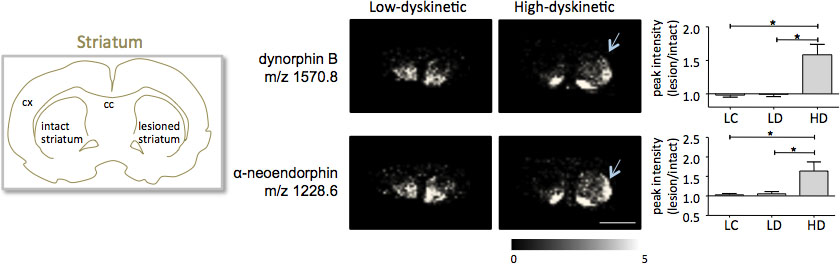
Bild 5. Dynorphin B und alpha-neoendorphin Peakintensitäten sind deutlich in der 6-OHDA erhöhte-Behandelten, Parkinson, Striatum von High-dyskinetische Tiere (HD, Pfeile) an die Low-dyskinetische (LD) und Läsion Kontrollgruppe (LC) verglichen. Peptid-Peak-Intensitäten als durchschnittlich fach-Wechsel der intakten Seite ± SEM (Läsion / intakt auf dieser Seite) ausgedrückt. * P <0,05; cx Kortex; cc Corpus callosum. Maßstab 5 mm.
Discussion
Es gibt mehrere Vorteile des Einsatzes von MALDI-Massenspektrometrie Bildgebung in der Studie von Neuropeptiden. Eine unvoreingenommene Analyse der MS-Daten können zeigen, dass nur bestimmte Gehirnkernen oder wie in den Ergebnissen hier, wo nur in den dorsolateralen Teil des Striatums mit einer bestimmten pathophysiologischen Zustands zugeordnet ist dargestellt. Durch die Beibehaltung des räumlichen Informationen ist es dann möglich, Regions of Interest neu zu definieren, um statistische Analysen mit höherer Empfindlichkeit und geringere Variabilität im Vergleich mit der Analyse des gesamten Gehirns Teile oder mit traditionellen Peptidomics Studien über Peptid-Extrakte durchzuführen. Darüber hinaus ist es wichtig zu erkennen, MALDI IMS ohne weiteres bisher unbekannte post-translationale Modifikationen erkennen, aber Strukturanalysen folgen, um die genauen Aminosäurepositionen, die sich ändern zu bestimmen.
Häufige Probleme bei der Visualisierung der MALDI IMS-Daten umfassen Abbilden der maximale Peak-Intensität zu einer linearen optischen Skala von black (0%) der Farbe (100%) für jeden Abschnitt in der Versuchsreihe (3) anstelle des Abbildens alle auf eine gemeinsame Absolutskala wobei 100% die maximale Peak-Intensität aller Abschnitte (5) . Die letztere Methode erlaubt den Vergleich der Gruppe Daten und die Visualisierung der Unterschiede zwischen den Behandlungsgruppen.
Ein großes Hindernis bei der MALDI-IMS-Analyse ist die Zuordnung von Peptiden auf bestimmte Massenpeaks. On-Gewebe-Tandem-Massenspektrometrie ist es manchmal möglich, aber erweist sich oft als recht schwierig 13,14. Wir finden, dass eine herkömmliche Ansatz mit einer präparativen Fraktionierung auf einer starken Kationenaustauschchromatographie, durch Umkehrphasen-LC-MS/MS gefolgt erfolgreich Sequenz vielen Neuropeptiden und insbesondere Opioid Peptide können verwendet werden. Es ist noch nicht ungewöhnlich, dass gute Qualität MS / MS-Spektren, die nicht übereinstimmen alle Datenbankeinträge mit gängigen Suchmaschinen wie MASCOT zu erhalten. In diesen Fällen De-novo-Sequenzierung von Hand ist die ONLy-Option. Der endgültige Beweis für Peak Identität kann durch MALDI IMS von Gewebeschnitten aus der entsprechenden Knockout-Maus erhalten werden, aber dies ist nicht immer verfügbar oder möglich ist. Eine Alternative ist, um die Ergebnisse durch eine diametral andere Methode zu validieren, zum Beispiel durch Western Immunoblotting oder Immunhistochemie. Diese können oft von Antikörpern und eine erhebliche Menge an Arbeit Validierung der neuen Antikörper.
Die allgemeine Strategie in diesem Protokoll vorgestellt wird, für groß angelegte Neuropeptid MALDI IMS Experimente, darunter mehrere Abschnitte und experimentellen Bedingungen optimiert. Das Protokoll wurde speziell für Opioidpeptide optimiert worden und wird große Auswirkungen in zukünftigen Studien haben, wie es in den unterschiedlichsten Bereichen der Forschung, einschließlich der Mechanismen, die Schmerzen und die körpereigene Reaktion auf Medikamente von Sucht beschäftigt.
Disclosures
Die Autoren haben nichts zu offenbaren.
Acknowledgments
Wir bedanken uns bei Hanna Warner für einen Beitrag die Daten für die Abbildung 3 und Prof. Jonas Bergquist für wertvolle Anregungen. Schwedischer Forschungsrat (Grant 522-2006-6416 (MA), 521-2007-5407 (MA); der AKE Wiberg Stiftung (MA, JH), Die Königlich Schwedische Akademie der Wissenschaften (MA, JH), und die schwedische Chemical Society (JH) sind dankbar für finanzielle Unterstützung.
References
- Obeso, J. A., Olanow, C. W., Nutt, J. G. Levodopa motor complications in Parkinson's disease. Trends Neurosci. 23, S2-S7 (2000).
- Caprioli, R. M., Farmer, T. B., Gile, J. Molecular imaging of biological samples: localization of peptides and proteins using MALDI-TOF MS. MALDI-TOF MS. Anal. Chem. 69, 4751-4760 (1997).
- Obeso, J. A. The evolution and origin of motor complications in Parkinson's disease. Neurology. 55, S13-S20 (2000).
- O, W. H. Noncommunicable Diseases and Mental Health Cluster, Noncommunicable Disease Prevention and Health Promotion Department, Ageing and Life Course. Active Ageing: A Policy framework. , (2002).
- Schapira, A. H. Movement disorders: advances in cause and treatment. Lancet Neurology. , 6-7 (2010).
- Obeso, J. A., Rodriguez-Oroz, M. C., Rodriguez, M., DeLong, M. R., Olanow, C. W. Pathophysiology of levodopa-induced dyskinesias in Parkinson's disease: problems with the current model. Ann. Neurol. 47, S22-S32 (2000).
- Cenci, M. A., Lee, C. S., Bjorklund, A. L-DOPA-induced dyskinesia in the rat is associated with striatal overexpression of prodynorphin- and glutamic acid decarboxylase mRNA. Eur. J. Neurosci. 10, 2694-2706 (1998).
- Andersson, M., Hilbertson, A., Cenci, M. A. Striatal fosB expression is causally linked with l-DOPA-induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson's disease. Neurobiol Dis. 6, 461-474 (1999).
- Hanrieder, J. Alterations of striatal neuropeptides revealed by imaging mass spectrometry. Molecular & Cellular Proteomics. , (2011).
- Cornett, D. S., Reyzer, M. L., Chaurand, P., Caprioli, R. M. MALDI imaging mass spectrometry: molecular snapshots of biochemical systems. Nat. Methods. 4, 828-833 (2007).
- Ljungdahl, Imaging Mass Spectrometry Reveals Elevated Nigral Levels of Dynorphin Neuropeptides in L-DOPA-Induced Dyskinesia in Rat Model of Parkinson's Disease. PLoS ONE. 6, e25653 (2011).
- Groseclose, M. R., Andersson, M., Hardesty, W. M., Caprioli, R. M. Identification of proteins directly from tissue: in situ tryptic digestions coupled with imaging mass spectrometry. J. Mass. Spectrom. 42, 254-262 (2007).
- Andersson, M., Groseclose, M. R., Deutch, A. Y., Caprioli, R. M. Imaging mass spectrometry of proteins and peptides: 3D volume reconstruction. Nat. Methods. 5, 101-108 (2008).
- Deininger, S. -O. Imaging Mass Spectrometry. Setou, M. , Springer. Japan. 199-208 (2010).
- Norris, J. L. Processing MALDI Mass Spectra to Improve Mass Spectral Direct Tissue Analysis. Int. J. Mass. Spectrom. 260, 212-221 (2007).
- Ihaka, R., Gentleman, R. R. A Language for Data Analysis and Graphics. Journal of Computational and Graphical Statistics. 5, 299-314 (1996).
- Mass Spectrometry Binning Software GAB. , Vanderbilt Center for Quantitative Sciences. Nashville, TN. Available from: http://www.vicc.org/biostatistics/software.php (2012).
- Tusher, V. G., Tibshirani, R., Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 98, 5116-5121 (2001).
- Bergstrom, L., Christensson, I., Folkesson, R., Stenstrom, B., Terenius, L. An ion exchange chromatography and radioimmunoassay procedure for measuring opioid peptides and substance P. Life. Sci. 33, 1613-1619 (1983).
- Falth, M. Neuropeptidomics strategies for specific and sensitive identification of endogenous peptides. Mol. Cell. Proteomics. 6, 1188-1197 (2007).
- Falth, M. SwePep, a database designed for endogenous peptides and mass spectrometry. Mol. Cell. Proteomics. 5, 998-1005 (2006).
